
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study on the performance of micro-flow injection preconcentration method on-line coupled to thermospray flame furnace AAS using MWCNTs wrapped with polyvinylpyridine nanocomposites as adsorbent
César Ricardo Teixeira Tarley *ab,
Kristiany Moreira Diniza,
Fabio Antonio Cajamarca Suquilaa and
Mariana Gava Segatellia
*ab,
Kristiany Moreira Diniza,
Fabio Antonio Cajamarca Suquilaa and
Mariana Gava Segatellia
aUniversidade Estadual de Londrina (UEL), Departamento de Química, Centro de Ciências Exatas, Rodovia Celso Garcia Cid, PR 445, km 380, Londrina, PR 86050-482, Brazil. E-mail: tarley@uel.br; Fax: +55 43 3371 4286; Tel: +55 43 3371 4366
bInstituto Nacional de Ciência e Tecnologia (INCT) de Bioanalítica, Universidade Estadual de Campinas (UNICAMP), Instituto de Química, Departamento de Química Analítica, Cidade Universitária Zeferino Vaz, s/n, Campinas, SP 13083-970, Brazil
First published on 31st March 2017
Abstract
This work describes the synthesis and characterization of a new adsorbent nanocomposite based on multi-walled carbon nanotubes (MWCNTs) and polyvinylpyridine and its use in the development of a highly sensitive micro-flow injection preconcentration method coupled to TS-FF-AAS for the determination of very low levels of Cd. The characterization of the nanocomposite was performed by FT-IR, TGA, SEM, TEM and textural data measurements using the BET and BJH methods. The optimized conditions of the micro-flow injection preconcentration method involved a preconcentrated sample (8.8 mL) at pH 8.0 flowed through 50.0 mg of nanocomposite packed into a mini-column at a flow rate of 4.4 mL min−1. The on-line elution of Cd(II) ions towards the TS-FF-AAS detector was carried out in countercurrent at a flow rate of 0.6 mL min−1 using 1.0 mol L−1 HCl. The analytical curve ranged from 0.12 to 6.0 μg L−1 (r = 0.997), and an enhancement factor of 19.5, sample throughput of 15 h−1 and consumptive index of 0.45 mL were achieved. In terms of sensitivity, the uniquely high adsorption capacity of the nanocomposite was confirmed by the low limit of detection (36 ng L−1) achieved after implementing the preconcentration step in TS-FF-AAS. In terms of selectivity, the proposed method was shown to be tolerable to several foreign ions and applicable to different kinds of waters (tap, mineral, lake, and synthetic seawater), cigarettes, a food sample (powder chocolate), a medicinal herb (Ginkgo biloba) and a certified reference material (fish protein DORM-3).
1. Introduction
Cd (Cd) is a very toxic element for animals and humans, even at low concentrations, and acts as a human carcinogen.1,2 Due to the increase in human activities, the transfer of Cd into human beings has become increasingly common and occurs through food, plants, drinking water and air.3 Therefore, due to its toxic characteristics, the maximum level of Cd in potable water has been set as 5.0 μg L−1 by the US Environmental Protection Agency (US EPA)4 and Brazilian Health Surveillance Agency (ANVISA).5 Another well-known source of Cd uptake by humans beings is from cigarette smoke since tobacco leaves accumulate high levels of Cd.6 Some reports have demonstrated that medicinal herbs such as Gingko biloba as well as chocolate, mainly chocolate rich in cocoa, might be a significant source of Cd ingestion.7,8 Thus, the determination of Cd in different kinds of samples is of particular interest for quality control and environmental monitoring. However, due to the very low Cd content along with the differences between matrices in the samples, the development of reliable methods capable of directly analyzing Cd in different kinds of samples is usually a difficult task.9 Hence, the pretreatment of samples using preconcentration methods is highly recommended for reliable analysis, especially if one considers the use of analytical techniques with high sensitivity, such as flame atomic absorption spectrometry (FAAS).10–12Among the preconcentration methods including solid phase extraction (SPE), co-precipitation and micro liquid–liquid extraction, the first is still the most used method for metal ion extraction and is mainly used in combination with atomic spectrometric techniques.13,14 The advantages of SPE stem from its simplicity to perform on-line or off-line, high preconcentration factor and flexibility due to the availability of commercial adsorbents such as octadecyl–C18–silica, Amberlite XAD-2, polyurethane foam, Amberlyst 36, Amberlite XAD-16, Chromosorb-106.15
Although widely used, these adsorbents suffer from some drawbacks, such as lack of selectivity, the need to use auxiliary chelating agents, low chemical stability in a large pH range, swelling effect in different fluids, low regeneration ability and the need for toxic organic solvents as eluents in the preconcentration system.16–18 Therefore, many efforts in the field of separation science have been made to synthesize new adsorbent materials with better adsorptive and selective features towards metal ion adsorption. Carbonaceous nanoadsorbent materials, including carbon nanotubes,19 carbon black20 and graphene;21 metal–organic frameworks (MOFs);22 nanostructured ion-imprinted polymers (IIPs);23 inorganic nanoadsorbents;24 nanostructured mixed oxides;25 magnetic adsorbents;26 and nanocomposites based on nanocarbonaceous materials and polymers27 represent a class of materials that has been widely investigated for metal ion adsorption. The basic advantage of these nanoadsorbents is that most of the binding sites responsible for the high chemical activity and adsorption capacity towards metal ions are on the surface of the nanomaterial.25
Carbon nanotubes (CNTs) are one class of carbonaceous nanoadsorbent materials that is currently considered the most exploited for metal ion adsorption; however, the adsorption properties of these materials depend upon their functional groups, dispersibility in aqueous media, site density and surface area.27 From this perspective, nanocomposite materials based on CNTs and chelating polymers are promising for solving these drawbacks by improving the dispersion of the carbonaceous phase and enhancing the ratio of polymer binding sites to volume due to the nanoscale polymers synthetized on the surface of the carbonaceous phase.28,29 Furthermore, the preparation of CNT/polymer nanocomposites may overcome the poor accessibility of the analyte to the binding sites of the polymer and the slow dynamic adsorption compared to bulk polymer.30
Although very interesting, the studies dedicated to combining CNTs and polymers have been performed mostly to improve the mechanical strength of the polymer in the nanocomposite;31 few have examined metal adsorption. To the best of our knowledge, the few studies reported on this subject involve the use of CNT/polyaniline (PANI) and poly(3,4-dioxythiophene) (PEDOT) nanocomposites for Au adsorption,32 CNT/PAMAN for Ni, Zn, As and Co adsorption,27 CNT/poly(2-amino thiophenol) for Cd and Pb adsorption,33 and CNT/polypyrrole for Pb, Ni, and Cd adsorption.28 Additionally, one should note that no preconcentration studies have been reported from the analytical point of view on the assessment of the performance of CNT/polymer composites as adsorbents for Cd ions using micro-flow injection as a preconcentration method.
Therefore, in this study, we have developed a CNT/polyvinylpyridine nanocomposite for the preconcentration of Cd using a micro-flow injection method on-line coupled to thermospray FAAS (TS-FF-AAS). Polyvinylpyridine was chosen as the chelating polymer due to the presence of nitrogen in the pyridine ring, which has a strong ability to bind Cd ion.34 Moreover, apart from its intrinsic sensibility for volatile metal ions, the interest in the on-line coupling of the preconcentration method with TS-FF-AAS results from its low flow rate. Therefore, micro-column packed with CNT/polyvinylpyridine nanocomposite seems to be very interesting for operating at low flow rates.
The nanocomposite was characterized by FTIR, TGA, SEM, TEM and textural data measurements using the BET and BJH methods, and the analytical performance of the proposed method for real analysis was checked by the analysis of different kinds of samples (water, chocolate, medicinal herbs, and cigarettes) and a certified reference material (fish protein DORM-3).
2. Experimental
2.1. Apparatus
Absorption measurements were performed using a Shimadzu AA-7000 flame atomic absorption spectrometer (Kyoto, Japan) equipped with a hollow cathode lamp for Cd and a deuterium lamp for background correction. The hollow cathode lamp was operated at 8.0 mA, and the wavelength was set at 229.0 nm. The flame composition was operated with an acetylene flow rate of 2.0 L min−1 and an airflow rate of 10.0 L min−1. The flow adsorbent preconcentration system was constructed using a peristaltic pump (Gilson Minipuls evolution; Middleton, USA) furnished with Tygon® tubes to propel all samples and reagent solutions. A homemade minicolumn made of polyethylene (3.0 cm in length and 0.5 cm in internal diameter at the top) packed with 50.0 mg of nanocomposite was coupled to a homemade injector commutator for preconcentration and elution procedures. A piece of cotton was fixed at the extremities of the minicolumn by inserting the upper end of a conical tip into the minicolumn cylindrical edge to avoid the loss of material. The TS-FF-AAS apparatus included a ceramic capillary (10 cm) nickel tube made of non-porous Al2O3 ceramic (Friatec, Mannhein, Germany) and a nickel tube (Inconel 600 tube with 72% Ni, 14–17% Cr, 6–10% Fe, 0.15% C, 1% Mn, and 0.5% SI; Camacam, Brazil) with a length of 10 cm and an i.d. of 1.0 cm and containing six holes with i.d. = 2.5 mm. Scanning electron microscopy (SEM) analyses were performed using a Model Quanta 200-Philips-FEI microscope (Amsterdam, Netherlands), and the samples were coated with a thin layer of carbon to minimize charging under the incident electron beam. For transmission electron microscopy (TEM), a JEOL® JEM-1400 microscope with an accelerating voltage of 120 kV was used. For TEM sample treatment, the material was dispersed in ethanol by sonication for 20 min, and the suspension was dried under vacuum on copper grids.A Shimadzu FTIR 8300 spectrometer (Kyoto, Japan) operating in transmission mode between 4000 and 400 cm−1 was used to elucidate the functional groups present in the nanocomposites. The surface area and average pore diameter were obtained by the multipoint BET and BJH methods, respectively, based on nitrogen adsorption experiments using a physical adsorption method with a Quantachrome Model Nova 1200e automatic nitrogen gas adsorption instrument (Boynton Beach, FL, USA). Thermogravimetric analysis (TGA) was carried using a 4000 Perkin Elmer thermobalance (Waltham, USA). For TGA analysis, ∼10 mg of sample was heated from 30 °C to 900 °C at a rate of 10 °C min−1 under a nitrogen flow of 20 mL min−1. The digestion of a certified reference material and cigarette samples was carried out using a microwave laboratory system (Microwave Milestone).
2.2. Reagents
All reagents were of analytical grade. All solutions and their respective dilutions were prepared with water purified using a Millipore Milli-Q purification system (Bedford, MA, USA). Before use, all glassware was kept for 24 h in a bath of HNO3 (10% v/v) to avoid metal contamination. The reagents and solvents used in the synthesis were trimethylolpropane trimethacrylate (TRIM) (99.9%), 2,20-azoisobutyronitrile (AIBN) (98%), vinyltrimethoxysilane (VTMS) (≥97.5%), toluene (HPLC grade; ≥99.9%), dimethylformamide (DMF; ≥99.8%), and hydroquinone (≥99%), all acquired from Sigma-Aldrich (Steinheim, Germany). MWCNTs were supplied by CNTs Co. Ltd. Yeonsu-Gu, Incheon, Korea (93%, diameter = 10–40 nm; length = 5–20 μm). Buffer solutions (phosphate, Tris–HCl, ammoniacal and borate; Merck, Massachusetts, USA) were used without further purification. The desired pH was adjusted with sodium hydroxide or hydrochloric acid solutions. A standard Cd(II) solution (5.0 μg L−1) was prepared from a standard Cd(II) solution (1000.0 mg L−1; Spec sol, Jacareí, São Paulo, Brazil) using appropriate dilutions. Standard solutions of Co(II), Cu(II), Fe(III), Pb(II), Mn(II), Zn(II), and Ca(II) used in interference studies were prepared daily by the appropriate dilution of stock solutions containing 1000 mg L−1 (Vetec and Specsol, Brazil). Humic acid powder was acquired from Sigma-Aldrich and dissolved in 1.0 mol L−1 KOH solution. The acids, bases and salts (HNO3, NaOH, NH4OH, H2O2 and KCl) were obtained from Sigma-Aldrich and used without further purification.2.3. Synthesis of MWCNT–polyvinylpyridine nanocomposites
To create hydroxyl groups on the MWCNT surface capable of reacting with vinyltriethoxysilane, MWCNTs were oxidized according to the literature with minor modifications.11 For this task, 1000 mg of MWCNT were mixed with 133.3 mL of a HNO3/H2SO4 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixture and refluxed at 65 °C for 2 h. Afterwards, the oxidized MWCNTs were successively washed with deionized water (pH 7.0) to remove acid excess, dried at 60 °C and stored at room temperature. Subsequently, 500.0 mg of oxidized MWCNTs were dispersed in 40.0 mL of toluene in a three-neck, round-bottom flask and kept in an ultrasonic bath (40 kHz) for 20 min. Then, 12.4 g of vinyltrimethoxysilane previously dissolved in 67.0 mL of toluene containing 0.12 g of hydroquinone was slowly added to the mixture under stirring. One of the three necks was sealed, the central neck was coupled to a reflux condenser, and the other neck was purged with nitrogen (N2) for 10 min. Afterwards, the neck was sealed, and the mixture was heated at 100 °C in an oil bath for 6 h under reflux. The obtained functionalized MWCNTs were washed with ethanol and dried at 50 °C for 6 h. For the synthesis of nanocomposite, 40.0 mg of functionalized MWCNTs were dispersed in 15.0 mL of DMF in a flat-bottomed flask and kept in an ultrasonic bath (40 kHz) for 20 min. In the next step, 800 μL of the functional monomer 4-vinylpyridine (7.42 mmol), 800 μL of crosslinking agent (TRIM; 2.51 mmol) and 80.0 mg of the radical initiator (AIBN; 0.48 mmol) were added to the mixture. The mixture was transferred to a round-bottom flask, purged with N2 for 10 min, sealed, and heated at 60 °C for 24 h under constant stirring. After synthesis, the final material was removed from the flask, rinsed with Milli-Q water to remove excess reagent, and dried at 50 °C for 24 h. Fig. 1 shows a schematic representation of the synthesis of MWCNT and polyvinylpyridine nanocomposites.
:1, v/v) mixture and refluxed at 65 °C for 2 h. Afterwards, the oxidized MWCNTs were successively washed with deionized water (pH 7.0) to remove acid excess, dried at 60 °C and stored at room temperature. Subsequently, 500.0 mg of oxidized MWCNTs were dispersed in 40.0 mL of toluene in a three-neck, round-bottom flask and kept in an ultrasonic bath (40 kHz) for 20 min. Then, 12.4 g of vinyltrimethoxysilane previously dissolved in 67.0 mL of toluene containing 0.12 g of hydroquinone was slowly added to the mixture under stirring. One of the three necks was sealed, the central neck was coupled to a reflux condenser, and the other neck was purged with nitrogen (N2) for 10 min. Afterwards, the neck was sealed, and the mixture was heated at 100 °C in an oil bath for 6 h under reflux. The obtained functionalized MWCNTs were washed with ethanol and dried at 50 °C for 6 h. For the synthesis of nanocomposite, 40.0 mg of functionalized MWCNTs were dispersed in 15.0 mL of DMF in a flat-bottomed flask and kept in an ultrasonic bath (40 kHz) for 20 min. In the next step, 800 μL of the functional monomer 4-vinylpyridine (7.42 mmol), 800 μL of crosslinking agent (TRIM; 2.51 mmol) and 80.0 mg of the radical initiator (AIBN; 0.48 mmol) were added to the mixture. The mixture was transferred to a round-bottom flask, purged with N2 for 10 min, sealed, and heated at 60 °C for 24 h under constant stirring. After synthesis, the final material was removed from the flask, rinsed with Milli-Q water to remove excess reagent, and dried at 50 °C for 24 h. Fig. 1 shows a schematic representation of the synthesis of MWCNT and polyvinylpyridine nanocomposites.
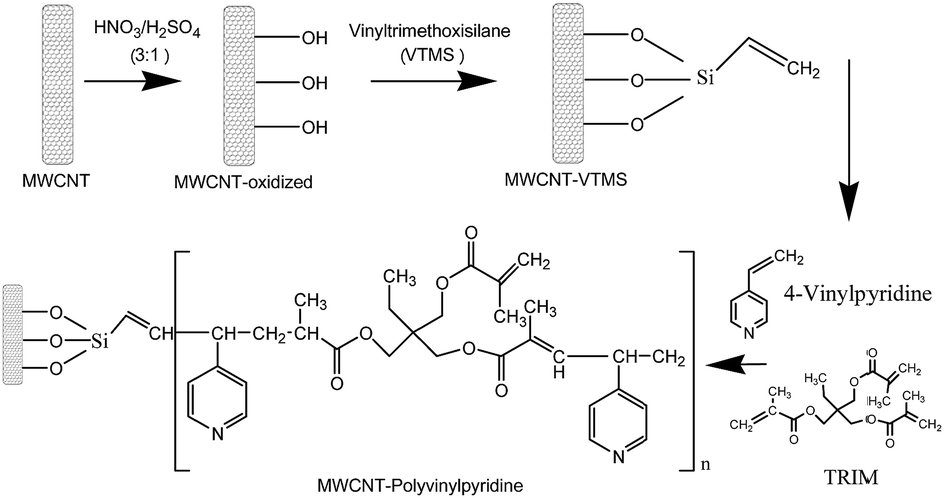 | ||
| Fig. 1 Schematic representation of the synthesis of nanocomposites based on multi-walled carbon nanotubes and 4-vinylpyridine. | ||
2.4. System of preconcentration for Cd(II) on-line coupled to TS-FF-AAS
The on-line preconcentration system was based on loading 8.8 mL of sample buffered with 0.01 mol L−1 ammoniacal buffer (pH 8.0) through 50.0 mg of MWCNT–polyvinylpyridine nanocomposite packed into a mini-column (3.0 cm in length and 0.5 cm in upper internal diameter) made of polyethylene at a flow rate of 4.4 mL min−1 (Fig. 2). To build the homemade cylindrical mini-column, a small piece of cotton tissue was placed at each end of the mini-column to hold the nanocomposite. The pieces of cotton tissue were fixed at the extremities of the mini-column by inserting the upper end of a conical tip into the mini-column cylindrical edge. The on-line elution step was coupled to the TS-FF-AAS detector and performed in countercurrent at a flow rate of 0.6 mL min−1 using 1.0 mol L−1 HCl.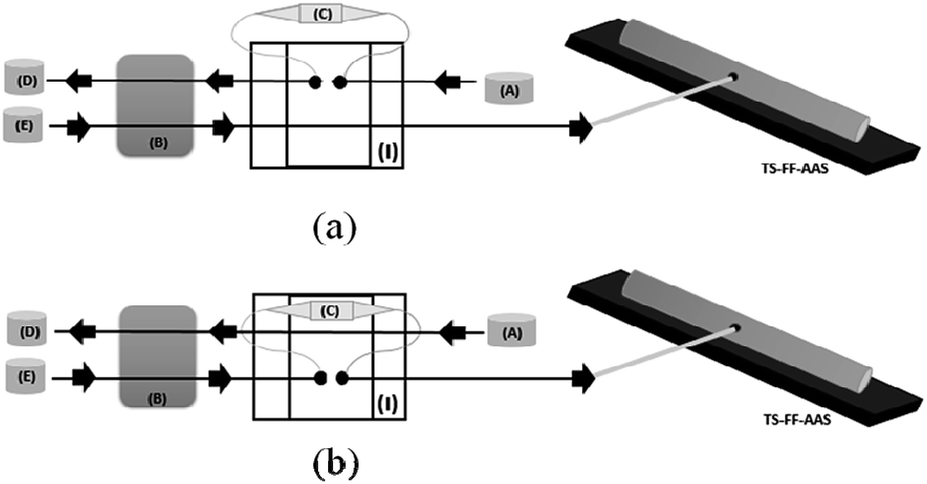 | ||
| Fig. 2 Diagram of the micro-flow injection preconcentration method on-line coupled to thermospray flame furnace-AAS: preconcentration step (a) and elution step (b). A = sample, B = peristaltic pump, C = minicolumn, D = waste, E = eluent. | ||
2.5. Sample preparation
Tap and mineral waters were acquired from the Campus of State University of Londrina and a local supermarket, respectively. After sampling, the pH of the sample was adjusted to 8.0 with 0.01 mol L−1 ammoniacal buffer, and the sample was immediately analyzed by the proposed method. Synthetic seawater was prepared according to ref. 35, and lake water samples were collected from Igapó Lake and treated according to our previous publication.34For the digestion of solid samples, cigarette (500 mg), powdered chocolate (50% cocoa; 700 mg), leaves of Ginkgo biloba (210 mg) and a certified reference material (fish protein DORM-3; 173 mg) were weighted into Teflon flasks and decomposed with 10.0 mL of concentrated HNO3 and 4.0 mL 30% (v/v) H2O2. The mixture was kept overnight followed by digestion in a closed microwave with the following sequence: heat to 80 °C for 6 min; maintain for 6 min at this temperature; heat from 80 °C to 120 °C for 7 min; maintained for 5 min at 120 °C; heating to 210 °C for 15 min; and maintain for 20 min at 210 °C.34 The digested samples were then heated on a hot plate to near dryness and cooled at room temperature. The samples were transferred to 50 mL volumetric flasks and then diluted to the appropriate concentration with ammoniacal buffer (pH 8.0).
3. Results and discussion
3.1. Characterization of MWCNTs and nanocomposites
Fig. 3a and b show the FT-IR spectra of MWCNTs (pristine, oxidized and functionalized) along with MWCNT–polyvinylpyridine and polyvinylpyridine. As can be seen from Fig. 3a, the spectra of pristine and oxidized MWCNTs are similar, except for the band at 1046 cm−1 attributed to δCH. The band at 3450 cm−1 is assigned to O–H stretching vibrations.11 The signals at 2920 and 2830 cm−1 correspond to symmetric and asymmetric C–H stretching, the band at 1630 cm−1 can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the carbon nanotube backbone, and the band at 1390 cm−1 may be attributed to C–C bond stretching.36 For MWCNTs functionalized with VTMS, the bands at 3190 and 3150 cm−1, which are assigned to the CH bonds, and the band at 1390 cm−1 were significantly diminished, indicating the grafting process of VTMS on the carbon nanotube backbone. Additional evidence of the grafting process is provided by the presence of the band at 1080 cm−1 in the spectrum of functionalized MWCNTs, which is characteristic of Si–O–Si stretching, and the bands at 780 and 650 cm−1, which are assigned to Si–O stretching.11 As shown in Fig. 3b, the spectral profile of MWCNT–polyvinylpyridine is similar to that of polyvinylpyridine. The band at 1715 cm−1 can be assigned to CO stretching from crosslinking reagent (TRIM).34 The signal at 1630 cm−1 can be attributed to overlapping bands of CC stretching from vinyl groups, OH deformation (water) or CN stretching from the pyridine ring. The band at 1380 cm−1 is ascribed to C–N stretching from the pyridine ring, which indicates the presence of the organic monomer 4-vinylpyridine in the polymeric matrix.37 Furthermore, the signal at 1460 cm−1 is ascribed to CC stretching from the 4-vinylpyridine ring. The intense band at 1150 cm−1 is attributed to C–O stretching from the ester group in the TRIM structure.29
C stretching of the carbon nanotube backbone, and the band at 1390 cm−1 may be attributed to C–C bond stretching.36 For MWCNTs functionalized with VTMS, the bands at 3190 and 3150 cm−1, which are assigned to the CH bonds, and the band at 1390 cm−1 were significantly diminished, indicating the grafting process of VTMS on the carbon nanotube backbone. Additional evidence of the grafting process is provided by the presence of the band at 1080 cm−1 in the spectrum of functionalized MWCNTs, which is characteristic of Si–O–Si stretching, and the bands at 780 and 650 cm−1, which are assigned to Si–O stretching.11 As shown in Fig. 3b, the spectral profile of MWCNT–polyvinylpyridine is similar to that of polyvinylpyridine. The band at 1715 cm−1 can be assigned to CO stretching from crosslinking reagent (TRIM).34 The signal at 1630 cm−1 can be attributed to overlapping bands of CC stretching from vinyl groups, OH deformation (water) or CN stretching from the pyridine ring. The band at 1380 cm−1 is ascribed to C–N stretching from the pyridine ring, which indicates the presence of the organic monomer 4-vinylpyridine in the polymeric matrix.37 Furthermore, the signal at 1460 cm−1 is ascribed to CC stretching from the 4-vinylpyridine ring. The intense band at 1150 cm−1 is attributed to C–O stretching from the ester group in the TRIM structure.29
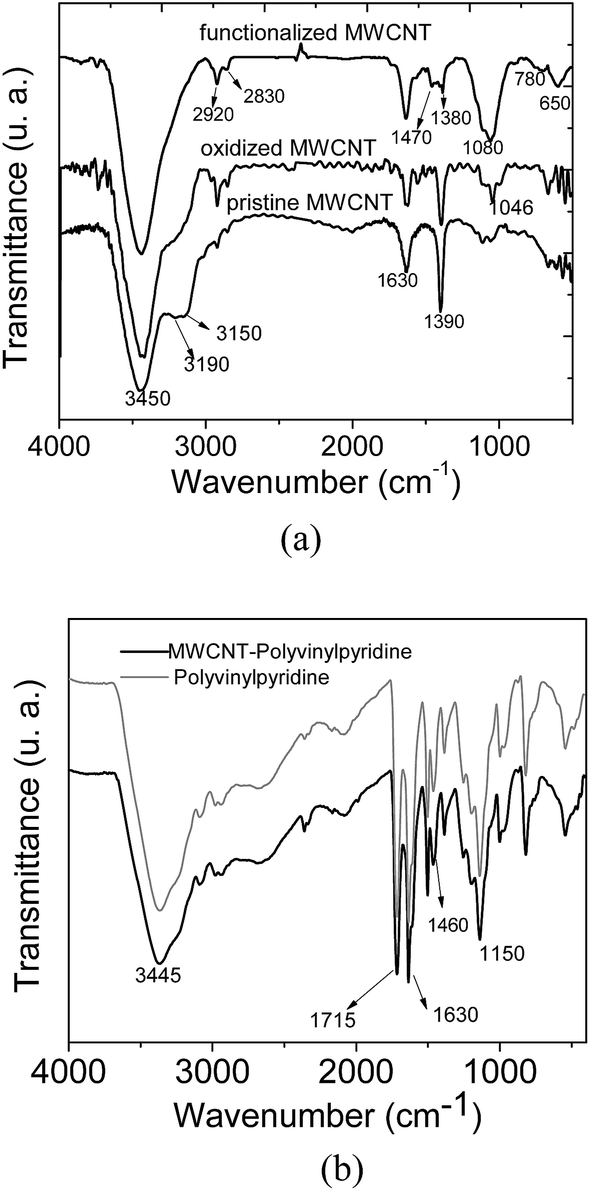 | ||
| Fig. 3 FT-IR spectra of (a) MWCNTs and (b) nanocomposite (MWCNT–polyvinylpyridine) and polyvinylpyridine. | ||
Fig. 4 shows the TGA curves of the MWCNTs (pristine, oxidized and functionalized), MWCNT–polyvinylpyridine composite and polyvinylpyridine. It is possible to observe similar thermal profile for MWCNTs with high thermal stability. A weight loss of about 13% was noted at 900 °C, demonstrating that the grafting of VTMS occurred but not a polymerization reaction based on polycondensation.38 For the MWCNT–polyvinylpyridine composite and polyvinylpyridine, the observed 20% and 4% weight losses, respectively, in the 30–100 °C temperature range are attributed to moisture loss.
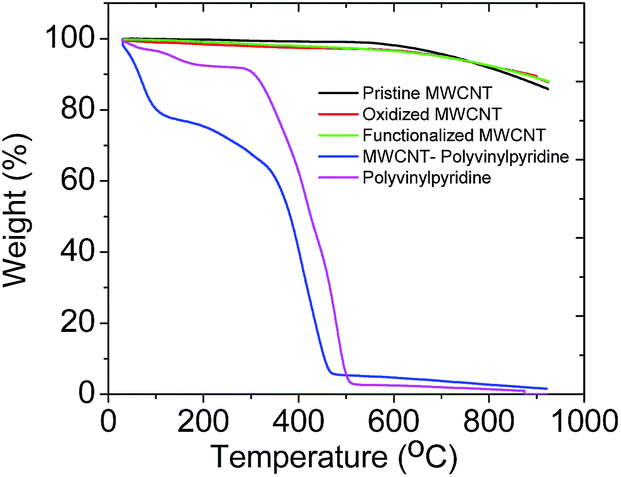 | ||
| Fig. 4 Thermogravimetric curves of MWCNTs, nanocomposite (MWCNT–polyvinylpyridine) and polyvinylpyridine. | ||
The TGA curve of polyvinylpyridine demonstrated that the polymer was stable to 294 °C, when the polymeric chain was degraded. A significant weight loss was observed starting at 347 °C for the MWCNT–polyvinylpyridine composite; this is attributed to the degradation of the polymeric chain, indicating a protective effect of MWCNTs on the thermal stability of the polymeric chain.
The SEM images of MWCNTs are shown in Fig. 5. The images of the pristine and oxidized MWCNTs are similar and show highly tangled nanotubes. This indicates that MWCNT fragmentation did not occur, and that the cylindrical structures of the MWCNTs were not open-ended, corroborating the FT-IR data. This result was expected since the treatment with oxidizing agents in this study was employed primarily to create hydroxyl groups in the MWCNTs. On the other hand, the functionalization of MWCNTs with VTMS gives rise to strong aggregation attributed to grafting process.
 | ||
| Fig. 5 SEM images of (a) pristine MWCNTs, (b) oxidized MWCNTs and (c) functionalized MWCNTs. Magnification = 50000×. | ||
SEM images of the MWCNT–polyvinylpyridine nanocomposite and polyvinylpyridine are shown in Fig. 6. The linkage of polyvinylpyridine on the MWCNTs can be clearly seen by the tubular shapes of the CNTs impregnated into the polyvinylpyridine network. The image of polyvinylpyridine (Fig. 6b) shows that the particles are more cohesive with a higher degree of aggregation, which is not favorable for adsorption.
 | ||
| Fig. 6 SEM images of (a) MWCNT–polyvinylpyridine nanocomposites and (b) polyvinylpyridine. | ||
The TEM images confirm the bonding of polyvinylpyridine with MWCNT, where polymer spots wrap some sections of MWCNT (Fig. 7). Additionally, it can be observed that the polymerization reaction of polyvinylpyridine also occurred in solution and in addition to surface polymerization.
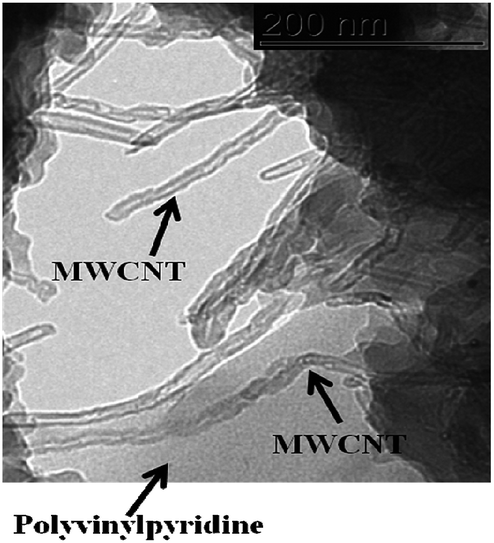 | ||
| Fig. 7 TEM image of MWCNT–polyvinylpyridine nanocomposite. | ||
Table 1 shows the textural data obtained for the materials. As expected, the surface areas of the pristine and functionalized MWCNTs are higher than those of polyvinylpyridine and the nanocomposite. The lower surface area of the nanocomposite resulted from the low pore volume, which can be explained by the penetration of MWCNTs into the polyvinylpyridine network to occupy the pores of the polymer. On the other hand, the textural data of polyvinylpyridine are similar to those of other polymers synthesized by bulk methods,39 and the particles have irregular shapes with rough surfaces.
| Materials | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| Pristine MWCNT | 203.4 | 1.3 | 26.3 |
| Functionalized MWCNT | 239.5 | 2.1 | 34.2 |
| MWCNT–polyvinylpyridine nanocomposite | 1.7 | 5.7 × 10−3 | 14.2 |
| Polyvinylpyridine | 56.7 | 8.0 × 10−2 | 7.5 |
Although polyvinylpyridine shows a higher surface area than the MWCNT–polyvinylpyridine nanocomposite, it should be noted that the nanocomposite adsorbs higher amounts of Cd, as will be demonstrated from the sensitivity of analytical curves. This suggests that the binding sites of the polyvinylpyridine network that interact with Cd ions are dispersed in the nanocomposite and are much more available on the surface of material; in contrast, in polyvinylpyridine, the binding sites are most likely to be inside the polymeric network.
3.2. Optimization of the micro-flow injection preconcentration method on-line coupled to TS-FF-AAS
The parameters that play an important role in the preconcentration system, including sample pH (4.5–9.0), preconcentration flow rate (1.4–4.4 mL min−1), concentration and type of buffer solution were investigated.To study the effect of pH, 26.8 mL of Cd solution at a concentration of 5.0 μg L−1 were preconcentrated in the nanocomposite at a flow rate of 6.7 mL min−1. The elution step was performed with 1.0 mL L−1 HCl at a flow rate of 0.6 mL min−1. Fig. 8 shows that the optimal pH for Cd preconcentration in the nanocomposite is 8.0. According to literature data, the pKa of the monomer 4-vinylpyridine is 5.46,40 while the pKa of polyvinylpyridine determined by potentiometric titration in aqueous medium is 4.0.41 Therefore, in acidic medium, the nitrogen from the pyridine ring is protonated, leading to electrostatic repulsion with the surface of the material. On the other hand, at pH 9.0, the adsorption in the material decreases, most likely due to the precipitation of Cd ions in the hydroxide form. Thus, for further experiments, pH 8.0 was used.
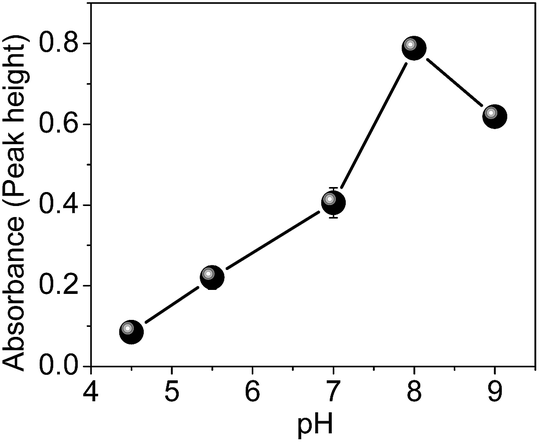 | ||
| Fig. 8 Influence of Cd adsorption on sample pH. | ||
After establishing the best pH for Cd adsorption, the effects of different kinds of buffer solutions (ammoniacal, phosphate and Tris–HCl) were evaluated.
A significant effect was noticed when phosphate was used as a buffer; this result can be rationalized on the basis of the strong interaction of phosphate salt with Cd ions. For ammoniacal and Tris–HCl buffers, higher analytical signals were observed, and no differences were noticed. Ammoniacal buffer was chosen over Tris–HCl due to its lower cost.
The results regarding the effect of buffer concentration (0.001–0.5 mol L−1) showed that 0.01 and 0.1 mol L−1 provide adequate buffer capacity and the best analytical signals for Cd ions. For very low (0.001 mol L−1) and high (0.5 mol L−1) buffer concentrations there was a significant decrease in the analytical signal due to the reduced buffer capacity and strong interaction between ammoniacal buffer and Cd ions. Therefore, 0.01 mol L−1 was chosen for subsequent experiments.
The preconcentration flow rate is a very important parameter to optimize when kinetics exert an influence on adsorption; this variable is strongly related to the sensitivity and sample throughput of the method. In this experiment, 8.8 mL aliquots of Cd solution were preconcentrated in the nanocomposite under the optimized conditions. Lower preconcentration flow rates (1.4 and 2.8 mL min−1) resulted decreased analytical signal, indicating the poor accessibility of Cd ions due to hydrodynamic limitation. On the other hand, the preconcentration increased with the highest flow rate (4.4 mL min−1), which clearly also suggest fast kinetic of mass transfer of Cd toward adsorbent surface. Additionally, the higher the sample flow rate, the greater the sample throughput for the method. Thus, a flow rate of 4.4 mL min−1 was chosen as the optimum value. The elution flow rate using 1.0 mol L−1 was chosen as 0.6 mL min−1 as the best value to produce a stable and continuous aerosol.
3.3. Effect of potentially interfering ions
To discover the degree to which the proposed method is prone to concomitant ions and macromolecules (humic acid), experiments were carried out in the presence of Ca(II), Mg(II), Zn(II), Pb(II), Cu(II), Fe(II), Hg(II), Co(II), Ca(II) and Mg(II). Binary solutions containing different ratios of analyte to interferent [1:1, 1:10, 1:50, 1:100 and 1:500 (w/w)] were subjected to the proposed method with a Cd concentration of 5.0 μg L−1. The influence of humic acid was studied at concentrations up to 5.0 mg L−1, the normal concentration of humic substances in natural water samples,26 to simulate dissolved organic matter in natural waters. Interference was defined to be significant when a relative error of ±10% in the analytic signal was observed. No interference was observed for the analyte: interferent ratios investigated, indicating that Cd preconcentration can be carried free of interference in a complex matrix.
3.4. Analytical characteristics of the proposed method
The analytical curves obtained by TS-FF-AAS without preconcentration and with preconcentration step using the MWCNT–polyvinylpyridine nanocomposites or polyvinylpyridine are illustrated in Fig. 9 and represented by the following linear equations: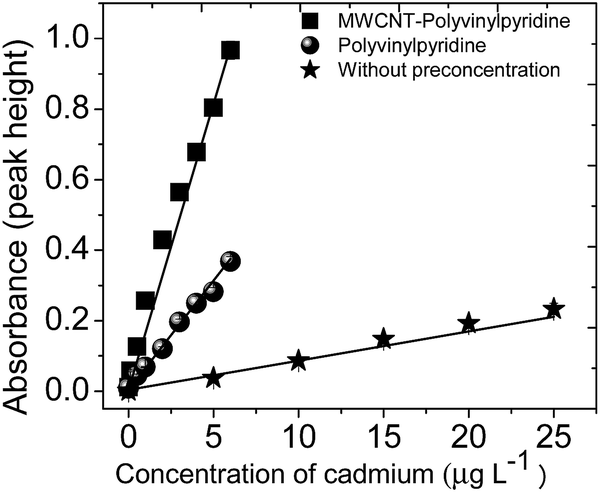 | ||
| Fig. 9 Analytical curves obtained without and with preconcentration steps using the MWCNT–polyvinylpyridine nanocomposite and polyvinylpyridine as adsorbents. | ||
Without preconcentration:
| Abs = 0.0037 + 0.0083(±6.13 × 10−4) [Cd(II), μg L−1]; r = 0.9851, | (1) |
With preconcentration using polyvinylpyridine:
| Abs = 0.0037 + 0.0620(±3.27 × 10−3) [Cd(II), μg L−1]; r = 0.9901, | (2) |
With preconcentration using MWCNT–polyvinylpyridine:
| Abs = 0.0037 + 0.1621(±3.68 × 10−3) [Cd(II), μg L−1]; r = 0.9979. | (3) |
A comparison of the curves shows that the sensitivity of the method was sharply enhanced by the preconcentration step, particularly when the MWCNT–polyvinylpyridine nanocomposite was used as the adsorbent. The enhancement factors (EFs) were determined as the ratio of the slopes of the analytical curves built with and without the preconcentration step. The EFs obtained using the MWCNT–polyvinylpyridine nanocomposite and polyvinylpyridine were 19.5 and 7.46, respectively. These results show that the insertion of MWCNTs into the polymeric network of polyvinylpyridine improves the sensitivity for Cd ion determination by 2.6 times. This result can be considered satisfactory because only 40 mg of MWCNTs were used in the synthesis, which exalts the synergic effect of MWCNTs and the polymeric matrix in the nanocomposite. The sensitivity could be increased by using a larger adsorbent mass and a higher preconcentration volume. For 8.8 mL of sample, the limit of detection (LOD) and limit of quantification (LOQ), determined according to IUPAC recommendations,42 were found to be 36 and 121 ng L−1, respectively. Other important parameters in the development of solid-phase preconcentration methods, such as consumption index (CI), which is defined as the volume required to attain a unit of EF, and the concentration efficiency (CE), defined as the EF obtained by operating the preconcentration system for 1 min, were also calculated.43 CI and CE were found to be 0.40 mL and 9.76 min−1, respectively. The sample throughput of the proposed method was 15 h−1. The intra-day precision was evaluated by preconcentrating (n = 10) Cd solutions at 1.0 and 5.0 μg L−1, giving rise to relative standard deviations (RSDs) of 2.10% and 1.81%, respectively. The inter-day precision for three consecutive working days using ten independent measurements yielded RSD values of 0.61% and 1.60% for the same concentrations of 1.0 and 5.0 μg L−1, respectively.
Compared with other solid-phase preconcentration methods for Cd with TS-FF-AAS determination using Amberlite XAD-4 modified with 3,4-dihydroxybenzoic acid,44 fullerene modified with APDC,45 Amberlite XAD-2 modified with TAN,46 polyurethane foam modified with DDTP,47 oxidized MWCNTs48 and poly(2-amino thiophenol)/MWCNT nanocomposites,33 the proposed method does not require toxic organic solvents in the flow system, presents low sample consumption, has high sample throughput, and provides a similar LOD.
3.5. Application and accuracy in real samples
The preconcentration method was applied to Cd determination in different kinds of water, cigarettes, a food sample (powder chocolate) and a medicinal herb (Ginkgo biloba), as shown in Tables 2 and 3. As Cd was not found in the analyzed water samples, a procedure to spike the samples with known amounts of Cd was implemented. According to the recovery values, which ranged from 91% to 109%, the proposed method allows interference-free Cd determination, even for samples containing high salt contents.| Samples | Cd added (μg L−1) | Cd foundb (μg L−1) | Recovery (%) |
|---|---|---|---|
| a <LOD = below of limit of detection.b Results are expressed as mean value ± standard deviation based on three replicates (n = 3). | |||
| Tap water | 0.0 | <LODa | — |
| 1.0 | 1.09 ± 0.09 | 109 | |
| 3.0 | 3.04 ± 0.07 | 101 | |
| Lake water | 0.0 | <LOD | — |
| 1.0 | 1.00 ± 0.08 | 100 | |
| 3.0 | 2.74 ± 0.09 | 92 | |
| Mineral water (brand 1) | 0.0 | <LOD | — |
| 1.0 | 0.97 ± 0.09 | 98 | |
| 3.0 | 2.75 ± 0.09 | 92 | |
| Mineral water (brand 2) | 0.0 | <LOD | — |
| 1.0 | 0.96 ± 0.02 | 97 | |
| 3.0 | 2.83 ± 0.16 | 94 | |
| Synthetic seawater | 0.0 | <LOD | — |
| 1.0 | 0.90 ± 0.03 | 91 | |
| 3.0 | 2.82 ± 0.04 | 94 | |
| Samples | Cd added (μg g−1) | Cd founda (μg g−1) | Recovery (%) |
|---|---|---|---|
| a Results are expressed as mean value ± standard deviation based on three replicates (n = 3). | |||
| Cigarette (brand 1) | 0.00 | 0.16 ± 0.20 × 10−2 | — |
| 0.15 | 0.27 ± 0.01 | 91 | |
| Cigarette (brand 2) | 0.00 | 0.32 ± 0.03 | — |
| 0.30 | 0.65 ± 0.01 | 105 | |
| Powder chocolate | 0.00 | 0.11 ± 0.60 × 10−2 | — |
| 0.10 | 0.19 ± 0.40 × 10−2 | 91 | |
| Ginkgo biloba | 0.00 | 0.10 ± 2.00 × 10−2 | — |
| 0.10 | 0.18 ± 7.00 × 10−2 | 90 | |
| Certified reference material (DORM-3) | Certified value (μg g−1) | Found valuea (μg g−1) |
|---|---|---|
| 0.29 ± 0.02 | 1.26 ± 0.02 |
The feasibility of applying this method for the determination of trace levels of Cd in different kinds of solid samples (cigarettes, powder chocolate, and medicinal herbs) subjected to acid digestion is assured by the high recovery values (90–105%; Table 3). As expected, Cd was found in cigarettes,49 powder chocolate8 and Gingko biloba7 samples, which represent sources of Cd uptake by human beings. The accuracy of the method was also verified by the analysis of a certified reference material DORM-3 (fish protein); the result achieved by the proposed method was statistically equal to the certified value based on a t-test at a confidence level of 95% (Table 3).
4. Conclusions
The synthesis of a nanocomposite and its application in analytical preconcentration was reported herein for the first time. The results demonstrate the great benefits of incorporating MWCNTs into the polymeric network of polyvinylpyridine as an adsorbent material for metal ions, which is of paramount importance for obtaining new analytical preconcentration methodologies. The preconcentration method on-line coupled to TS-FF-AAS has good accuracy and repeatability, low sample consumption, high sample throughput, no need for toxic organic solvents in the flow system, and broad applicability for different types of samples for the determination of trace levels of Cd.Acknowledgements
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Grant No. 481669/2013-2, 305552/2013-9, 472670/2012-3), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (25/2014), Fundação Araucária do Paraná, Secretaria da Ciência, Tecnologia e Ensino Superior do Paraná (SETI), SANEPAR, Laboratório de Espectroscopia da Central de Multiusuário da PROPPG and Instituto Nacional de Ciência e Tecnologia de Bioanalítica (INCT) (Grant No. 573672/2008-3) for their financial support and fellowships.References
- H.-T. Fan, J. Li., Z.-C. Li and T. Sun, Appl. Surf. Sci., 2012, 258, 3815 CrossRef CAS.
- Y. Zhai, Y. Liu, X. Chang, S. Chen and X. Huang, Anal. Chim. Acta, 2007, 593, 123 CrossRef CAS PubMed.
- Y. Liu, C. Xijun, S. Wang, Y. Guo, B. Din and S. Meng, Anal. Chim. Acta, 2004, 519, 173 CrossRef CAS.
- US EPA, National Primary Drinking Water Standards, Maximum Contaminant Level, United States Environmental Protection Agency Office of Water, 2003. http://water.epa.gov/drink/contaminants/index.cfm#List, accessed on July, 2015 Search PubMed.
- ANVISA (Brazilian Health Surveillance Agency), Consultation Paper No. 2.914, December 12, 2011, Available at (in Portuguese), http://bvsms.saude.gov.br/bvs/saudelegis/gm/2011/anexo/anexo_prt2914_12_12_2011.pdf, accessed January, 17 2016 Search PubMed.
- L. Yue, Biomed. Environ. Sci., 1992, 5, 53 CAS.
- E. D. Caldas and L. L. Machado, Food Chem. Toxicol., 2004, 42, 599 CrossRef CAS PubMed.
- J. E. L. Villa, R. R. A. Peixoto and S. Cadore, J. Agric. Food Chem., 2014, 62, 8759 CrossRef CAS PubMed.
- M. Rajabi, A. Rezaie and M. Ghaedi, RSC Adv., 2015, 5, 89204 RSC.
- F. A. C. Amorim and M. A. Bezerra, Microchim. Acta, 2007, 159, 183 CrossRef CAS.
- M. Z. Corazza, B. S. Fabrin, M. G. Segatelli and C. R. T. Tarley, J. Hazard. Mater., 2012, 243, 326 CrossRef CAS PubMed.
- M. Behbahani, A. Aliakbari, M. M. Amini, A. S. Behbahani and F. Omidi, RSC Adv., 2015, 5, 68500 RSC.
- S. L. C. Ferreira, J. B. de Andrade, M. G. Korn, M. G. Pereira, V. A. Lemos, W. N. L. dos Santos, F. M. Rodrigues, A. S. Souza, H. S. Ferreira and E. G. P. da Silva, J. Hazard. Mater., 2007, 145, 358 CrossRef CAS PubMed.
- E. Yilmaz and M. Soylak, Environ. Monit. Assess., 2014, 189, 5461 CrossRef PubMed.
- C. R. T. Tarley, N. C. Farias, G. F. Lima, F. M. de Oliveira, R. Bonfílio, D. C. Dragunski, D. N. Clausen and M. G. Segatelli, J. AOAC Int., 2014, 97, 605 CrossRef PubMed.
- S. Radi, S. Tighadouini, M. Bacquet, S. Degoutin, L. Janus and Y. N. Mabkhot, RSC Adv., 2016, 6, 8250 Search PubMed.
- K. M. Diniz, F. A. Gorla, E. S. Ribeiro, M. B. O. Nascimento, R. J. Corrêa, C. R. T. Tarley and M. G. Segatelli, Chem. Eng. J., 2014, 239, 233 CrossRef CAS.
- H. Watanabe, K. Goto, S. Taguchi, J. W. McLaren, S. S. Berman and D. S. Russell, Anal. Chem., 1981, 53, 738 CrossRef CAS.
- M. Tuzen, K. O. Saygi and M. Soylak, J. Hazard. Mater., 2008, 152, 632 CrossRef CAS PubMed.
- D. M. Nanicuacua, M. G. Segatelli, M. Z. Corazza and C. R. T. Tarley, Anal. Methods, 2016, 8, 2820 RSC.
- Y. Jin-Gang, Y. Lin-Yan, Y. Hua, L. Qi, C. Xiao-Hong, J. Xin-Yu, C. Xiao-Qing and J. Fei-Peng, Sci. Total Environ., 2015, 502, 70 CrossRef PubMed.
- M. Babazadeh, R. Hosseinzadeh-Khanmiri, J. Abolhasani, E. Ghorbani-Kalhor and A. Hassanpour, RSC Adv., 2015, 5, 1988 RSC.
- L. D. Marestoni, M. D. P. T. Sotomayor, M. G. Segatelli, L. R. Sartori and C. R. T. Tarley, Quim. Nova, 2013, 36, 1194 CrossRef CAS.
- P. Z. Ray and H. J. Shipley, RSC Adv., 2015, 5, 29885 RSC.
- M. Khajeh, S. Laurent and K. Dastafkan, Chem. Rev., 2013, 113, 7728 CrossRef CAS PubMed.
- K. M. Diniz and C. R. T. Tarley, Microchem. J., 2015, 123, 185 CrossRef CAS.
- B. Hayati, A. Maleki, F. Najafi, H. Daraei, F. Guaribi and G. McKay, J. Mol. Liq., 2016, 224, 1032 CrossRef CAS.
- R. Kanthapazham, C. Ayyaru and D. Mahendiradas, Desalin. Water Treat., 2016, 57, 16871 CAS.
- E. S. Moretti, F. M. Oliveira, G. L. Scheel, L. H. Dall'Antônia, D. Borsato, L. T. Kubota, M. G. Segatelli and C. R. T. Tarley, Electrochim. Acta, 2016, 212, 322 CrossRef CAS.
- J. Fu, L. Chen, J. Li and Z. Zhang, J. Mater. Chem. A, 2015, 3, 13598 CAS.
- H. Ago, K. Petritch, M. S. P. Shaffer, A. H. Windle and R. Friend, Adv. Mater., 1999, 11, 1281 CrossRef CAS.
- M. R. Nabid, R. Sedghi, R. Hajimirza, H. A. Oskooie and M. M. Heravi, Microchim. Acta, 2011, 175, 315 CrossRef CAS.
- M. R. Nabid, R. Sedghi, A. Bagheri, M. Behbahani, M. Taghizadeh, H. A. Oskooie and M. M. Heravi, J. Hazard. Mater., 2012, 203–204, 93 CrossRef CAS PubMed.
- C. R. T. Tarley, M. Z. Corazza, F. M. Oliveira, B. F. Somera, C. C. Nascentes and M. G. Segatelli, Microchem. J., 2017, 131, 57 CrossRef CAS.
- M. Zougagh, P. C. Rudner, A. G. de Torres and J. M. C. Pavon, J. Anal. At. Spectrom., 2000, 15, 1589 RSC.
- D. S. Ahmeda, A. J. Haiderb and M. R. Mohammad, Energy Procedia, 2013, 36, 1111 CrossRef.
- D. D. Maksin, S. O. Kljajevic, M. B. Dolic, J. P. Markovic, B. M. Ekmescic, A. E. Onjia and A. Nastasovic, Hem. Ind., 2012, 66, 795 CrossRef CAS.
- E. S. Moretti, J. F. Giarola, M. Kuceki, M. C. Prete, A. C. Pereira and C. R. T. Tarley, RSC Adv., 2016, 6, 28751 RSC.
- C. R. T. Tarley, M. Z. Corazza, B. F. Somera and M. G. Segatelli, J. Colloid Interface Sci., 2015, 450, 254 CrossRef CAS PubMed.
- Accessed on January 11, 2017. https://chemicalize.com/#/calculation.
- M. Satoh, E. Yoda, T. Hayashi and J. Komiyama, Macromolecules, 1989, 22, 1808 CrossRef CAS.
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712 CrossRef.
- Z. Fang, S. Xu and S. Zhang, Anal. Chim. Acta, 1987, 200, 35 CrossRef CAS.
- V. A. Lemos, M. A. Bezerra and F. A. Amorim, J. Hazard. Mater., 2008, 15, 613 CrossRef PubMed.
- M. G. Pereira, E. R. Pereira-Filho and M. A. Z. Arruda, Spectrochim. Acta, 2004, 59, 515 CrossRef.
- F. A. C. Amorim and M. A. Bezerra, Microchim. Acta, 2007, 159, 183 CrossRef CAS.
- C. R. T. Tarley and M. A. Z. Arruda, Anal. Sci., 2004, 20, 961 CrossRef CAS PubMed.
- C. R. T. Tarley, A. F. Barbosa, M. G. Segatelli, E. C. Figueiredo and P. O. Luccas, J. Anal. At. Spectrom., 2006, 21, 1305 RSC.
- C. D. Zappielo, D. M. Nanicuacua, W. N. L. dos Santos, D. L. F. da Silva, L. H. Dall'Antônia, F. M. de Oliveira, D. N. Clausen and C. R. T. Tarley, J. Braz. Chem. Soc., 2016, 27, 1715 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |