
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peptide-binding induced inhibition of chemokine CXCL12†
Hongyang Duan abd,
Ling Zhuad,
Jiaxi Pengad,
Mo Yangad,
Hanyi Xieabd,
Yuchen Linad,
Wenzhe Liabd,
Changliang Liuad,
Xiaojin Lic,
Hua Guoc,
Jie Mengc,
Haiyan Xu*c,
Chen Wang*ad and
Yanlian Yang*ad
abd,
Ling Zhuad,
Jiaxi Pengad,
Mo Yangad,
Hanyi Xieabd,
Yuchen Linad,
Wenzhe Liabd,
Changliang Liuad,
Xiaojin Lic,
Hua Guoc,
Jie Mengc,
Haiyan Xu*c,
Chen Wang*ad and
Yanlian Yang*ad
aCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Key Laboratory of Biological Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: wangch@nanoctr.cn; yangyl@nanoctr.cn; Tel: +86-10-82545561 Tel: +86-10-82545559
bAcademy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, P. R. China
cInstitute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100005, P. R. China. E-mail: xuhy@pumc.edu.cn; Tel: +86-10-69156437
dUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 18th April 2017
Abstract
The chemokine CXCL12, and its receptor CXCR4, have been recognized to be involved in various instances of cancer metastasis. The CXCL12/CXCR4 axis has emerged as a potential target for cancer therapy. Here, we demonstrate a designed peptide (W4) targeting CXCL12 with high binding affinity, and describe its significant inhibitory effect on the CXCL12/CXCR4 axis. We show that W4 has comparable binding affinity (KD = 5.7 × 10−8 M) to that of the antibody of CXCL12 (KD = 3.0 × 10−9 M) using the surface plasma resonance (SPR) technique. Upon introduction of W4, the circular dichroism (CD) spectra show that the α-helical structure of CXCL12 gradually transformed into a β-sheet and random coil. These effects lead to the significant inhibitory effects on the CXCL12/CXCR4 axis using the CXCR4-positive breast cancer cell lines MCF-7 and MDA-MB-231 and the leukemia cell lines HL-60 and U937 as models. The results show that W4 significantly inhibits CXCL12-induced cell migration of MCF-7, MDA-MB-231, HL-60 and U937 even to 20.0% when the mole ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, completely abolishing the effect of CXCL12. These effects may provide evidence of the modulating ligand–receptor interactions of peptides as anti-ligand molecules that differ from the traditional receptor antagonists leading to therapeutic agents.
:1, completely abolishing the effect of CXCL12. These effects may provide evidence of the modulating ligand–receptor interactions of peptides as anti-ligand molecules that differ from the traditional receptor antagonists leading to therapeutic agents.
1 Introduction
Cancer metastasis is a major cause of morbidity and mortality.1 In the process of metastasis, cancer cells locally invade the surrounding tissue, enter the lymph and blood circulation, survive in the circulation, exit at distant organs, and adapt to the new microenvironments there.2 CXCL12, also known as stromal derived factor-1 (SDF-1), belongs to the family of CXC chemokines that bind to and activate the seven-transmembrane G protein-coupled receptor CXCR4.3 The CXCL12/CXCR4 axis is reported to be involved in various cellular behaviors such as cell migration, adhesion and invasion.4 Under normal conditions, the CXCL12/CXCR4 axis helps leukocyte recruitment and is critical for homing and retention of hematopoietic cells to the bone marrow. In addition to its physiological roles, CXCR4 has also been found to be a prognostic marker which is shown to regulate site-specific distant metastasis in various types of human tumors including breast,5,6 prostate7,8 and colon tumors9,10 and multiple myeloma.11 The high expression level of CXCR4 in tumor cells enables them to migrate to target tissues (like lung, liver and bone) that have an abundant source of the ligand CXCL12, which allows tumor cells to form directional migration via the CXCL12 chemotactic gradient. The influx of leukocytes into tissues has similarities with the invasion of tissues by metastasizing tumor cells, as well as hematopoietic tumors. For example, myeloid12,13 and lymphoid14,15 leukemia cells also have a high level of CXCR4 expression, which induces leukemia migration towards stromal cells secreting CXCL12, and as a result of this they reside in the microenvironment that protects them from chemotherapy-induced cell death.16,17As noted, increasing efforts have been made in impeding the function of the CXCL12/CXCR4 axis by targeting either the ligand or the receptor using small molecules,18 peptides19,20 and antibodies.21,22 Most of these antagonists are still at the stage of preclinical study.23 Previous studies mostly focused on the blockade of CXCR4 with CXCR4 antagonists, however, targeting the other part of this axis, the chemokine CXCL12, represents an attractive alternative approach. It has been reported that saccharides hinder the migration of human tumor cells via blocking of the interaction between CXCL12 and CXCR4, and these findings were supported by the structural analysis of CXCL12’s interactions with saccharides.24,25 Furthermore, the treatment of human CD4+ T lymphocytes with a series of chalcones inhibited CXCL12-induced migration.26 By analogy to the effect of neutralizing antibodies, these molecules behave as neutralizing compounds that may prove to have valuable pharmacological and therapeutic potentials. However, peptides are rarely reported binding to the chemokine itself to neutralize its biological activity. Considering that peptides are readily synthesized, metabolically cleaved and rapidly cleared from the body without introducing serious immune responses and toxic side effects, it is very necessary and of great significance to develop peptides that target CXCL12 to provide more therapeutic options in the treatment of tumor metastasis.
Allosteric modulation of protein–substrate interactions results from binding of an allosteric ligand to a distinct site on the protein, in a way that does not directly compete with the binding site of its cognate substrate.27 Binding of the allosteric ligand may increase or decrease the affinity to the cognate substrate through minor modifications to the conformation of the protein, thus opening or closing the active site and resulting in positive or negative protein activity.28 Here, we have identified a CXCL12 recognition peptide W4 with high binding affinity to target and modulate the secondary structure of CXCL12, and we described its significant inhibitory effect on the CXCL12/CXCR4 axis using the CXCR4-positive breast cancer cell lines MCF-7 and MDA-MB-231, and leukemia cell lines HL-60 and U937. We speculate a potential allosteric coupling at the binding site of CXCL12 to W4 and the conformational change caused might transmit the signal to the active site of CXCL12, which dramatically reduces the interaction of CXCL12 with its receptor CXCR4. These effects may provide a feasible strategy for modulating the ligand–receptor interactions of peptides leading to therapeutic anti-ligand agents.
2 Experimental section
2.1 Peptide W4
The peptide W4 with the sequence of GGRGDLDWIQRYLRDA was selected from screening of a collection of de novo designed peptides based on the amyloid proteins and fragments of various proteins. A surface plasmon resonance (SPR)-based selection process was performed for screening peptide ligands with high binding affinity. This collection of peptides was examined using scanning probe microscopy (SPM) in our previous studies for unraveling various interactions, such as the hydrogen bonding, electrostatic and hydrophobic interactions between the side chains, and peptide assemblies.29–31 The peptide W4 used in this study (purity 98%), with and without fluorescein isothiocyanate (FITC) labeling, was purchased from GL Biochem Ltd, Shanghai, China and used without further purification.2.2 Cell culture
The human promyelocytic leukemia HL-60 cells and myelomonocytic leukemia U937 cells were purchased from the Chinese Academy of Medical Science & Peking Union Medical College, Beijing, China. The human breast cancer MCF-7 cells and MDA-MB-231 cells were kindly provided as a gift from Professor Chunying Chen at the National Center for Nanoscience and Technology, Beijing, China. The MCF-7, HL-60 and U937 cells were cultured in RPMI-1640 culture medium (Gibco, USA) and the MDA-MB-231 cells in DMEM culture medium (Gibco, USA). The culture media were supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, USA) and 1% streptomycin–penicillin. The cells were placed in a dish in the cell culture incubator with 5% CO2 at 37 °C.2.3 Surface plasmon resonance (SPR)
The binding affinity and binding dynamics parameter of W4 and anti-CXCL12 with CXCL12 were investigated using an SPR system (Kx5, Plexera, USA) at 25 °C. CXCL12 was in the flowing phase, and the peptide and antibody were immobilized on the commercial SPR chips with modification of the self-assembled layer of the carboxyl group-containing thiol molecules via a covalent bond on the gold (50 nm) coated glass (Plexera, USA). CXCL12 (R&D Systems, USA) was dissolved in phosphate buffer saline (PBS; Hyclone, USA) at a series of concentrations (50, 100 and 200 nM), and 1 mg mL−1 W4 and anti-CXCL12 (RayBiotech, USA) dissolved in triple distilled water were dropped on the chip. Then the surface was washed with water twice. The real-time binding signals were recorded and analyzed by a Data Analysis Module (DAM, Plexera, USA). The binding analysis was performed using BIAevaluation 4.1 software (Biocore, GE Healthcare, USA).2.4 ELISA assay
0.1 μg of CXCL12 diluted in 50 μL of coating buffer (0.1 M sodium bicarbonate, pH 9.6) was added to each well of a 96-well microplate. Then the plate was incubated for 2 hours at 37 °C, washed three times with PBS containing 0.01% Tween 20 (PBST) and blocked for 2 h at 37 °C with 5% skim milk in PBST. The plate was then washed three times with PBST and 50 μL of anti-CXCL12 diluted in 5% skim milk in PBST with different concentrations of W4 was added and incubated for 1 h at 37 °C. The plate was washed three times with PBST and 100 μL of horseradish peroxidase (HRP)-conjugated anti-IgG (Cell signaling Technology, USA) diluted in 5% skim milk in PBST was added and incubated for 1 h at 37 °C. The plate was washed three times with PBST and developed using 3,3,5,5-tetramethylbenzidine (TMB). The reaction was stopped with 100 μL of 2 M H2SO4 and the absorbance of each well read using a microplate reader (Infinite M200, Tecan, Switzerland) at a test wavelength of 450 nm.2.5 Dot blot assay
0.1 μg of CXCL12 diluted in 5 μL of PBS was spotted onto a nitrocellulose membrane (0.45 μm, Pierce, USA) and dried at room temperature. Then the membrane was blocked with 5% skim milk in Tris-buffered saline (TBS) containing 0.01% Tween 20 (TBST) for 1 h at room temperature, washed three times with TBST and incubated with anti-CXCL12 diluted in 5% skim milk in TBST with different concentrations of W4 overnight at 4 °C. The membrane was washed with TBST three times and incubated for 1 h with HRP-conjugated anti-IgG in 5% skim milk in TBST at room temperature. Following five washes with TBST, the membrane was developed with an ECL chemiluminescence system.2.6 Circular dichroism (CD) spectroscopy assay
The CD experiments were performed using a quartz cell with 0.1 cm path length using a spectropolarimeter (J-1500, JASCO, Japan) equipped with a microcomputer. The CXCL12 solution at 5 μM was prepared in triple distilled water with different concentrations of W4. The CD spectra of CXCL12 were recorded between 190 nm and 260 nm with a scan speed of 200 nm min−1, spectral bandwidth of 2 nm, and data pitch of 0.5 nm. Every CD spectrum data of CXCL12 used was the average of three scans with the background of the peptide and the solvent subtracted.2.7 Molecular docking
The NMR structure of the chemokine CXCL12 was retrieved from the RSCB Protein Data Bank (PDB ID: 2SDF). The structure of W4 was obtained by PyMol, and the formation converted and 3D structure generated by Open Babel. The rotatable bonds within the peptide were defined using AutoDockTools. AutoDock Vina was used for the automated docking simulation. The standard parameters were created with Raccoon, a script program in the AutoDock software package, and used. Calculations were performed with a 26-node PC cluster in which each node has a 16-core central processing unit (2.8 GHz, Pentium-D, Intel, USA). The lowest binding free energy was recorded as the binding affinity between W4 and CXCL12.2.8 Wound healing assay
The MCF-7 and MDA-MB-231 cells were plated in a 6-well plate at a concentration of 50 × 104 and 30 × 104 cells per well, respectively, and allowed to form a confluent monolayer overnight. Then the monolayer was scratched with a 10 μL pipette tip, washed 3 times with PBS to remove the floating cells, and treated with different concentrations of W4 in culture medium in the presence or absence of 100 ng mL−1 CXCL12. The cells were photographed (IX71, OLYMPUS, Japan) at 3 randomly selected sites per well and incubated for 24 h at 37 °C. The cells were then photographed again at the selected sites. The actual migration of the cells was analyzed according to the original and final wound zone.2.9 Transwell assay
The transwell assay was applied using millicell hanging cell culture inserts (5 μm for U937, 8 μm for MCF-7, MDA-MB-231 and HL-60; Millipore, Switzerland). The MCF-7 (10 × 104 cells per chamber), MDA-MB-231 (10 × 104 cells per chamber), HL-60 (20 × 104 cells per chamber) and U937 (20 × 104 cells per chamber) cells were plated into the upper chambers of the inserts in culture medium. Then the inserts were placed into a well of a 24-well plate containing 800 μL culture medium in the presence or absence of CXCL12 treated with different concentrations of W4. The concentrations of CXCL12 were 100 ng mL−1 for the MCF-7 and MDA-MB-231 cells, 200 ng mL−1 for the HL-60 cell and 50 ng mL−1 for the U937 cell (at these concentrations, the cells reached the maximum of migration, data not shown). After a 24 h incubation at 37 °C, for the MCF-7 and MDA-MB-231 cells, the top surface of the insert was scraped using a cotton swab, and the cells on the lower surface of the membrane were fixed and stained with crystal violet. The cells that had migrated to the bottom of the membrane were visualized and counted using a microscope (DMI3000B, Leica, Germany). For each replicate, the cells in five randomly selected fields were counted and averaged. For the HL-60 cells and U937 cells, the cells migrating to the lower chambers were counted.To confirm the absence of off-target effects of W4 on cell migration, the HL-60 and U937 cells were plated into the upper chambers of the inserts and then the inserts were placed in a well containing 800 μL of culture medium in the presence of CXCL12 and 10 μg mL−1 anti-CXCL12 prior to the addition of W4. After 24 h incubation at 37 °C, the cells migrating to the lower chambers were counted.
2.10 Realtime fluorescence quantitative PCR
The MCF-7, MDA-MB-231, HL-60 and U937 cells were maintained overnight in medium containing 0.5% FBS at 1 × 106 cells per well in a 6-well plate. After that, CXCL12 (100 ng mL−1 for MCF-7 and MDA-MB-231, 200 ng mL−1 for HL-60 and 50 ng mL−1 for U937) with and without W4 at the mole ratio of 1:1 was added into the medium for 15 min at 37 °C. The total RNA was extracted using Trizol (Life Technologies, USA), and then reverse transcribed with a QuantScript RT Kit (TIANGEN, China) for 1 h at 37 °C, according to the instructions of the manufacturer. The following primers (Sangon Biotech, China) were used for the subsequent PCR: Vimentin (forward: 5′ GAA CGC CAG ATG CGT GAA ATG 3′; reverse: 5′ CCA GAG GGA GTG AAT CCA GAT TA 3′), N-cadherin (forward: 5′ GAG GAG TCA GTG AAG GAG TCA 3′; reverse: 5′ GGC AAG TTG GAG GGA TG 3′), MMP2 (forward: 5′ ACC CTC AGA GCC ACC CCT AA 3′; reverse: 5′ AGC CAG CAG TGA AAA GCC AG 3′), and MMP9 (forward: 5′ TCC CTG GAG ACC TGA GAA CC 3′; reverse: 5′ CGG CAA GTC TTC CGA GTA GTT 3′). The reverse transcription was checked with primers for the GAPDH gene (forward: 5′ GAG AAG GCT GGG GCT CAT TT 3′; reverse: 5′ AGT GAT GGC ATG GAC TGT GG 3′) as a control. Then the cDNA was subjected to PCR with a Realplex 4 (Eppendorf, Germany) and the cycling conditions were: 5 min at 95 °C, 40 cycles of 15 s at 95 °C, 15 s at 60 °C, 200 s at 72 °C, and finally an elongation step of 5 min at 72 °C.
2.11 Western blot analysis
The MCF-7, MDA-MB-231, HL-60 and U937 cells were maintained overnight in medium containing 0.5% FBS at 1 × 106 cells per well in a 6-well plate. After that, CXCL12 (100 ng mL−1 for MCF-7 and MDA-MB-231, 200 ng mL−1 for HL-60, and 50 ng mL−1 for U937) with and without W4 at a mole ratio of 1:1 was added into the medium for 15 min at 37 °C. Then, adding excess PBS, the cells were washed and lysed in RIPA lysis buffer (Pierce, USA) supplemented with protease inhibitors and a phosphatase inhibitor mixture on ice for 1 h. The lysates were clarified by centrifugation at 14000g for 10 min. Equal protein (10 μg) was loaded and separated on 10% Bis–Tris gels (Invitrogen, USA), and transferred to PVDF membranes (0.45 μm, Millipore, USA). The phosphorylated and total proteins were probed with specific primary antibodies (Akt, phosphorylated-Akt, Erk, phosphorylated-Erk, p38, phosphorylated-p38, and GAPDH; Cell signaling Technology, USA) overnight at 4 °C and the secondary antibodies conjugated to HRP for 1 h at room temperature. The immunocomplex on the membrane was visualized using a ChemiDoc® Touch Imaging System (Bio-Rad, USA) with SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Scientific, USA).
2.12 Confocal microscopy analysis
The MCF-7 and MDA-MB-231 cells were seeded at 10 × 104 cells per dish and incubated for 24 h. Then the cells were washed 3 times, followed by supplementation with 1 mL of culture medium containing 1 μM rhodamine B isothiocyanate (RBITC)-labeled CXCL12 (Abace Biology, China), 1 μM FITC-labeled W4, and 1 μM RBITC-labeled CXCL12 with FITC-labeled W4. The cells were incubated for 1 h at 37 °C. Next the cells were washed and incubated with Hoechst 33342 (Sigma-Aldrich, USA) for 10 min. Images were taken using a laser confocal microscope (LSM760, Zeiss, Germany).2.13 Flow cytometry assay
The MCF-7, MDA-MB-231, HL-60 and U937 cells were washed once with washing buffer (PBS containing 2% FBS) and then resuspended with culture medium to 5 × 106 cells per mL. 50 μL of the cell suspension was added into the tube and centrifuged at 500g for 1 min. Then the supernatant was removed carefully. 60 μL of RBITC-labeled CXCL12, FITC-labeled W4, and RBITC-labeled CXCL12 with FITC-labeled W4 at 1 μM in culture medium were added into the tube. The cells were incubated at room temperature for 1 h and then washed twice with 1 mL of washing buffer. Then the supernatant was removed and the cells were resuspended with 500 μL of washing buffer. 1 × 104 cells were analyzed by a C6 Accuri flow cytometer (BD Biosciences, USA).2.14 Statistical analysis
All the experiments were carried out in triplicate and Student’s t-test was performed to assess the statistical significance of the results (*P < 0.05 and **P < 0.01).3 Results and discussion
3.1 Binding affinity between W4 and CXCL12
To validate the binding parameters (the equilibrium dissociation constant KD) between W4 and CXCL12, an SPR assay was employed, which allowed for estimation of the kinetic and binding constants for the interactions between them. A series of concentrations (50, 100 and 200 nM) of CXCL12 were used to investigate the KD of the binding and the antibody of CXCL12 was also determined as a positive control. We could tell that the SPR signal intensity increased with the rise in the concentration (Fig. S1†). The sensor grams were recorded for calculation of the quantitative binding kinetic parameters, such as the association rate constant ka and dissociation rate constant kd, as shown in Table 1. The KD values were calculated from the measured ka and kd (KD = kd/ka). As listed, W4 showed comparable binding affinity (5.7 × 10−8 M) to that of anti-CXCL12 (3.0 × 10−9 M) binding to CXCL12.| Kinetic parameters | ||||
|---|---|---|---|---|
| ka (M−1 s−1) | kd (s−1) | KA (M−1) | KD (M) | |
| Anti-CXCL12 | 2.5 × 104 | 7.6 × 10−5 | 3.3 × 108 | 3.0 × 10−9 |
| W4 | 1.4 × 104 | 8.0 × 10−4 | 1.8 × 107 | 5.7 × 10−8 |
Using ELISA and dot blot assay, we next studied the competitive binding between W4 and the antibody of CXCL12 to CXCL12. Early analysis of the binding affinity between W4 and CXCL12 revealed comparable binding capability of W4 with anti-CXCL12, leading to the hypothesis that W4 would compete with anti-CXCL12 to target CXCL12. In the ELISA assay, for the anti-CXCL12 supplemented assay that didn’t contain the peptide, the absorbance of HRP-anti-IgG was at a relatively high level. After being treated with W4, the absorbance was decreased and tested in a concentration-dependent manner, exhibiting that the antibody binding to CXCL12 could be partly replaced by W4 (Fig. 1A). Stronger experimental evidence for the competitive binding between W4 and anti-CXCL12 was shown in the dot blot images. The antibody bound to the protein CXCL12, which led to the development of HRP-anti-IgG positive spots. However, this effect was regulated by treatment with W4 at a mole ratio of 10:1 with anti-CXCL12, which showed a light spot (Fig. 1B).
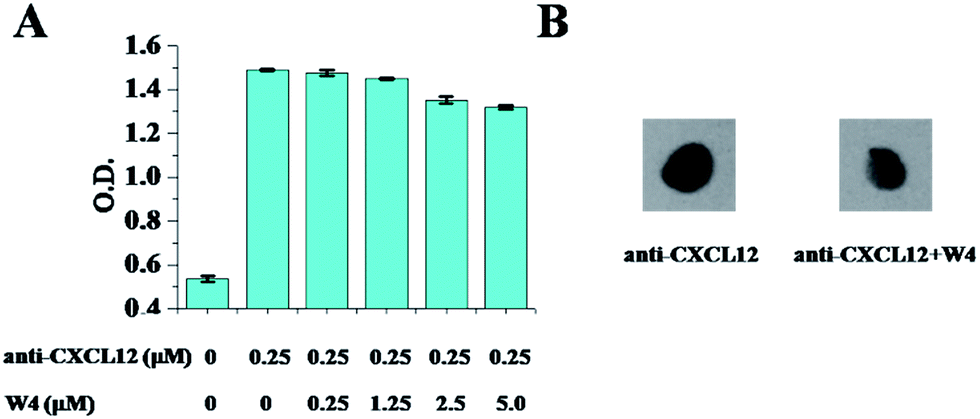 | ||
| Fig. 1 Competitive binding between anti-CXCL12 and W4. (A) ELISA assay is used to measure the binding levels of anti-CXCL12 to CXCL12 after treatment of different concentrations of W4. The error bars represent the standard deviation (n = 3). (B) The dot blot analysis of the binding amount of anti-CXCL12 to CXCL12 after treatment with W4. The mole ratio of anti-CXCL12 to W4 was 1:10. | ||
These results clearly indicate that W4 has comparable binding affinity to that of anti-CXCL12, indicating that W4 could be a possible candidate for targeting CXCL12 for interfering with the binding of CXCL12 to the receptor CXCR4.
3.2 Transformation of the secondary structure of CXCL12 induced by W4
Considering that chemokines are rather small proteins, questions concerning the structural change at the binding site for chemokines are raised.32 Proteins can fold into several well-defined secondary structures, originating mainly from the strong intramolecular hydrogen bonds, such as α-helixes, β-sheets and random coils.33 The conformation of proteins can be changed due to interaction with other molecules, which can influence the formation of the intramolecular hydrogen bonds in the protein. In order to investigate the conformation change of CXCL12 upon W4 addition, the circular dichroism (CD) spectra of CXCL12 with and without W4 interaction were obtained in water. As shown in Fig. 2, the CD spectrum of CXCL12 in solution exhibited a maximum value near 190 nm and negative values around 200 nm, corresponding to the mixed structures of α-helix, β-sheet and random coils, which was consistent with previous reports.34 Upon introduction of W4 into the CXCL12 solution, the characteristic peaks near 190 nm and 200 nm were weakened, and the positive band shifted to the right little by little, indicating that the α-helical structure of CXCL12 gradually transformed into a β-sheet structure and random coils after interacting with W4. Analysis within the software supported the transformation of the secondary structures of CXCL12 by mixing with W4 at different mole ratios (Table 2), which was in agreement with the CD spectra. The percentages of the α-helix, β-sheet and random coils structures of CXCL12 were 18.1%, 40.5% and 41.4%, respectively. After mixing with the peptide, the secondary structure of CXCL12 was induced into the β-sheet structure and random coils, manifesting so that the percentage of β-sheet and random coils increased to 53.2% and 46.8%, while the α-helical structure disappeared when the mole ratio of CXCL12 and W4 was 1:5. Therefore, the CD spectra revealed that the addition of W4 could lead to conformation change of CXCL12.
 | ||
| Fig. 2 Transformation of the secondary structures of CXCL12 induced by W4. Every CD spectrum data used was the average of three scans and corrected for the background of the peptide and solvent. | ||
| Ratio (%) | |||||
|---|---|---|---|---|---|
| n(CXCL12):n(W4) |
|||||
| CXCL12 | 1:0.2 |
1:0.5 |
1:1 |
1:5 |
|
| α-Helix | 18.1 | 14.5 | 7.6 | 3.5 | 0 |
| β-Sheet | 40.5 | 43.2 | 48.3 | 51.9 | 53.2 |
| Random coils | 41.4 | 42.3 | 44.1 | 44.6 | 46.8 |
| Total | 100 | 100 | 100 | 100 | 100 |
To further reveal the interaction mode between W4 and CXCL12, the automated docking simulation was performed. The crystal structure of human CXCL12 (PDB ID: 2SDF) was utilized as the target structure, whereas the peptide W4 was used as a docking probe. The molecular docking simulation initiated with AutoDock Vina (Fig. 3) showed the designed recognition peptides were mostly binding to the α-helix and random coil domains of CXCL12. According to the illustration, W4 formed intermolecular hydrogen bonds with the CXCL12 proteins, like Arg12, Pro32, Asn33, Leu36, Pro53 and Asn67. More importantly, W4 formed three continuous hydrogen bonds with the Gln59 and Glu63 region of CXCL12, which formed an α-helix structure in the C-terminal domain. These interactions appeared to efficiently change the α-helical structure of CXCL12 after binding with W4 with high affinity.
 | ||
| Fig. 3 Molecular modeling simulations of CXCL12 (2SDF) with W4 (aqua sticks). The red, yellow and green parts in the CXCL12 protein structure represent the α-helix, β-sheet and random coil domains, respectively. The zoomed-in part shows the possible hydrogen bonds between Gln59 and Glu63 in the α-helix domain of CXCL12 and the N-terminal of W4. The molecular docking studies were initiated with AutoDock Vina. | ||
3.3 Inhibition of CXCL12-induced migration of both breast cancer cells and acute myelocytic leukemia cells by W4
Preclinical studies on different tumor models have revealed that CXCR4 activation mediates tumor cell migration towards the CXCL12 expressing organs, which is a central player in tumor progression.3,35 Therefore, in the following chemotaxis experiments, the wound healing assay and transwell assay were utilized to determine whether W4 could weaken the binding of CXCL12 to CXCR4 and then inhibit CXCL12-induced migration of the CXCR4-positive cell lines. The four CXCR4-positive cell lines, MCF-7, MDA-MB-231, HL-60 and U937, were chosen as models and the expression levels of CXCR4 in the four cell lines were determined to be 61.4%, 88.1%, 97.6% and 92.5%, respectively [analyzed by flow cytometry (FCM) using the CXCR4 antibody, Fig. S2†].The wound healing assay is shown in Fig. 4. 24 h after the cell monolayers were wounded, ∼30.0% of MCF-7 cells and ∼20.0% of MDA-MB-231 cells without CXCL12 treatment had filled in the cleared areas. After CXCL12 was supplemented, the adhesive cells were enhanced to migrate into the wounded area (set at 100% as a control). When treated with W4, cell migration was significantly inhibited in the two breast cell lines, and the inhibitory effect was associated with the concentration. With the increase in the mole ratio of W4 to CXCL12, the migration rates gradually reduced, especially when the mole ratio was 1:1, where the optimal inhibitory effects reached 30.0% and 35.0% for MCF-7 and MDA-MB-231 respectively.
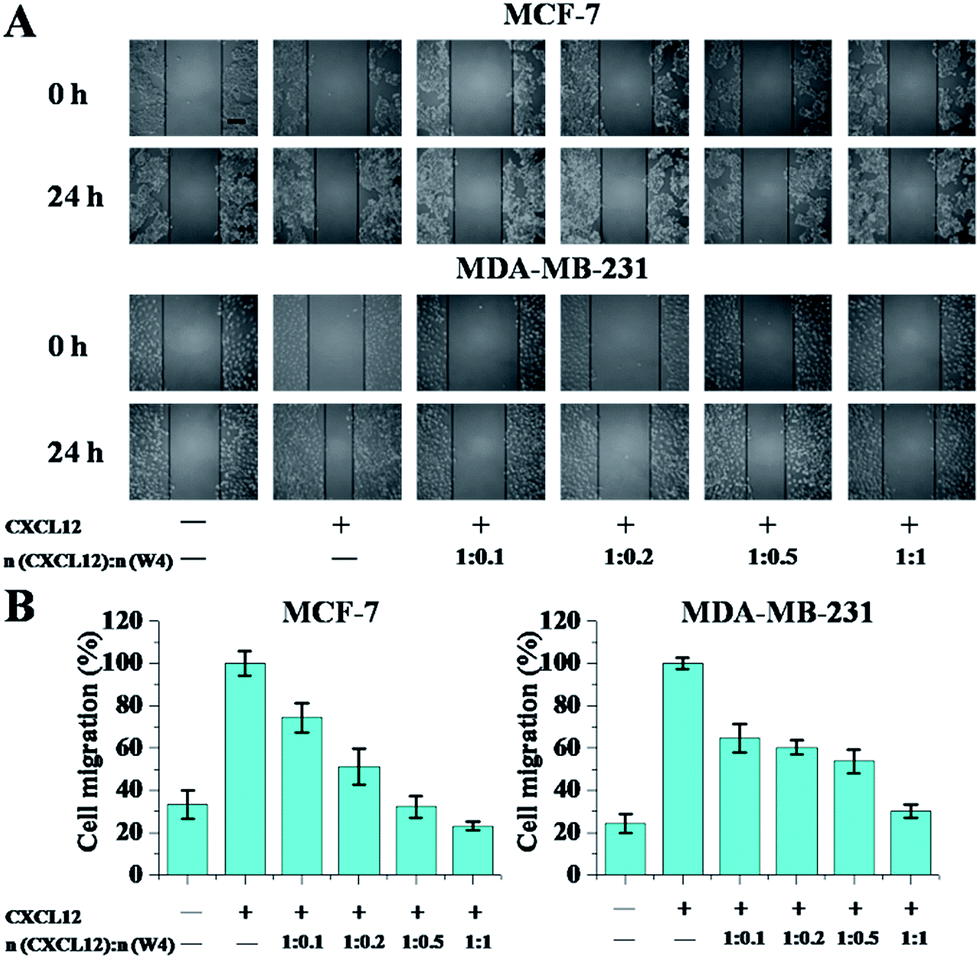 | ||
| Fig. 4 The wound healing assay showed the inhibitory effect of W4 on cell migration. (A) Representative images showed the migration of two adhesive cancer cells before (0 h) and after (24 h) W4 treatment in the presence or absence of CXCL12. The scale bar represents 200 μm. (B) The areas of the migrated cells were counted at the three selected sites. The CXCL12 supplemented one without W4 was set at 100% as the control. The error bars represent the standard deviation (n = 3). | ||
In the transwell assay, CXCL12 was treated with W4 prior to being added to the lower chamber. In the absence of CXCL12, the migration of cells was ∼40.0% for MCF-7 and ∼20.0% for MDA-MB-231, HL-60 and U937 (Fig. 5). When supplied with CXCL12, the cells were enticed to migrate to the lower chamber. By visualizing the cells that migrated to the bottom of the culture inserts, when treated with W4 at different mole ratios with CXCL12, the migration was markedly inhibited in all cell lines but the migration percentages were relatively high when the concentration of W4 was at a lower level. When the mole ratio reached 1:1, the migration percentages were substantially reduced to their minimum of ∼20.0% for MCF-7, ∼30.0% for MDA-MB-231, ∼45.0% for HL-60 and ∼40.0% for U937, completely abolishing the effect of CXCL12.
 | ||
| Fig. 5 A transwell assay shows the inhibitory effect of W4 on cell migration. (A) Representative images show the migration of two adhesive cancer cells after W4 treatment for 24 h in the presence or absence of CXCL12. The scale bar represents 100 μm. (B) The migrated cells are visualized and counted in five randomly selected fields using a microscope. A CXCL12 supplemented assay without W4 and set to 100% is used as the control. (C) To confirm the absence of off-target effects of W4 on cell migration, inserts with HL-60 and U937 cells were placed into a well containing 800 μL of culture medium in the presence of CXCL12 and 10 mg mL−1 anti-CXCL12, prior to the addition of W4. The molar ratio of CXCL12 to W4 was 1:1. The error bars represent the standard deviation (n = 3). | ||
To confirm the absence of off-target effects of W4 on cell migration, inserts with HL-60 and U937 cells were placed into a well containing culture medium in the presence of CXCL12 and anti-CXCL12, prior to the addition of W4. It turned out that after binding with anti-CXCL12, CXCL12 could not be inhibited by W4 any more, indicating that W4 targeted CXCL12 specifically and thus was functioning (Fig. 5C). Overall, these changes indicate that W4 could more effectively inhibit CXCL12-induced cell migration of both the breast cancer cell lines and acute myelocytic leukemia cell lines that are overexpressing CXCR4.
We have provided biological data to show that a peptide is able to bind to the chemokine CXCL12, and thus alter the functional consequences of the interaction between CXCL12 and its receptor CXCR4, as demonstrated here by the inhibition of in vitro chemotaxis. Considering that chemokines are rather small proteins, our observations raise questions concerning the molecular mechanism of the neutralizing molecule W4. It has been reported that CXCL12 binds to CXCR4 in a two-step process. First, the extracellular flexible N-terminal domain of the receptor recognizes and binds to the surface of CXCL12, and then CXCL12 “docks” its own flexible N-terminal domain into the receptor resulting in activation and downstream signaling.36 The first step, however, represents a viable alternative target and a prototypic example of protein–protein interactions, mediated by a binding surface area covering a large portion of the chemokine. From the molecular docking results, we could see that W4 wraps nearly halfway around the chemokine, creating an interface that prevents the binding between CXCL12 and the CXCR4 N-terminus (Fig. 3). Noteworthy, the reported binding affinity of CXCL12 for CXCR4 (KD ∼ 2 to 4 × 10−9 M)37,38 is one order of magnitude higher than that of W4 for CXCL12 (KD = 5.7 × 10−8 M, Table 1). Compared with the significant inhibitory effects of W4 on cell migration (Fig. 4 and 5), we speculated that competitive inhibition might not be the sole reason for blocking of the CXCL12/CXCR4 axis.
On the other hand, it has been described that minor modifications to the conformation of a protein are nevertheless sufficient to affect the protein’s ability to bind to, or properly interact with, its substrates, which is a common form of allosteric regulation.39,40 Volkman and his colleagues found the allosteric behavior of the chemokine CXCL12 with a CXCR4 peptide increased the CXCL12 dimerization affinity by allosteric coupling at its binding site.32 In our work, the CD spectrum of CXCL12 in solution shows that its conformation was changed upon interaction with W4 as the α-helical structure disappeared and transformed into β-sheet and random coil structures (Fig. 2 and 3). We could propose a hypothesis that the coordinated movement of the segments of CXCL12 might transmit the allosteric signal to the active site on the N-terminal tail of CXCL12, which is deformed, and dramatically reduce the interaction of CXCL12 with its receptor CXCR4.
However, with the increase of the mole ratio of W4 to CXCL12, the inhibitory effects detected on the four cell lines all slightly decreased (Fig. S3 and S4†), perhaps because W4 had a mild tendency of aggregation over 24 h, which led to a lower effective drug concentration being obtained at the target protein (Fig. S5†).
3.4 Down-regulation of CXCL12-induced expression of genes and proteins by W4
CXCR4/CXCL12 interaction has been demonstrated to increase the expression of the Vimentin, N-cadherin, MMP-2 and MMP-9 genes, which accounts for the invasion and metastasis of tumor cells.8,41 To clarify whether W4 inhibits cell migration through affecting the intracellular signaling of CXCL12/CXCR4, we performed realtime fluorescence quantitative PCR for activation of the Vimentin, N-cadherin, MMP-2 and MMP-9 genes in the four cell lines. The cells were treated with CXCL12 alone, and CXCL12 with W4 at a mole ratio of 1:1. As shown in Fig. 6, CXCL12 treatment enhanced the expression levels of the Vimentin, N-cadherin, MMP-2 and MMP-9 genes in reference to that of the control, while treatment with W4 significantly diminished the CXCL12-induced gene expression. These results clearly indicated that W4 could interact with CXCL12 and then inhibit cancer cells from responding to CXCL12 stimulation by suppressing the expression of the Vimentin, N-cadherin, MMP-2 and MMP-9 genes.
 | ||
| Fig. 6 Effects of W4 on the CXCL12-induced activation of genes including Vimentin, N-cadherin, MMP2 and MMP9 determined by realtime fluorescence quantitative PCR. GAPDH mRNA was used to normalize the variability in template loading. The mole ratio of CXCL12 to W4 was 1:1. The error bars represent the standard deviation (n = 3). | ||
For upstream proteins, the stimulation of CXCL12 on CXCR4 results in the activation of Akt, Erk and p38.42–44 We next conducted western blot analysis for activation of Akt (phospho-Akt), Erk (phospho-Erk) and p38 (phospho-p38) in the four kinds of CXCR4-positive cell lines to further clarify whether W4 inhibits cell migration through affecting the intracellular signaling of CXCL12/CXCR4. The results displayed in Fig. 7 show that CXCL12 treatment enhanced the phosphorylation levels of Akt, Erk and p38 (lane b) in reference to that of the control, while W4 dramatically inhibited the robust phosphorylation of Akt, Erk and p38 in the cells stimulated by CXCL12 (lane c). These results clearly indicate that W4 inhibits CXCR4-positive cells from responding to CXCL12 stimulation by suppressing the expression of p-Akt, p-Erk and p-p38.
 | ||
| Fig. 7 Effects of W4 on CXCL12-induced Akt, Erk and p38 activation of cancer cells by western blot analysis. (a) Control, (b) CXCL12 treatment only, and (c) treated with CXCL12 and W4 at a mole ratio of 1:1. | ||
3.5 W4 binds to CXCL12 but not to the receptor CXCR4
The evidence supporting W4 binding to CXCL12 was provided by analysis using confocal microscopy. The results of the two adhesive breast cancer cell lines are displayed representatively in Fig. 8A. When the cells were incubated with RBITC-labeled CXCL12 alone, we found brighter red fluorescence dispersed on the whole cell membrane uniformly. If W4 was used to interact with CXCL12, the signals of red fluorescence were observed to decrease on the cell membrane, which indicated that the amount of CXCL12 bound to the cell membrane reduced after interacting with W4. However, when the cells were incubated in medium containing FITC-W4, the green fluorescence on the cell membrane was faint in both cells (Fig. S6†). After treatment with CXCL12, the intensity on the cell surface was attenuated and even negligible for the green fluorescence, which proved that W4 didn’t bind to the cell surface. In order to illustrate the amounts of CXCL12 bound to the cell surface, we performed FCM assay. The results showed that lower mean fluorescent intensity (MFI) could be observed after interaction with W4 (Fig. 8B). These data revealed that W4 interacted with CXCL12 and neutralized its binding capacity to its receptor CXCR4, but did not act directly on CXCR4.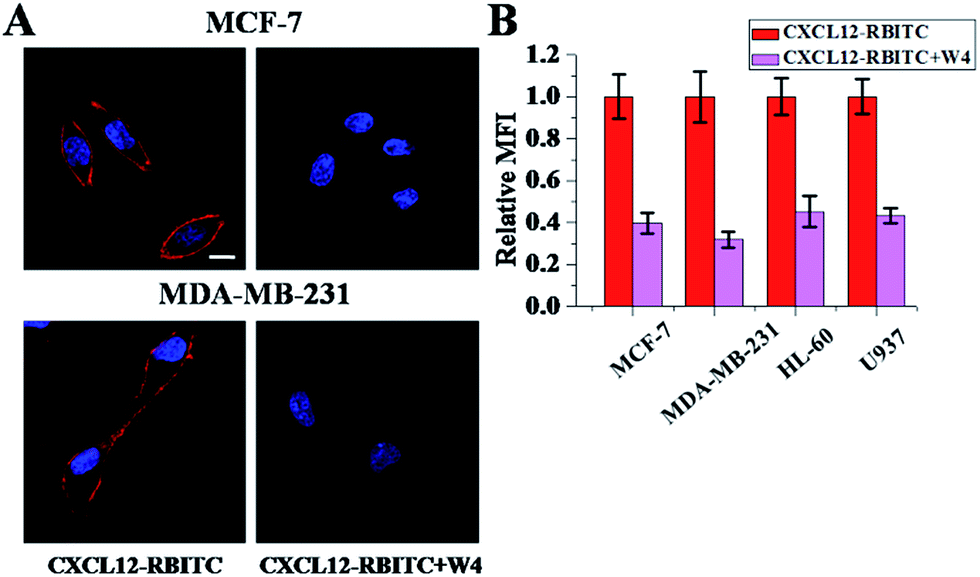 | ||
| Fig. 8 The amounts of CXCL12 bound to the cells after treatment with W4. (A) Confocal microscopy images of CXCL12 (left line) and CXCL12 with W4 (right line) bound to two adhesive cancer cells were obtained using RBITC labeled CXCL12, and the nucleus was revealed with Hoechst 33342 (blue fluorescence). The scale bar represents 10 μm. (B) The amounts of CXCL12 bound to the cell surface after treatment with W4 were measured by FCM analysis. The CXCL12 treated group was set as 1. The error bars represent the standard deviation (n = 3). The mole ratio of CXCL12 to W4 was 1:1. | ||
The CXCR4/CXCL12 axis is a target for therapeutics that block their interaction to inhibit downstream intracellular enzyme activities, as well as to interfere with the stromal cell-mediated tumor cell drug-resistance. In view of the great importance of the CXCL12/CXCR4 axis in the spread of different types of tumors, multiple antagonists targeting the ligand or receptor have been developed. Considering that W4 exhibits potent anti-metastasis activity upon binding to CXCL12 but not to CXCR4, its mechanism of action markedly differs from that of other pharmacological agents acting upon binding to the receptor CXCR4, such as the small molecule AMD3100 (ref. 23 and 45) and peptide T140.19,20 The analysis of the functional properties of these antagonists on constitutively active mutants of CXCR4 reveals that, at high doses, AMD3100 is a weak partial agonist and T140 is an inverse agonist of the receptor functions.46 As a consequence, both AMD3100 and T140 are cautiously considered to be anti-metastatic agents in many cancer types involving CXCR4.47 In contrast to the CXCR4 antagonists, the anti-ligand molecules do not alter the basal function of the receptor CXCR4.
It is notable that to date, the peptide antagonists for CXCL12 itself have been rarely published. Therefore, it is necessary and of significance to develop peptide antagonists targeting CXCL12 for use in anti-metastasis, so that more therapeutic options can be provided to patients. Contrary to those small molecules,24,26 W4 is designed according to the sequence features of human CXCL12 and chemically synthesized, rather than derived from any natural compounds, which makes it more easily, flexibly and economically produced. Another attractive feature of this peptide targeting the chemokine is that it has no cytotoxicity to cells up to a high concentration of 30 μM (Fig. S7†). It is thus interesting to consider anti-ligand peptides as pharmacological agents with mechanisms of action that differ from traditional competitive antagonists binding to the receptor.
4 Conclusion
We demonstrate a peptide W4 that interacts with CXCL12 with high binding affinity (5.7 × 10−8 M). As a result, the conformation change of CXCL12 can be identified, thereby the CXCL12/CXCR4 axis can be blocked leading to impacts on the subsequent biological functions, manifesting such that W4 inhibits the CXCL12-induced cell migration by down-regulating the activation of the Akt, Erk and p38 signaling pathway. We speculate allosteric coupling at the binding site of CXCL12 to W4 and the conformational change caused might transmit the signal to the active site of CXCL12, which dramatically reduces the interaction of CXCL12 with its receptor CXCR4. These results may provide an alternative means of drug design to target ligand–receptor interactions using anti-ligand peptides as therapeutic agents.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21273051, 21673055). The Beijing Natural Science Foundation (Grant No. 2162044) is also gratefully acknowledged. Financial support from CAS Key Laboratory of Standardization and Measurement for Nanotechnology is also gratefully acknowledged.References
- F. Y. Jin, U. Brockmeier, F. Otterbach and E. Metzen, Mol. Cancer Res., 2012, 10, 1021–1031 CrossRef CAS PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed.
- B. A. Teicher and S. P. Fricker, Clin. Cancer Res., 2010, 16, 2927–2931 CrossRef CAS PubMed.
- J. A. Burger and T. J. Kipps, Blood, 2006, 107, 1761–1767 CrossRef CAS PubMed.
- A. Muller, B. Homey, H. Soto, N. F. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verastegui and A. Zlotnik, Nature, 2001, 410, 50–56 CrossRef CAS PubMed.
- M. C. P. Smith, K. E. Luker, J. R. Garbow, J. L. Prior, E. Jackson, D. Piwnica-Worms and G. D. Luker, Cancer Res., 2004, 64, 8604–8612 CrossRef CAS PubMed.
- R. S. Taichman, C. Cooper, E. T. Keller, K. J. Pienta, N. S. Taichman and L. K. McCauley, Cancer Res., 2002, 62, 1832–1837 CAS.
- S. Singh, U. P. Singh, W. E. Grizzle and J. W. Lillard, Lab. Invest., 2004, 84, 1666–1676 CrossRef CAS PubMed.
- S. Brand, J. Dambacher, F. Beigel, T. Olszak, J. Diebold, J. M. Otte, B. Goke and S. T. Eichhorst, Exp. Cell Res., 2005, 310, 117–130 CrossRef CAS PubMed.
- J. Kim, H. Takeuchi, S. T. Lam, R. R. Turner, H.-J. Wang, C. Kuo, L. Foshag, A. J. Bilchik and D. S. Hoon, J. Clin. Oncol., 2005, 23, 2744–2753 CrossRef CAS PubMed.
- Y. Alsayed, H. Ngo, J. Runnels, X. Leleu, U. K. Singha, C. M. Pitsillides, J. A. Spencer, T. Kimlinger, J. M. Ghobrial, X. Y. Jia, G. W. Lu, M. Timm, A. Kumar, D. Cote, I. Veilleux, K. E. Hedin, G. D. Roodman, T. E. WitZig, A. L. Kung, T. Hideshima, K. C. Anderson, C. P. Lin and I. M. Ghobrial, Blood, 2007, 109, 2708–2717 CAS.
- Z. H. Zeng, Y. X. Shi, I. J. Samudio, R. Y. Wang, X. Y. Ling, O. Frolova, M. Levis, J. B. Rubin, R. R. Negrin, E. H. Estey, S. Konoplev, M. Andreeff and M. Konopleva, Blood, 2009, 113, 6215–6224 CrossRef CAS PubMed.
- S. Tavor, I. Petit, S. Porozov, A. Avigdor, A. Dar, L. Leider-Trejo, N. Shemtov, V. Deutsch, E. Naparstek, A. Nagler and T. Lapidot, Cancer Res., 2004, 64, 2817–2824 CrossRef CAS PubMed.
- J. A. Burger, M. Burger and T. J. Kipps, Blood, 1999, 94, 3658–3667 CAS.
- D. A. Sipkins, X. B. Wei, J. W. Wu, J. M. Runnels, D. Cote, T. K. Means, A. D. Luster, D. T. Scadden and C. P. Lin, Nature, 2005, 435, 969–973 CrossRef CAS PubMed.
- J. A. Burger and A. Burkle, Br. J. Haematol., 2007, 137, 288–296 CrossRef CAS PubMed.
- A. Zepeda-Moreno, R. Saffrich, T. Walenda, V. T. Hoang, P. Wuchter, S. Sanchez-Enriquez, A. Corona-Rivera, W. Wagner and A. D. Ho, Exp. Hematol., 2012, 40, 666–674 CrossRef CAS PubMed.
- B. Debnath, S. L. Xu, F. Grande, A. Garofalo and N. Neamati, Theranostics, 2013, 3, 47–75 CrossRef CAS PubMed.
- M. Burger, T. Hartmann, M. Krome, J. Rawluk, H. Tamamura, N. Fujii, T. J. Kipps and J. A. Burger, Blood, 2005, 106, 1824–1830 CrossRef CAS PubMed.
- Z. X. Liang, T. Wu, H. Lou, X. W. Yu, R. S. Taichman, S. K. Lau, S. M. Nie, J. Umbreit and H. Shim, Cancer Res., 2004, 64, 4302–4308 CrossRef CAS PubMed.
- A. Ottaiano, A. di Palma, M. Napolitano, C. Pisano, S. Pignata, F. Tatangelo, G. Botti, A. Acquaviva, G. Castello, P. Ascierto, R. Iaffaioli and S. Scala, Cancer Immunol. Immunother., 2005, 54, 781–791 CrossRef CAS PubMed.
- M. R. Kuhne, T. Mulvey, B. Belanger, S. Chen, C. Pan, C. L. Chong, F. Cao, W. Niekro, T. Kempe, K. A. Henning, L. J. Cohen, A. J. Korman and P. M. Cardarelli, Clin. Cancer Res., 2013, 19, 357–366 CrossRef CAS PubMed.
- J. A. Burger and A. Peled, Leukemia, 2009, 23, 43–52 CrossRef CAS PubMed.
- W. W. Wen, S. Xie, X. L. Xin, M. Y. Geng, J. Ding and Y. Chen, Acta Pharmacol. Sin., 2013, 34, 1554–1559 CrossRef CAS PubMed.
- C. Laguri, F. Arenzana-Seisdedos and H. Lortat-Jacob, Carbohydr. Res., 2008, 343, 2018–2023 CrossRef CAS PubMed.
- M. Hachet-Haas, K. Balabanian, F. Rohmer, F. Pons, C. Franchet, S. Lecat, K. Y. C. Chow, R. Dagher, P. Gizzi, B. Didier, B. Lagane, E. Kellenberger, D. Bonnet, F. Baleux, J. Haiech, M. Parmentier, N. Frossard, F. Arenzana-Seisdedos, M. Hibert and J. L. Galzi, J. Biol. Chem., 2008, 283, 23189–23199 CrossRef CAS PubMed.
- I. Kalatskaya, Y. A. Berchiche, S. Gravel, B. J. Limberg, J. S. Rosenbaum and N. Heveker, Mol. Pharmacol., 2009, 75, 1240–1247 CrossRef CAS PubMed.
- R. A. Laskowski, F. Gerick and J. M. Thornton, FEBS Lett., 2009, 583, 1692–1698 CrossRef CAS PubMed.
- X. Mao, Y. Guo, C. Wang, M. Zhang, X. Ma, L. Liu, L. Niu, Q. Zeng, Y. Yang and C. Wang, ACS Chem. Neurosci., 2011, 2, 281–287 CrossRef CAS PubMed.
- X.-B. Mao, C.-X. Wang, X.-K. Wu, X.-J. Ma, L. Liu, L. Zhang, L. Niu, Y.-Y. Guo, D.-H. Li, Y.-L. Yang and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19605–19610 CrossRef CAS PubMed.
- C. Wang, X. Mao, A. Yang, L. Niu, S. Wang, D. Li, Y. Guo, Y. Wang, Y. Yang and C. Wang, Chem. Commun., 2011, 47, 10638–10640 RSC.
- J. J. Ziarek, A. E. Getschman, S. J. Butler, D. Taleski, B. Stephens, I. Kufareva, T. M. Handel, R. J. Payne and B. F. Volkman, ACS Chem. Biol., 2013, 8, 1955 CrossRef CAS PubMed.
- O. Koch, M. Bocola and G. Klebe, Proteins: Struct., Funct., Bioinf., 2005, 61, 310–317 CrossRef CAS PubMed.
- J. W. Murphy, Y. Cho, A. Sachpatzidis, C. Fan, M. E. Hodsdon and E. Lolis, J. Biol. Chem., 2007, 282, 10018–10027 CrossRef CAS PubMed.
- X. Q. Sun, G. C. Cheng, M. G. Hao, J. H. Zheng, X. M. Zhou, J. A. Zhang, R. S. Taichman, K. J. Pienta and J. H. Wang, Cancer Metastasis Rev., 2010, 29, 709–722 CrossRef CAS PubMed.
- E. W. Smith, A. M. Nevins, Z. Qiao, Y. Liu, A. E. Getschman, S. L. Vankayala, M. T. Kemp, F. C. Peterson, R. Li, B. F. Volkman and Y. Chen, J. Med. Chem., 2016, 59, 4342–4351 CrossRef CAS PubMed.
- M. P. Crump, J. H. Gong, P. Loetscher, K. Rajarathnam, A. Amara, F. Arenzana-Seisdedos, J. L. Virelizier, M. Baggiolini, B. D. Sykes and I. Clark-Lewis, EMBO J., 1997, 16, 6996–7007 CrossRef CAS PubMed.
- K. Balabanian, B. Lagane, S. Infantino, K. Y. Chow, J. Harriague, B. Moepps, F. Arenzana-Seisdedos, M. Thelen and F. Bachelerie, J. Biol. Chem., 2005, 280, 35760–35766 CrossRef CAS PubMed.
- I. A. Shumilin, C. Zhao, R. Bauerle and R. H. Kretsinger, J. Mol. Biol., 2002, 320, 1147–1156 CrossRef CAS PubMed.
- E. Latz, A. Verma, A. Visintin, M. Gong, C. M. Sirois, D. C. Klein, B. G. Monks, C. J. McKnight, M. S. Lamphier and W. P. Duprex, Nat. Immunol., 2007, 8, 772–779 CrossRef CAS PubMed.
- Y. Zhu, P. Yang, Q. Wang, J. Hu, J. Xue, G. Li, G. Zhang, X. Li, W. Li, C. Zhou, M. Zhao and D. Wang, Anat. Rec., 2013, 296, 1850–1856 CrossRef CAS PubMed.
- B. Lv, X. Yang, S. Lv, L. Wang, K. Fan, R. Shi, F. Wang, H. Song, X. Ma, X. Tan, K. Xu, J. Xie, G. Wang, M. Feng and L. Zhang, Mol. Neurobiol., 2015, 52, 1263–1268 CrossRef CAS PubMed.
- P. Kukreja, A. B. Abdel-Mageed, D. Mondal, K. Liu and K. C. Agrawal, Cancer Res., 2005, 65, 9891–9898 CrossRef CAS PubMed.
- L. J. Bendall, R. Baraz, J. Juarez, W. Shen and K. F. Bradstock, Cancer Res., 2005, 65, 3290–3298 CAS.
- E. De Clercq, Mol. Pharmacol., 2000, 57, 833–839 CAS.
- W.-b. Zhang, J.-M. Navenot, B. Haribabu, H. Tamamura, K. Hiramatu, A. Omagari, G. Pei, J. P. Manfredi, N. Fujii and J. R. Broach, J. Biol. Chem., 2002, 277, 24515–24521 CrossRef CAS PubMed.
- F. Balkwill, Semin. Cancer Biol., 2004, 14, 171–179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01735a |
| This journal is © The Royal Society of Chemistry 2017 |