DOI:
10.1039/C7RA00879A
(Paper)
RSC Adv., 2017,
7, 19280-19287
In situ DRIFT investigation on the photocatalytic NO oxidation mechanism with thermally exfoliated porous g-C3N4 nanosheets
Received
20th January 2017
, Accepted 18th March 2017
First published on 31st March 2017
Abstract
Bulk g-C3N4 suffers from a low surface area and high charge recombination rate. To advance the photocatalytic efficiency of g-C3N4, porous g-C3N4 nanosheets were prepared using a simple thermal exfoliation method. The effects of thermal exfoliation time on the microstructure and photocatalytic performance of g-C3N4 was investigated. Porous g-C3N4 nanosheets treated for 4 h (C3N4-4h) exhibited a highly enhanced NO removal ratio of 51.2%, which is 3.2 times higher than that of bulk g-C3N4. The enhanced activity of C3N4-4h can be ascribed to an increased surface area and promoted charge separation. An in situ DRIFT investigation was applied to monitor the time-dependent NO adsorption–photocatalysis process. Based on the observed reaction intermediates, a molecular-level mechanism for photocatalytic NO oxidation with porous g-C3N4 nanosheets was proposed.
1. Introduction
Graphitic carbon nitride (g-C3N4) as a stable and eco-friendly semiconducting photocatalyst, has attracted extensive attention due to its diverse potential applications in environmental remediation,1 water splitting,2 electrocatalysis3,4 and water treatment.5–7 However, the low specific surface area and the high recombination rate of charge carriers resulted in the relatively low visible light photocatalytic activity and thus limited its practical application.8,9 In order to enhance the photocatalytic performance of g-C3N4, many strategies have been developed and applied. For instance, metal deposition,10–14 metal/nonmetal doping,15,16 coupling with other semiconductors,17,18 and nanostructure engineering. Niu et al.19 developed a thermal exfoliation method using for porous graphene-like g-C3N4 with enhanced photocatalytic activity. Dong et al.20 investigated the effect of exfoliation temperature on the microstructure and photocatalytic performance of porous g-C3N4. It was found that the ultrathin exfoliated porous g-C3N4 nanosheets were favorable for charge transfer and reactants diffusion.
Recently, g-C3N4-based photocatalysts have been extensively reported the removal of NO. Dong et al.21,22 immobilized the g-C3N4 photocatalysts on Al2O3 ceramic foam for efficient photocatalytic NO removal. The holes, superoxide and hydroxyl radicals were found to be the main reactive species for NO oxidation. Wang et al.23 prepared g-C3N4 with honeycomb structures and applied them for enhanced photocatalytic NO removal. Li et al.24 fabricated the g-C3N4/rutile-brookite TiO2−xNy composite photocatalysts for NO purification. However, the mechanism of photocatalytic NO oxidation with g-C3N4 has not been revealed as the reaction intermediates during photocatalysis are difficult for to be detected. Also, the effects of thermal exfoliation time on the microstructure of porous g-C3N4 are still unknown.
Herein, we adopted the thermal exfoliation approach to prepare ultrathin porous g-C3N4 nanosheets. The effect of exfoliation time on the microstructure and photocatalytic performance of g-C3N4 investigated. The as-prepared porous g-C3N4 nanosheets were applied in visible light photocatalytic NO removal and exhibited enhanced activity. In situ DRIFT was employed to dynamically monitor the NO adsorption–photocatalysis process on the ultrathin porous g-C3N4 nanosheets. Based on the time-dependent evolution of reaction intermediates, a precise mechanism of photocatalytic NO oxidation was proposed. The present work could provide new insights into g-C3N4 photocatalysis mechanism for air purification.
2. Experimental
2.1 Synthesis of photocatalyst
2.1.1 Synthesis of g-C3N4. All chemicals were analytical grade purity and used without further purification. 0.50 g of dicyanodiamine was placed in an alumina crucible with a cover and calcined at 550 °C for 2 h with a ramping rate of 15 °C min−1, and then cooled slowly to room temperature. The product was collected after grinding into powder in an agate mortar for further use.
2.1.2 Synthesis of reoxidized ultrathin g-C3N4. 0.5 g of the as-prepared g-C3N4 and 20 mL of deionized water were placed in an alumina crucible and calcined at 550 °C for different time (2, 4 and 6 h) with heating rate of 15 °C min−1 and then cooled slowly to room temperature. The product was collected as C3N4-2h, C3N4-4h, C3N4-6h after grinding.
2.2 Characterization
The prepared g-C3N4 samples were examined by XRD (XRD: model D/max RA, Japan). The morphology structures for the obtained products were characterized by transmission electron microscopy (TEM: JEM-2010, Japan). The optical absorption spectra were recorded on a UV-vis diffuse reflection spectrophotometer (UV-2450, Shimadzu, Japan) in the diffuse reflectance mode, using BaSO4 as reflectance sample. The nitrogen adsorption–desorption isotherms were investigated in a nitrogen adsorption apparatus (ASAP 2020, USA) with products degassed at 150 °C prior to measurements. The photocurrent responses of the prepared samples were investigated in computer-controlled electrochemical apparatus.
2.3 Visible light photocatalytic activity
The photocatalytic activity of the obtained products was evaluated by oxidation of NO at ppb level in a continuous flow reactor. The volume of the rectangular reactor, made of stainless steel and covered with Saint-Glass, was 4.5 L (30 cm × 15 cm × 10 cm). A commercial tungsten halogen lamp (150 W) coupled with a 420 nm cutoff filter is utilized as the light source. For the test, 0.20 g of the photocatalyst spread on two sample dishes (12.0 cm in diameter) was placed in the middle of the reactor. By mixing with flowing air, the NO concentration is diluted to 600 ppb with a flow rate of 2.4 mL min−1 set by a mass flow controller. Then it was allowed to achieve an adsorption–desorption equilibrium. A NOx analyzer was used to continuously measure the NO concentration during test. According to the equation (η%) = (1 − C/C0) × 100% (C and C0 represent the outlet and inlet NO concentrations, respectively), the NO removal rate (η) can be determined.
2.4 In situ DRIFTS study on photocatalytic NO oxidation process
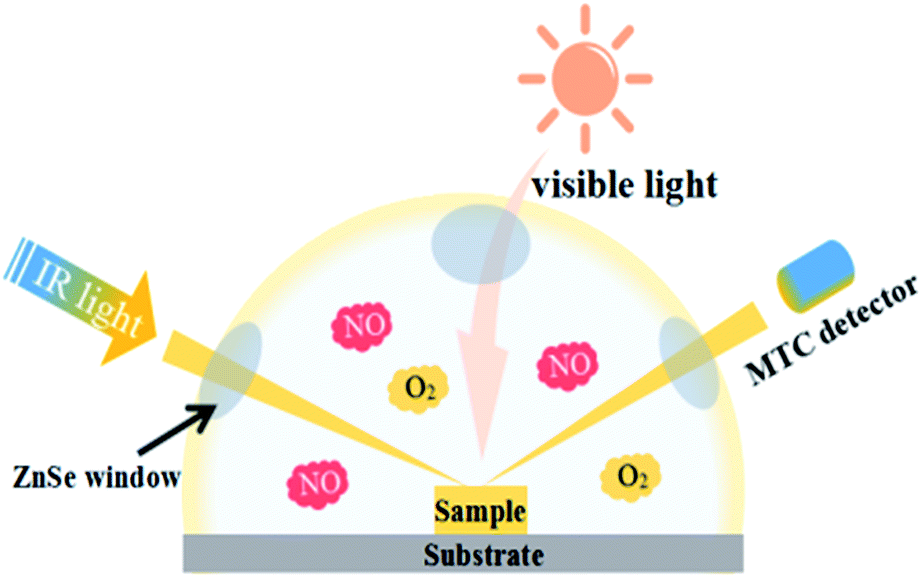
In situ DRIFTS measurements were conducted using the Tensor II FTIR spectrometer (Bruker) equipped with an in situ diffuse reflectance cell (Harrick), as shown in Fig. 1. Photocatalysts were put into the cell. First, He gas (50 mL min−1) was used to remove the residual hydrocarbons, H2O and CO2. The real-time FTIR spectrum after ventilation was utilized as background. Then, the reaction mixtures (25 mL min−1 NO, 25 mL min−1 O2) were introduced into the cell. The NO adsorption on the catalysts was carried out for 20 min. Next, photocatalysts were illuminated by visible light source (MVL-210, Japan) for 40 min. The real-time FTIR spectra were detected every eight minutes. Meanwhile the gas fluxes keep the same (25 mL min−1 NO, 25 mL min−1 O2). Finally, FTIR spectra were tested every two minutes with the same gas fluxes after turning off the light. The IR scanning range was 4000–600 cm−1 and 1900–1200 cm−1 was analyzed to present the photocatalytic oxidation process on porous g-C3N4 nanosheets.
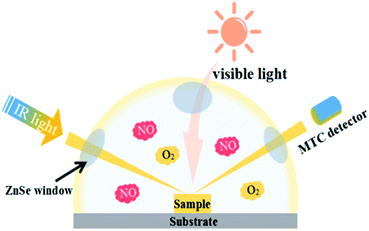 |
| | Fig. 1 Scheme of photoreaction cell used for in situ FT-IR data recording. | |
3. Results and discussion
3.1 Crystal structures
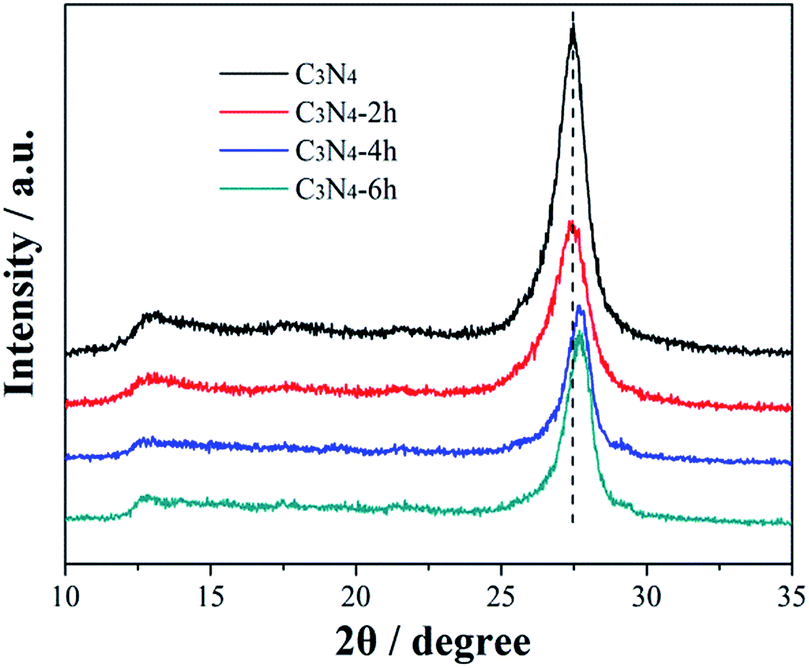
Fig. 2 shows the XRD patterns of the prepared g-C3N4 samples treated for different time. All materials present two diffraction peaks, in agreement with the g-C3N4 (JCPDS card no. 87-1526), suggesting that the thermal treated g-C3N4 have similar crystal structure as their pristine bulk g-C3N4. The strong peak, identified as the (002) peak of graphitic materials, can be attributed to the characteristic inter-layer stacking of the conjugated aromatic C–N units.25 This typical peak is shifted from 27.48° for C3N4 to 27.67° for C3N4-6h, which can be ascribed to the decreased inter-layer spacing due to thermal exfoliation effect. Another low-angle reflection peak is found around 13.1°, which is assigned to (100) peak and corresponds to an inter-planar distance of d = 0.68 nm from the in-plane structural stacking motif.25 Further, the (100) peak of C3N4-4h and C3N4-6h shifts to a lower angle of 12.7°, demonstrating that the planar size is increased during thermal exfoliation.26
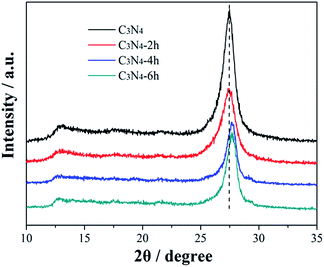 |
| | Fig. 2 XRD patterns of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h. | |
3.2 Morphological structures
The morphology of the samples treated at different time was analyzed by TEM as showed in Fig. 3. Fig. 3a shows that the bulk C3N4 has a bulk layered structure. The enlarged view in Fig. 3b shows that the bulk C3N4 exhibits orderly stacked layers and a typical nonporous architecture. Compared to the pristine bulk g-C3N4 samples, the thermally treated g-C3N4 samples appear as thin and porous structure (Fig. 3c to h). This can be understood as the pristine bulk g-C3N4 samples were exfoliated into thin layers during thermal treatment. The bulk layered structure of g-C3N4 has typical CN layers linked together by weak van der Waals forces and hydrogen bonds. The van der Waals forces and hydrogen bonds are not stable enough against thermal oxidation process in air, and the layers of the CN unit would be gradually oxidized away from the bulk material in a layer-by-layer manner.20 As showed in Fig. 3e to h, after thermal treatment for a long time, the C3N4-4h and C3N4-6h sample exhibit ultra-thin and porous layered structure. The AFM image (Fig. 4a) reflects that C3N4-4h has the nanosheets structure. As showed in Fig. 4b, the nanosheets thickness is 2.1 nm.
 |
| | Fig. 3 TEM images of C3N4 (a, b), C3N4-2h (c, d), C3N4-4h (e, f) and C3N4-6h (g, h). | |
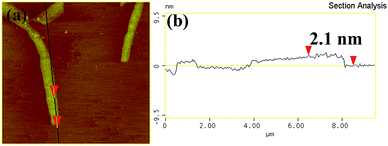 |
| | Fig. 4 The typical AFM image (a) and the corresponding thickness analysis results (b) of C3N4-4h. | |
3.3 BET surface areas and pore structure
The detailed information about the specific surface areas and porosity of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h was accessed by nitrogen absorption–desorption isotherms and Barrett–Joyner–Halenda (BJH) pore-size distribution are measured (Fig. 5 and Table 1). Fig. 5a shows that the above samples belong to type IV in BDDT classification shape of isotherms with a H3-type hysteresis loop in the IUPAC classification, suggesting the presence of slit-like mesopores. As showed in Fig. 5b, the C3N4-4h and C3N4-6h are abundant in large mesopores (22 to 60 nm) and small mesopores (3.79 nm).
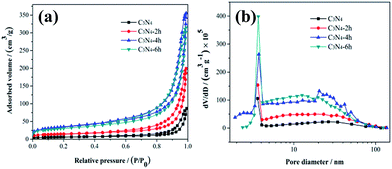 |
| | Fig. 5 N2 adsorption–desorption isotherms (a) and the pore-size distribution curves (b) of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h. | |
Table 1 The SBET, total pore volume, peak pore size and NO removal ratio for C3N4, C3N4-2h, C3N4-4h and C3N4-6h
| Sample name |
SBET (m2 g−1) |
Total pore volume (cm3 g−1) |
Peak pore size (nm) |
NO removal ratio (%) |
| C3N4 |
20 |
0.13 |
3.6/26.5 |
16.0 |
| C3N4-2h |
50 |
0.31 |
3.6/18.0 |
47.8 |
| C3N4-4h |
122 |
0.55 |
3.7/20.5 |
51.2 |
| C3N4-6h |
105 |
0.49 |
3.6/12.6 |
49.7 |
The specific surface area, pore volume, peak pore size, and NO removal ratios are summarized in Table 1. The C3N4-4h has a higher specific surface area (122 m2 g−1) and correspondingly larger pore volume (0.55 cm3 g−1) than the others. Generally, the introduction of mesoporosity into g-C3N4 leads to increased surface areas.20 The enlarged surface area would be beneficial for mass transfer and provide more active sites for photocatalysis reaction.20
3.4 Optical properties and band gap structure
As showed in the UV-vis DRS spectra (Fig. 6a), all the samples show an absorption edge (420–460 nm) in the visible light region. The absorption spectra of the samples thermal treated at different time exhibit an obvious blue shifted in comparison with the original g-C3N4. The band gap energy of C3N4, C3N4-2h, C3N4-4h and C3N4-6h are calculated to be 2.39, 2.46, 2.58 and 2.55 eV (Fig. 6b). The enlarged band gap for the thin layered samples can be attributed to the well-known quantum confinement effect by shifting the conduction and valence band edges in opposite directions.27 The increase in band gap energy is beneficial for the enhancement of redox ability of the charge carriers generated by g-C3N4.
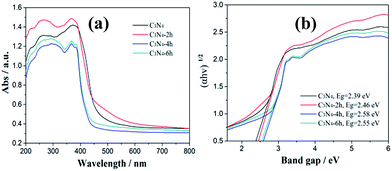 |
| | Fig. 6 UV-vis diffuse reflectance spectra (a) and band gap energies (b) of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h. | |
The separation efficiency of photogenerated carriers is investigated by the photocurrent measurement.28 As shown in the wavelength-dependent photocurrent density in Fig. 7, in contrast to the pristine C3N4 with a low photocurrent density under visible-light irradiation, the C3N4-4h shows a remarkably improved photocurrent density, which indicates the enhanced charge separation efficiency of C3N4-4h sample. This could be attributed to ultra-thin layered structure of C3N4-4h sample (Fig. 3e and f), which could promote the charge migration and separation.19,20
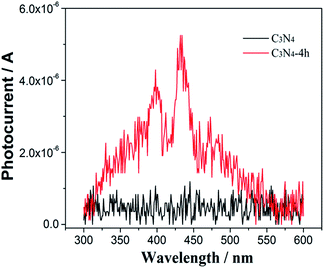 |
| | Fig. 7 Wavelength-dependent photocurrent of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h. | |
3.5 Visible light photocatalytic activity and stability for NO removal
To demonstrate the photocatalytic activities of all the obtained samples for air purification, visible light photocatalytic removal of NO in air in a continuous reactor was carried out.29–31 As showed in Fig. 8, the variation of NO concentration (C/C0/%) with irradiation time over the g-C3N4 samples. After irradiation for 30 min, the NO removal ratio of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h was 16.0%, 47.8% and 49.7%, respectively. Unprecedentedly, the C3N4-4h exhibited a dramatically enhanced NO removal ratio of 51.2%, 3.2 times higher than that of the bulk g-C3N4. The remarkably improved photocatalytic activities of the ultra-thin g-C3N4 demonstrated above can be explained as the synergistic effects of increased surface area and pore structure (Fig. 5b and Table 1), enhanced redox ability (Fig. 6b) and improvement in effective charge separation (Fig. 7).
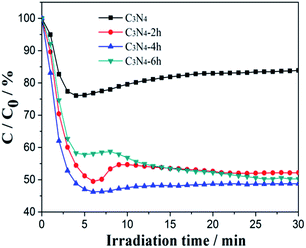 |
| | Fig. 8 Visible light photocatalytic activities of the C3N4, C3N4-2h, C3N4-4h and C3N4-6h. | |
The large surface area can increase the number of possible reactive sites for adsorption and diffusion of reactant molecules.19 The ultra-thin layered structure of C3N4-4h could promote carrier transfer and separation and thus more carriers can take part in the photocatalytic reaction. The band gap of the C3N4-4h is increased by 0.2 eV, which could enhance the redox ability of charge carriers generated in the C3N4-4h. The significant role of a marginally increased band gap in promoting photo catalytic activities has also been reported from Cs0.68Ti1.83O4−xNx and faceted anatase TiO2 crystals.32–34 The above favorable factors co-contribute to the substantially improved photocatalytic activity of the ultra-thin g-C3N4.
3.6 In situ DRIFT study on the adsorption and photocatalytic NO reaction on g-C3N4 under visible light
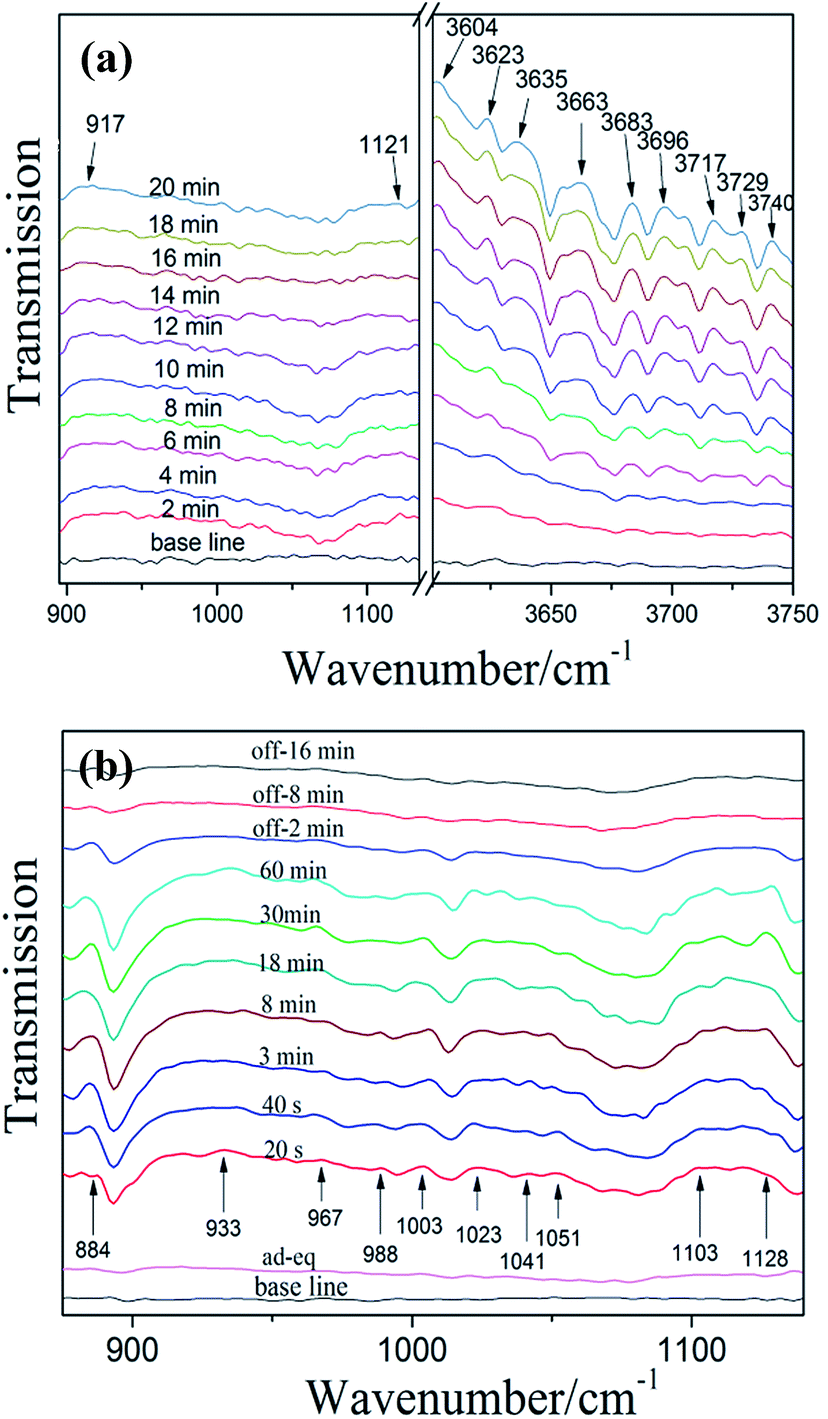
3.6.1 NO adsorption and reaction on bulk g-C3N4. Fig. 9a shows the IR spectra of NO adsorbed on bulk g-C3N4. In the region of 900–1200 cm−1 and 3600–3750 cm−1, several absorption bands were formed after the contact of bulk g-C3N4 with gaseous NO and O2 at 25 °C. In the first 2 min, the adsorption of NO on bulk g-C3N4 resulted in the formation of iso-N2O4 (917 cm−1)36 and N2O (1121 cm−1).36 In the high-frequency region (3600–3750 cm−1), plenty of adsorption bands can be observed after 4 min. All these bands (3604, 3623, 3635, 3663, 3683, 3696, 3717, 3729 and 3740 cm−1) could attribute to NO2.36 Oxidation of NO with O2 on bulk g-C3N4 caused the formation of NO2. As time passed, the intensity of these bands is increased, as more amount of NO are adsorbed on bulk g-C3N4 and quickly transformed to NO2. Especially, in the first 4 min, the adsorbed NO2 may be consumed and transformed to N2O4, resulting in no obvious changes of spectra in the region of NO2 (3600–3750 cm−1).37 Some investigators found that the band of N2O increased at the beginning of NO adsorption and then decreased over time.37 Noteworthy, similar results have been observed in this work. This may attribute to the NO of N–NO was further oxidation and formed NO2, causing consumption of N2O. The probable adsorption processes are proposed as follows (Table 2).36,38
| NO + e(CN)− → NO− + e(CN)− → NO2− |
| NO + O2 → NO3 + NO2 → N2O5 |
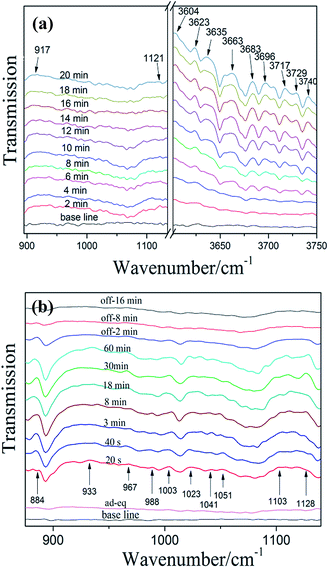 |
| | Fig. 9 In situ DRIFT spectra of adsorption (a) and photocatalytic reaction (b) on bulk g-C3N4 (“ad-eq” represents adsorption-equilibrium). | |
Table 2 Assignments of the FT-IR bands observed upon adsorption-reaction of NO on bulk g-C3N4 and C3N4-4h followed by visible irradiation
| Wavenumbers (cm−1) |
Assignment |
References |
| 884 |
NO2− |
36 |
| 933, 1003, 1009, 1023, 1041, 1051, 1002, 1038 |
NO3− |
36, 39 and 40 |
| 988 |
NO2− |
39 |
| 1099, 1103, 1113 |
NO−/NOH |
41 |
| 961, 967 |
sym-N2O3 |
36 |
| 1121, 1128, 1130 |
N2O |
| 917, 914 |
iso-N2O4 |
| 2821, 2881, 2945, 3463, 3603–3742 |
NO2 |
| 866, 902 |
N2O5 |
Fig. 9b shows the spectra of photocatalytic NO oxidation on bulk g-C3N4 during visible irradiation in time sequence. The spectrum of “ad-ep” is the same as that of “20 min” in Fig. 9a, at which NO and O2 adsorption is reached equilibrium after introducing NO and O2 in the dark. The irradiation with visible light for 20 s results in new absorption bands and causes significant changes in the region of 850–1150 cm−1. Obviously, several new adsorption bands appear. These bands belong to reaction intermediates (NO2−, N2O3 and NO−) and final products (NO2− and NO3−). This indicates that some kinds of photogenerated active species induce the photocatalytic reaction and lead to the conversion of NO to intermediates and end products. According to previous work, the main reactive species for g-C3N4 are superoxide and hydroxyl radicals.42 The absorption bands at 884, 967, 1103 and 1128 cm−1 can be indexed to NO2−, N2O3, NO− and N2O, respectively.36,41 The band at 933, 1003, 1023, 1041 and 1051 cm−1 can be assigned to nitrates (NO3−).36,39,40 In addition, the NO2− was detected at 988 cm−1.39 Noteworthy, the peaks of formative species gradually disappear after visible-light off. However, we can see from the spectrum of “off-16 min”, some weak adsorption peaks could be detected as some reaction species are still adsorbed on the surface of the photocatalyst.22,43 The mechanism of photocatalytic NO oxidation reactions are proposed as follows (NOx = NO2−, N2O3, NO− and N2O).22
| NO2 + ˙OH → NO2−/NO3− + H+ |
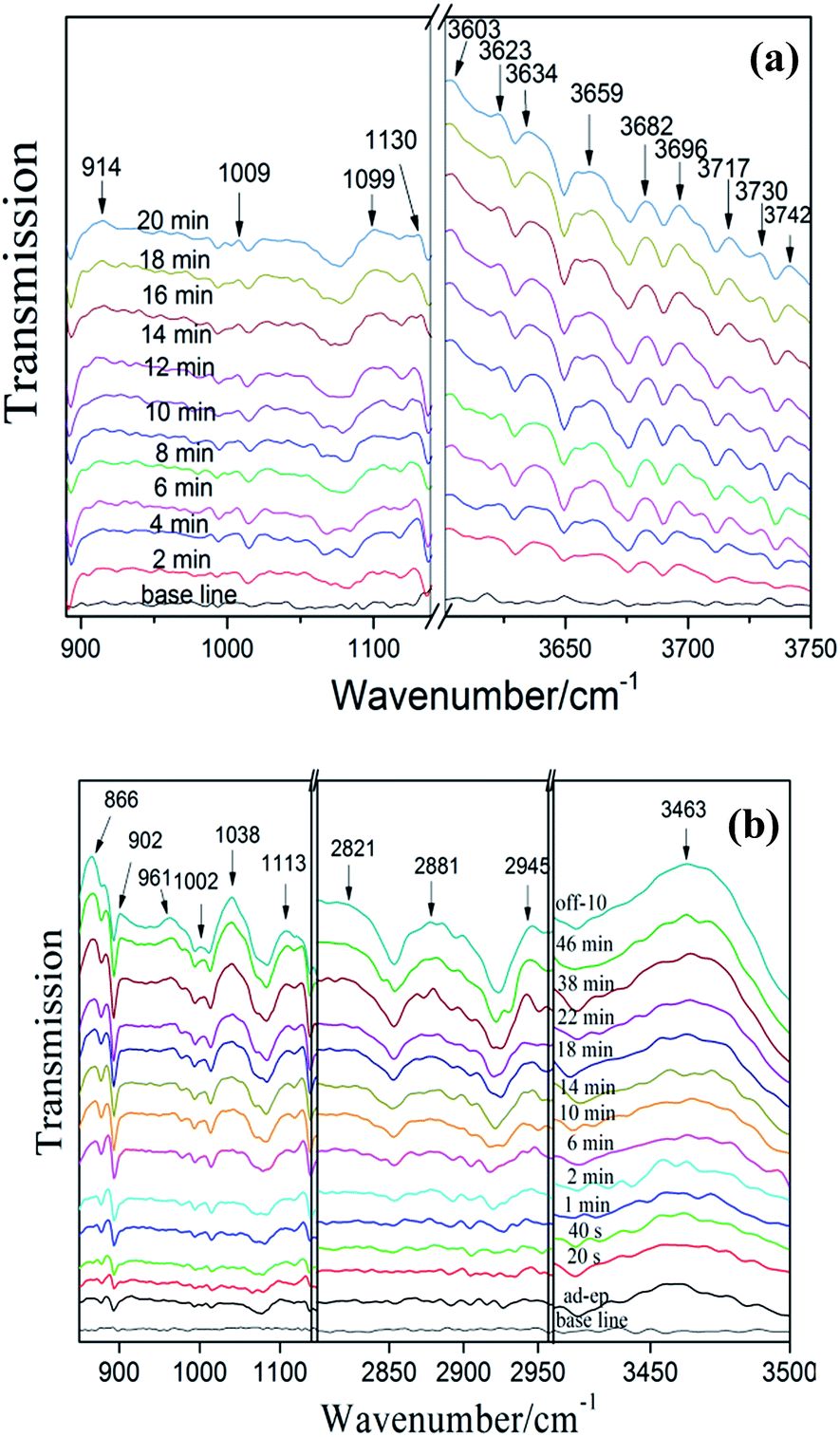
3.6.2 NO adsorption and reaction on exfoliated porous g-C3N4 nanosheets. Fig. 10a shows the spectra of NO adsorbed on exfoliated porous g-C3N4 nanosheets (C3N4-4h). The adsorption peaks in Fig. 9a appear in Fig. 10a as well. In addition, the new bands at 1009 and 1099 cm−1 were assigned to nitrates and NO−.36 Obviously, the bands assigned to NO2 were appeared after 2 min, which indicates that C3N4-4h possess stronger adsorption ability than bulk g-C3N4. This could be attributed to the enlarged surface areas of C3N4-4h, which accelerate the absorption process and enhance the absorption of NO.
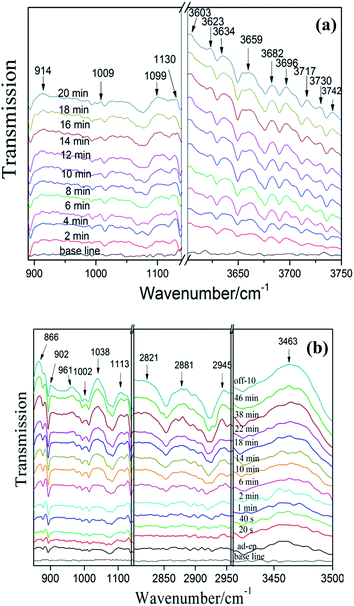 |
| | Fig. 10 In situ DRIFT spectra of adsorption (a) and photocatalytic reaction (b) on C3N4-4h. | |
Fig. 10b shows the spectra photocatalytic NO oxidation on C3N4-4h during visible irradiation in time sequence. The new adsorption bands of reaction intermediates (NO2 (at 2821, 2881, 2945 and 3463 cm−1),36 NOH (at 1113 cm−1),41 N2O5 (866 and 902 cm−1),36 N2O3 (961 cm−1)36) and final products nitrates (1002 and 1038 cm−1)36,40 can be detected. The photocatalytic reaction process on bulk g-C3N4 and C3N4-4h exhibited similar behaviour and the mechanism was also applied to C3N4-4h. Obviously, the intensity of nitrate adsorption bands is stronger than the bulk g-C3N4. This indicates the C3N4-4h has higher photocatalytic oxidation ability, which leads to the production of more active species, promoting NOx transformation from NO to final products of nitrates or nitrites.
To determine the stability of the C3N4-4h, we carried out the XPS and SEM measurements of C3N4-4h after recycled photocatalytic test is shown in Fig. 11. The XPS measurements (Fig. 11a–d) show that the similar presence of C, N and O in the C3N4-4h after photocatalytic test.35 Fig. 10 shows the typical high resolution XPS spectra for C, N and O elements, indicating that the chemical structure of C3N4-4h does not change after repeated irradiations. It can be observed from the SEM image (Fig. 11e) that the morphological structure of C3N4-4h is well-maintained.
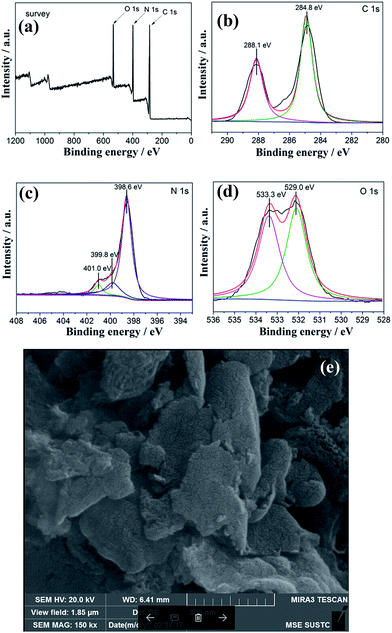 |
| | Fig. 11 XPS spectra of survey (a), C 1s (b), N 1s (c), O 1s (d) and SEM image (e) of C3N4-4h after recycled tests. | |
4. Conclusions
In summary, the thermally treated ultra-thin porous g-C3N4 nanosheets were synthesized by a facile method. The porous g-C3N4 nanosheets exhibited remarkably enhanced photocatalytic activity in NO removal under visible light irradiation owing to the cooperation of the increased surface area and pore structure, enhanced redox ability and improvement of effective charge separation. The in situ DRIFT was employed to monitor the evolution of reaction intermediates basing on the time-dependent photocatalytic reaction. A mechanism for photocatalytic NO oxidation with g-C3N4 was proposed. The porous g-C3N4 nanosheets demonstrated enhanced adsorption performance and oxidation ability for NO. The present work could provide new insights into the understanding of g-C3N4 photocatalysis for NO removal.
Acknowledgements
This research was financially supported by National Natural Science Foundation of China (21501016, 51478070, 51108487 and 21676037), the Innovative Research Team of Chongqing (CXTDG201602014), Natural Science Foundation of Chongqing (cstc2016jcyjA0481 and cstc2015jcyjA0061), and the project from Chongqing Education Commission (KJ1600625, KJ1500601 and Kj1500637).
Notes and references
- W. J. Ong, L. L. Tan, H. N. Yun, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith and M. Jaroniec, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS PubMed.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 124, 3958–3962 CrossRef.
- S. Zhang, H. Gao, X. Liu, Y. S. Huang, X. Xu, N. S. Alharbi, T. Hayat and J. X. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35138–35149 CAS.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 22116–22125 CAS.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, J. Mater. Chem. A, 2015, 3, 10119–10126 CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 46, 2150–2176 CrossRef PubMed.
- Q. Han, B. Wang, Y. Zhao, C. Hu and L. Qu, Angew. Chem., Int. Ed., 2015, 54, 11433–11437 CrossRef CAS PubMed.
- L. Yu, X. Zhang, C. Zhuang, L. Lin, R. Li and T. Peng, Phys. Chem. Chem. Phys., 2014, 16, 4106 RSC.
- S. Samanta, S. Martha and K. Parida, ChemCatChem, 2014, 6, 1453–1462 CAS.
- X. Bai, R. Zong, C. Li, D. Liu, Y. Liu and Y. Zhu, Appl. Catal., B, 2014, 147, 82–91 CrossRef CAS.
- O. Fontellescarceller, M. J. Mu ozbatista, M. Fernandezgarcia and A. Kubacka, ACS Appl. Mater. Interfaces, 2015, 6, 22–25 Search PubMed.
- X. Wang, G. Zhang, Z. A. Lan, L. Lin and S. Lin, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Z. Ding, X. Chen, A. Markus and X. Wang, ChemSusChem, 2011, 4, 274–281 CAS.
- Z. Li, C. Kong and G. Lu, J. Phys. Chem. C, 2015, 120, 56–63 Search PubMed.
- L. Gu, J. Wang, Z. Zou and X. Han, J. Hazard. Mater., 2014, 268, 216 CrossRef CAS PubMed.
- M. Xu, L. Han and S. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 12533–12540 CAS.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- F. Dong, Y. Li, Z. Wang and W. K. Ho, Appl. Surf. Sci., 2015, 358, 393–403 CrossRef CAS.
- Y. Sun, T. Xiong, Z. Ni, J. Liu, F. Dong, W. Zhang and W. K. Ho, Appl. Surf. Sci., 2015, 358, 356–362 CrossRef CAS.
- F. Dong, Z. Wang, Y. Li, W. K. Ho and S. C. Lee, Environ. Sci. Technol., 2014, 48, 10345–10353 CrossRef CAS PubMed.
- Z. Wang, W. Guan, Y. Sun, F. Dong, Y. Zhou and W. K. Ho, Nanoscale, 2015, 7, 2471–2479 RSC.
- H. Li, X. Wu, S. Yin, K. Katsumata and Y. Wang, Appl. Surf. Sci., 2016, 392, 531–539 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Yang, F. Qian, G. Zou, M. Li, J. Lu, Y. Li and M. Bao, Appl. Catal., B, 2016, 193, 22–35 CrossRef CAS.
- A. P. Alivisatos, Semiconductor clusters, nanocrystals, and quantum dots, Science, 1996, 271, 933–937 CAS.
- H. G. Kim, P. H. Borse, W. Choi and J. S. Lee, Angew. Chem., 2005, 44, 4585–4589 CrossRef CAS PubMed.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W. K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS.
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 6489–6496 CAS.
- F. Dong, Z. Wang, Y. Sun, W. K. Ho and H. Zhang, J. Colloid Interface Sci., 2013, 401, 70–79 CrossRef CAS PubMed.
- G. Liu, P. Niu, L. Wang, G. Q. Lu and H. M. Cheng, Catal. Sci. Technol., 2011, 1, 222–225 CAS.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. Lu and H. M. Cheng, Chem. Commun., 2010, 46, 755–757 RSC.
- J. Pan, G. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- C. Liu, Y. Zhang, F. Dong, X. Du and H. Huang, J. Phys. Chem. C, 2016, 10381–10389 CAS.
- P. Jussieu, Characterization of Nitrogen Oxides by Vibrational Spectroscopy, Inorg. Chem., 1982, 466–509 Search PubMed.
- J. C. S. Wu and Y. T. Cheng, J. Catal., 2006, 237, 393–404 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev., 2007, 42(1&2), 71–144 Search PubMed.
- K. Hadjiivanov, V. Avreyska, A. Dimitar Klissurski and T. Marinova, Langmuir, 2002, 18, 1619–1625 CrossRef CAS.
- T. Weingand, S. Kuba, K. Hadjiivanov and H. Knzinger, J. Catal., 2002, 209, 539–546 CrossRef CAS.
- M. Kantcheva, J. Catal., 2001, 204, 479–494 CrossRef CAS.
- W. Cui, X. Li, C. Gao, F. Dong and X. Chen, Catal.
Today, 2016, 67–76 Search PubMed.
- Z. Zhao, Y. Sun, Q. Luo, F. Dong, H. Li and W. K. Ho, Sci. Rep., 2015, 5, 14643 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Yanjuan Sun*a and
Fan Dong
b,
Yanjuan Sun*a and
Fan Dong