
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeveral orders of magnitude increase in the hydraulic permeability of flow-through capacitive deionization electrodes via laser perforations†
Eric N. Guyes ,
Anastasia Simanovski and
Matthew E. Suss*
,
Anastasia Simanovski and
Matthew E. Suss*
Faculty of Mechanical Engineering, Technion – Israel Institute of Technology, Technion City 1, Haifa 32000, Israel. E-mail: mesuss@technion.ac.il
First published on 13th April 2017
Abstract
Capacitive deionization (CDI) is a fast-emerging water desalination technology in which an applied potential of ∼1 V across a pair of porous carbon electrodes removes salt from the feedwater via electrosorption. In flow-through electrode (FTE) CDI cell architecture, feedwater is pumped through the macropores of the porous electrodes, enabling highly compact cells and rapid salt removal. However, FTE CDI requires specialized electrode materials possessing large macropores with diameters above 1 μm in order to allow for reasonable feed pressures. We herein present and demonstrate a low-cost, simple technique for laser perforating CDI electrodes, which delivers highly permeable electrodes from carbon materials with pore structures otherwise unsuitable for flow-through operation. We show that laser cutting channels with ∼200 μm aperture diameters through a commercial CDI electrode material with sub-micron macropores increases the electrode hydraulic permeability by approximately five orders of magnitude, thereby greatly reducing the feed pressure. Furthermore, we demonstrate that key performance metrics such as the cell's salt adsorption capacity and gravimetric capacitance are largely unaffected by the laser perforation technique.
Introduction
Brackish water desalination is considered a promising technique to boost freshwater supplies in areas affected by water scarcity. A commonly applied technology for brackish water desalination is reverse osmosis (RO), in which the salt-containing feedwater is pressurized to above its osmotic pressure in order to force water molecules through a semi-permeable membrane.1 A fast-emerging technology, capacitive deionization (CDI), can potentially allow for improved energy efficiency in brackish water desalination and wastewater purification.2,3 A typical CDI cell consists of two microporous carbon electrodes with a separator between them. A feedwater requiring desalination is pumped through the separator, and applying a voltage to the cell results in salt ion removal from the feed and storage within electric double layers (EDLs) that form in the electrodes' micropores. The desalted water is simultaneously pumped out of the cell. Once the electrodes are fully charged, the electrodes may be discharged by short-circuiting, which releases the stored ions into the flow and result in a waste brine stream. The typical CDI cell is a flow by or flow between electrodes (FB) cell architecture, but many alternative cell architectures have been developed.2,3 Some architectures utilize suspension electrodes such as slurries or fluidized beds, in which salt ions are stored in charged, flowing micron-scale particles.4–9 Other cell designs leverage intercalation processes to store salt ions rather than micropore EDLs.10–13Another CDI cell architecture is the flow-through electrode (FTE) architecture,14–20 where the feedwater flows directly through electrodes' macropores (see Fig. 1A). In FTE cells, the electric field and flow directions are parallel, allowing for convenient optimization of flow and ionic resistances,19 one-dimensional system modeling,20 and improved desalination rates and more compact cells.20 FTE cells also have drawbacks relative to FB cells, such as an observed enhanced rate of anode corrosion, though this can be mitigated by de-oxygenating the feedwater.17 Additionally, FTE cells can require greater feed pressures than FB cells.19 To maintain acceptably low feed pressure, which should be at least one order of magnitude under the feed osmotic pressure to prevent excessive pump energy requirements, FTE cells typically require use of specialized electrodes consisting of relatively large macropores. For example, Suss et al. utilized hierarchical carbon aerogel monolith electrodes with macropore diameters of roughly 2 μm,19 while Cohen et al. used woven carbon cloth electrodes with order 100 μm gaps between fibers as the flow channels.15 The requirement of large macropores significantly limits the number of CDI electrode materials that can be utilized as FTEs.
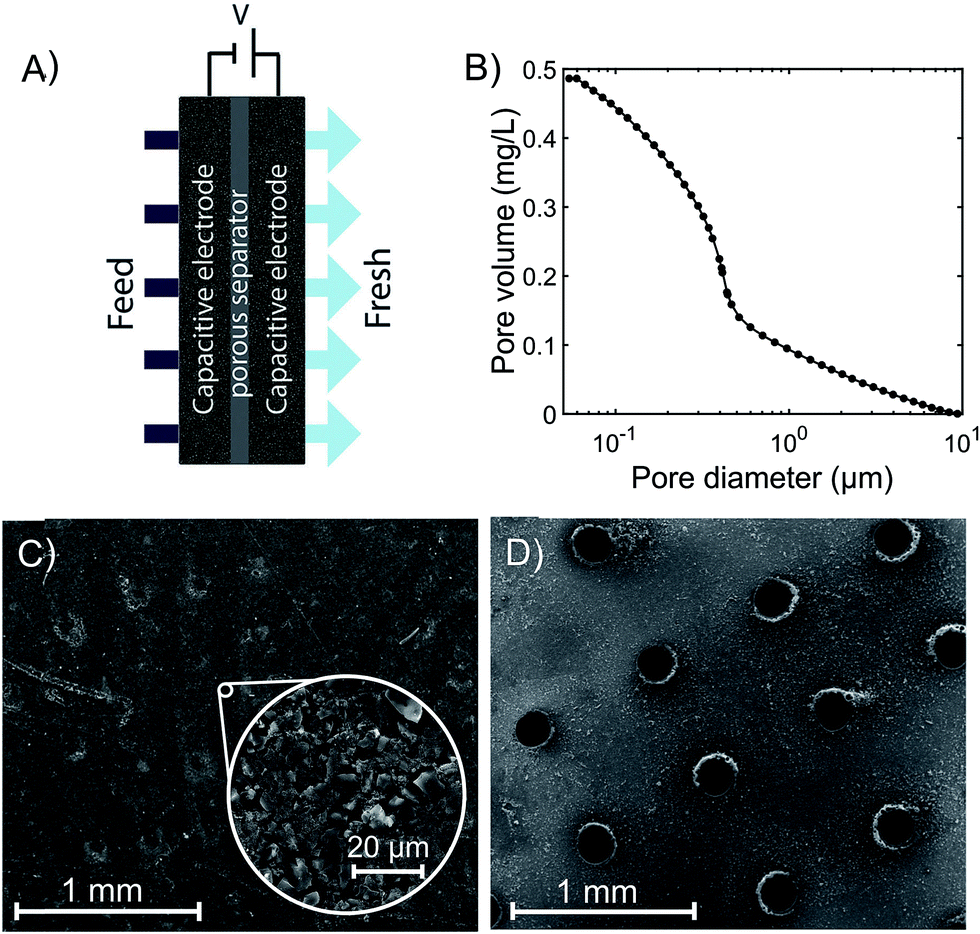 | ||
| Fig. 1 (A) Schematic of the flow-through electrode (FTE) CDI cell architecture. (B) Cumulative pore volume of the as-received commercial electrode in the range 50 nm to 10 μm determined via mercury porosimetry, showing a median macropore diameter of 0.380 μm. (C) SEM image of the as-received (unperforated) commercial electrode material, and (D) SEM image of the same commercial material after laser perforation, showing a regular array of channels with apertures roughly 200 μm in diameter. | ||
Here, we demonstrate and study laser perforating an array of flow channels into CDI electrodes in order to improve electrode hydraulic permeability and reduce feed pressure. Laser-perforated electrodes have been utilized in flow battery and fuel cell systems for the purposes of improving power density, cell stability, and cell performance,21–25 and it is predicted that perforated graphite anodes can increase ion transport rates in Li-ion batteries.26 However, laser perforations have not yet been explored as a technique for reducing pressure requirements in FTE CDI cells, and the effect of the perforations and laser-cutting process on desalination performance has not been previously measured. We here show that laser perforating commercial CDI electrodes, which would ordinarily not be appropriate for FTE CDI as they contain mostly sub-micron macropores, renders them suitable for use as FTEs. Specifically, we show that laser cutting dramatically increases the hydraulic permeability of these electrodes by roughly five orders of magnitude. Further, we show that laser perforating the electrodes does not affect key performance metrics such as electrode gravimetric capacitance and salt adsorption capacity (SAC).
Experimental
The experimental FTE CDI cell consists of a pair of commercial porous activated carbon electrodes (PAC MM 203, Materials & Methods, Irvine, CA, USA, ∼300 μm thickness each, 1.75 × 1.75 cm2). From nitrogen adsorption, the specific micropore volume is 0.34 mL g−1 (t-plot method). In Fig. 1B, we show results of mercury intrusion porosimetry measurements of the as-received electrode material. Results show that the total pore volume for pore diameters between 50 nm and 10 μm is about 0.5 mL g−1. The median diameter in this range is 0.380 μm, indicating that the material is likely not suitable for flow-through. Similar electrode material has been previously evaluated in FB CDI cells.27–29 A porous separator (GE Life Sciences, Whatman Grade 2 cellulose filter paper, 190 μm thickness, 2.4 × 2.4 cm2) electrically isolates the electrodes. Gaskets laser-cut from ePTFE (W. L. Gore & Associates, Gore-Tex NSG16X-GP, 1.4 mm uncompressed thickness, 5 × 5 cm2) seal the cell, and a square aperture in the gasket (1.55 × 1.55 cm2) permits feedwater to pass through the electrodes and separator. The electrodes fit tightly into grooves on the aperture perimeter, preventing feedwater from leaking around the electrode edges. The upstream anode current collector is laser-cut acrylic coated with silver paint (05002-AB, SPI, West Chester, PA, USA) and the downstream cathode collector is milled from impervious graphite (FC-GR, Graphitestore.com, Inc., Buffalo Grove, IL, USA). The current collectors both contain an array of cylindrical channels (6 × 6 grid, 1.5 mm diameter, 3 mm length) that allow water to pass through, and tabs that enable electrical contact with the voltage source. Water reservoirs are created upstream and downstream of the collectors via two ePTFE gaskets and an acrylic spacer (2 mm thickness) on the upstream side, and a single ePTFE gasket on the downstream side. The cell terminates on either side with endplates, of which the upstream endplate is milled from PVDF and the downstream endplate is 3D-printed from ABS (both endplates are 5 × 5 cm2). The cell is sealed with 8 bolts (M4 × 30 mm), which are tightened to 35 N cm. Each endplate includes one fluid flow line and one vent for removing air from the cell.In order to produce the laser-perforated electrodes, we cut a 21 × 21 array of channels in the electrode material with a CO2 laser (VLS 4.60, United Laser Systems, Scottsdale, AZ, USA) (see Fig. 1C and D). We designed the perforation cross section to be circular with a 200 μm diameter, which was approximately the smallest diameter attainable by our laser system. The channel density was chosen to provide close spacing between perforations while minimizing the mass loss of the electrode due to ablation to ∼5%. At the electrode surface farthest from the laser head during cutting, the resulting laser cut holes were roughly circular in cross section and with ∼200 μm diameter (see Fig. 1D and S1†). However, the perforations on the top surface were larger and exhibited a more elliptical shape, demonstrating that the laser holes are not perfectly cylindrical through the electrode (see Fig. S1†). From mass measurements of 21 perforated electrode samples and 22 unperforated samples, perforation removed an average of 6.1% of the electrode mass.
For CDI system feed pressure measurements, a pump drew deionized water from a bulk reservoir and introduced it into the CDI cell via semi-rigid polyethylene tubing. We removed the separator from the CDI cell in order to avoid adding an extra hydraulic resistance to the system. A pressure sensor (PX419 series, Omega Engineering, Stamford, CT, USA) was connected to the tubing approximately 10 cm upstream of the cell inlet. The sensor pressure without water flow was slightly offset from zero and was recorded before each experiment to provide a baseline pressure. For experiments using the as-received (unperforated) electrodes, we utilized a peristaltic pump (Masterflex 0755130, Cole-Parmer) that was able to withstand the high flow pressures. We measured the baseline pressure at zero flow and then increased the flow rate in increments of 0.1 mL min−1. The actual flow rates were determined to be ±2% of the set flow rates via effluent mass measurements. Pumping at each flow rate continued until the pressure no longer changed in time, except for small pressure oscillations of ∼5 kPa caused by the peristaltic pumping action. Each pressure was computed as a time average to account for the effect of the oscillations. High flow pressures due to flowing through the unperforated electrode material, which approached the maximum allowable pressure of our peristaltic pump, restricted the maximum flow rate to 1.0 mL min−1. For pressure measurements with perforated electrodes, we utilized a syringe pump with ±2% maximum flow rate error (Fusion 720, Chemyx, Stafford, TX, USA) that provides a constant flow rate without the aforementioned oscillations and is suited for the low pressures observed with these electrodes. A large-diameter cylindrical container filled with DI water served as the outlet reservoir, and the CDI cell outlet line rested slightly below the water's surface in order to minimize pressure oscillations caused by droplet formation. The large reservoir diameter ensured that the water level remained approximately constant over the course of an experiment. We increased the flow rate from 0 to 5.0 mL min−1 in increments of 0.5 mL min−1. Each flow rate was maintained until steady pressure measurements were achieved.
For desalination experiments, the peristaltic pump drew feedwater (NaCl 5.5 mM concentration) from a glass bulk reservoir and fed it into the CDI cell via the semi-rigid polyethylene tubing. The pump flow rate was set as 1.0 mL min−1 and the actual flow rate was determined from effluent mass measurements. The cell effluent entered a conductivity cell (5-ring, Metrohm, Inc., Switzerland) that contained a custom-milled insert to reduce the internal sensor volume, and then returned to the bulk reservoir. The bulk reservoir volume (∼2 L) was significantly larger than the volume of the remainder of the setup (∼20 mL) in order to maintain constant concentration in the reservoir throughout the experiments. During the charging half-cycle (30 min) of the desalination experiments, a voltage source (SourceMeter 2400, Keithley Instruments, Inc., Solon, OH, USA) applied a voltage of 1.0 V to the cell, while during the discharging half-cycle (30 min) it maintained a voltage of 0 V.
Results & discussion
Fig. 2 shows the feed pressures required to flow through the cell with as-received (unperforated) electrodes and with laser-perforated electrodes for various feedwater flow rates. Experimental results (circle and triangle markers) approximately show the expected linear behavior of pressure versus flow rate, consistent with a constant cell hydraulic resistance. The set of data for the cell with unperforated electrodes (circular markers) shows that the pressure required for pumping through these electrodes reaches nearly 200 kPa at 1 mL min−1 feed flow rate. For context, note that the osmotic pressure of the feed is estimated to be ∼20 kPa via the Van't Hoff equation. Thus, we can conclude that the electrodes as received are not suitable for FTE CDI, as the pump energy requirements alone at modest throughputs are well above the minimum thermodynamic energy required for desalination of the same feed volume by reverse osmosis.30 For the cell with laser-perforated electrodes, experimental results (triangle markers) show that feed pressure was reduced by roughly 4 orders of magnitude to about 0.02 kPa at 1 mL min−1, indicating that the electrodes are now suitable for FTE CDI.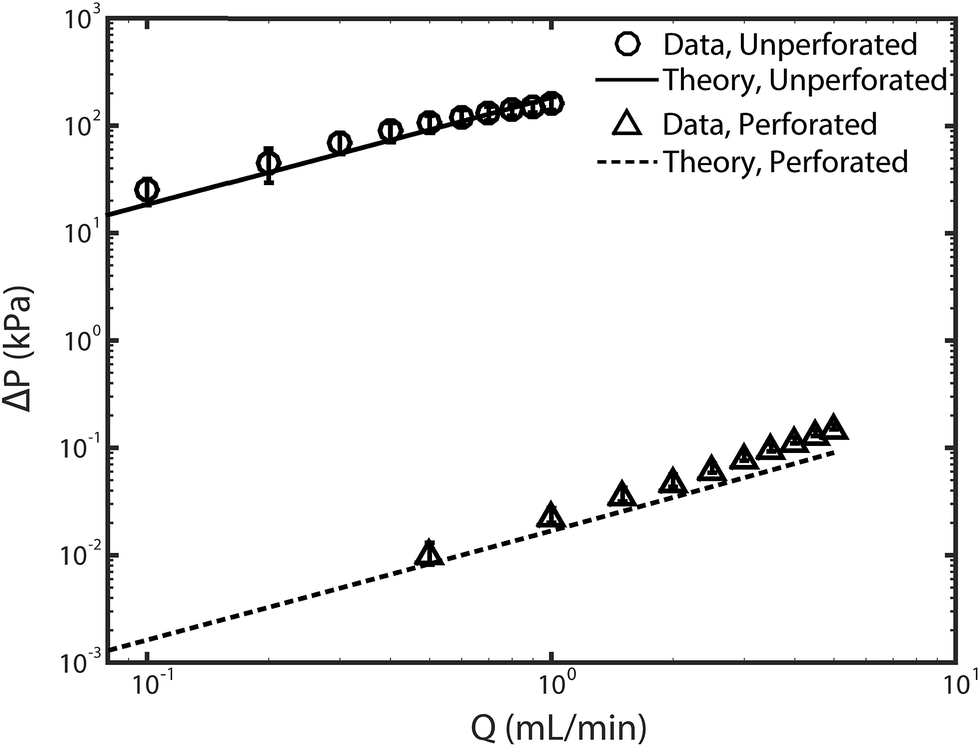 | ||
| Fig. 2 Plot of pressure versus pump flow rate for the as-received unperforated electrodes (circle markers) and the laser-perforated electrodes (triangle markers). Theory lines were obtained from eqn (1) for unperforated electrodes, and with eqn (2) for the perforated electrodes. Error bars represent the 95% confidence interval calculated from at least three realizations. | ||
In order to relate our feed pressure measurements to the electrodes' permeability, we developed models of the electrodes' hydraulic permeability, κ, and the expected pressure drop across the electrodes, ΔP. Flow through a porous medium can be described using Darcy's law as
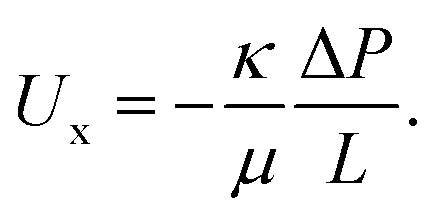 | (1) |
Rearranging eqn (1) with the given expression for κ yields the following equation for the pressure drop across both perforated electrodes,
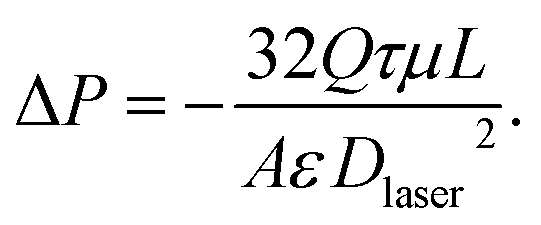 | (2) |
Here the velocity Ux is replaced by Q/A, the flow rate divided by the cross-sectional electrode area available for flow (2.4 cm2). We used eqn (2) to calculate the pressure drop across two perforated electrodes. Furthermore, as the pressure drop expected across the perforated electrodes is on the order of the pressure drop anticipated across other cell and system components, such as the reservoir channels and flow tubing, we also included these other components in a predictive model for feed pressure (see Section SI2 for further details). The model results for feed pressure for the system with perforated electrodes can be seen in Fig. 2 (dashed line), which only slightly under-predicts the measured data.
While laser perforation can significantly improve electrode hydraulic permeability, it is also important to evaluate the effect of the laser cutting process on electrode desalination performance. CDI electrode performance is primarily evaluated via the metric of maximum salt adsorption capacity (mSAC), which is the mass of salt that a pair of CDI electrodes can store upon reaching equilibrium after being charged with a constant voltage.2,33 The latter metric is obtained by integrating the cell's effluent concentration versus time curve during the desalination half cycle.2 The measured cell effluent concentration over full charge/discharge cycles are shown in Fig. 3A and B for the CDI cell with either unperforated or perforated electrodes. Ten full cycles are shown using a charge voltage of 1.0 V and discharge voltage of 0 V. The first few cycles show significant changes in the height of the peaks and depths of the valleys in the effluent concentration profile, where such changes are expected and widely observed as the CDI cell reaches its limit cycle.2 Past the fourth cycle, the cell demonstrates approximately uniform conductivity profiles for the remaining cycles. In Fig. 3C, we show the maximum salt adsorption capacity (mSAC) obtained from integrating the concentration versus time curves of Fig. 3A and B, for all curves past the fourth cycle. We also show the measured gravimetric capacitance of the electrodes, obtained from integrating the cell's current response to an applied voltage (see Fig. S3† for current vs. time plots). As can be seen, the cell's mSAC and gravimetric capacitance are nearly identical when using perforated or unperforated electrodes, demonstrating that the laser perforation process does not appreciably impact the remaining electrode material's charge or salt storage capability. The SEM images provide further evidence for this result, in which no apparent heat damage is observed to the remaining electrode material surrounding the laser perforations, aside from the apparent melted binder on the bottom side of the electrode (see Fig. S1†).
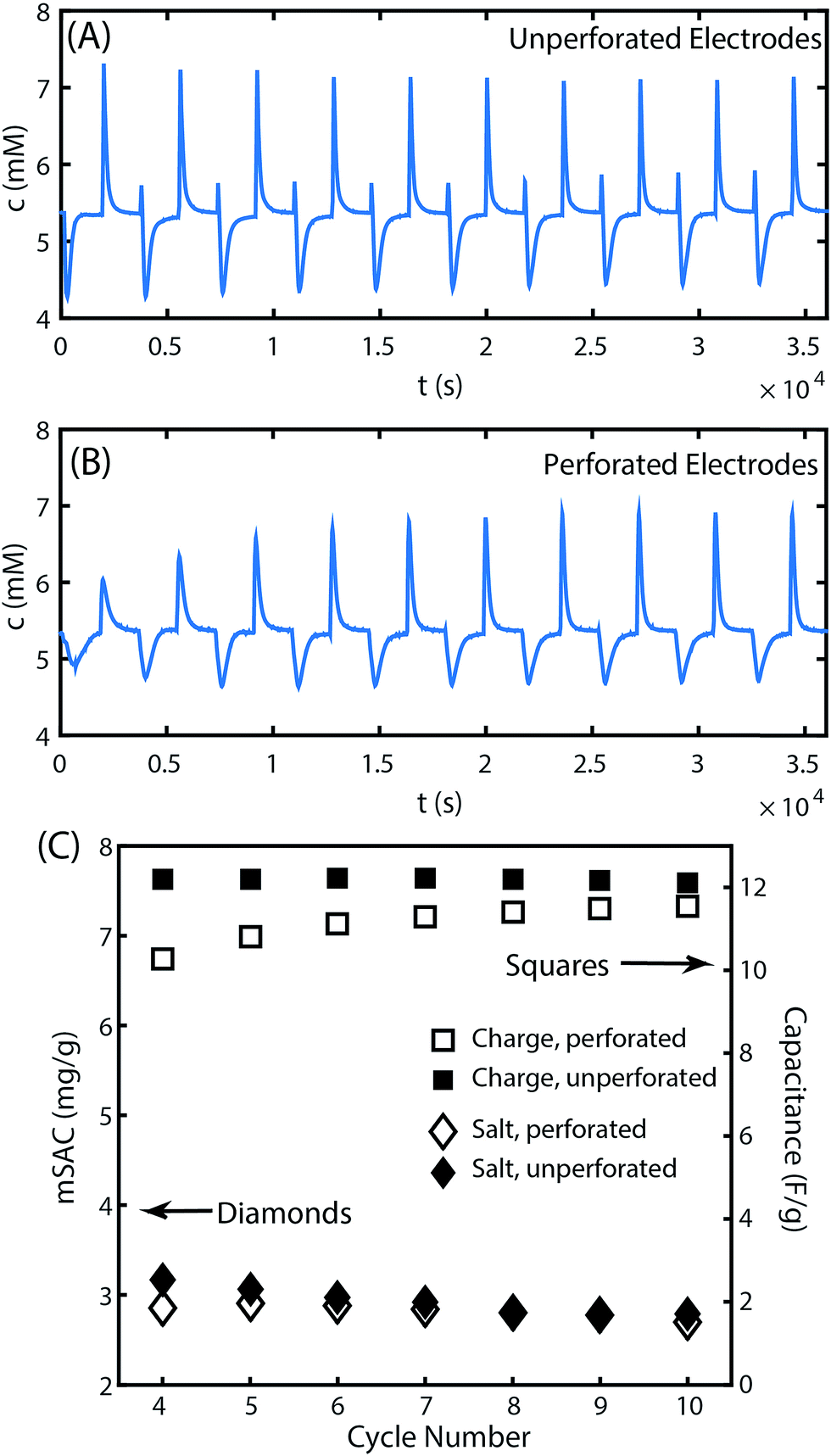 | ||
| Fig. 3 Measured cell effluent concentration versus time during cell charge (1.0 V) and discharge (0 V) cycling for (A) unperforated electrodes and (B) perforated electrodes. (C) The measured maximum salt adsorption capacity (mSAC) and gravimetric capacitance for cycles shown in (A) and (B). | ||
While mSAC measures the equilibrium salt storage in CDI electrodes, the metric of average salt adsorption rate (ASAR) quantifies the salt electrosorption kinetics. ASAR is defined as the salt adsorption capacity divided by the full cycle time, and can be affected by both electrode and cell parameters (e.g. by contact resistance and separator parameters).2,27 In the 10th cycle, ASAR is nearly identical between the cell with unperforated electrodes and perforated electrodes at 0.047 and 0.045 mg g−1 min−1, respectively. However, the ASAR averaged over a cycle reaching equilibrium does not give the total picture of the salt electrosorption kinetics. When comparing cell effluent versus time curves between the perforated and unperforated cells, we can see subtle differences in the dynamics (see Fig. S5† for a direct comparison). Generally, the features in the unperforated electrode case appear sharper than those in the perforated case, and a brief inversion peak is observed immediately upon cell charging only in the unperforated case. The latter observation is likely due to the introduction of an additional transport process in the system with perforated electrodes. In this system, salt must transport out of the laser-cut channels and into the ion-depleted porous electrode material during cell charging to effect desalination of the feed stream, and this transport is likely via molecular diffusion. In the perforated electrodes, the half-distance L between perforations is ∼500 μm, and the diffusion time over this distance, τD = L2/D, is roughly two minutes (where the mean ion diffusivity D is 1.6 × 10−5 cm s−1 for 1–10 mM NaCl solutions34). This additional timescale is small relative to the half-cycle time of 30 minutes in our experiments, but not insignificant. Thus, the introduced diffusion process may serve to slightly dampen the effluent concentration vs. time signal of the perforated case compared to the unperforated case. A future strategy to optimize the performance of laser-perforated FTE CDI electrodes may be to created denser channel arrays in order to further reduce this extra diffusion time.
To further delve into the presence of inversion peaks at the beginning of each charge step in Fig. 3A, we note that they appear at every cycle except the first despite the use of fresh electrodes for this experiment. We hypothesize that this electrode material possessed an initially high concentration of carboxyl groups in the micropores, which can cause inversion features due to enhanced co-ion expulsion from anode micropores.16,35,36 Such features are typically seen only after extended electrode cycling,37 but here are seen immediately upon use of the electrode. The low mSAC measured for this electrode of only ∼3 mg g−1 and a low calculated charge efficiency of ∼40% are also signs of excessive co-ion expulsion.2,27 In Fig. 3B, the inversion peak is not observed during charging for any of the 10 cycles. We attribute the latter observation to the aforementioned additional diffusion timescale in the perforated electrodes that may dampen a sharp inversion peak. The extensive co-ion expulsion in the perforated electrode cell is instead detected indirectly via this cell's low mSAC of ∼3 mg g−1 and charge efficiency of ∼40%, nearly identical to that of the unperforated electrode cell.
Conclusions
We here proposed and demonstrated the technique of laser perforating an array of channels into CDI electrodes to enhance electrode hydraulic permeability. We showed that applying this technique to commercial CDI electrodes with sub-micron macropores increases by five orders of magnitude the electrode permeability without adversely impacting the electrodes' salt adsorption capacity. In addition to reducing pressure requirements, this technique promises to enable a wider range of electrode materials for FTE CDI by adapting materials that would ordinarily require prohibitively high feed pressures. Future work can investigate optimizing the laser perforated channel array via the use of a higher channel density in order to further minimize the transport distance between perforations.Acknowledgements
The Israeli Ministry of National Infrastructures, Energy and Water Resources funded this work.References
- J. Zhou, V. W. Chang and A. G. Fane, Energy Environ. Sci., 2011, 4, 2267–2278 CAS.
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 CAS.
- S. Porada, R. Zhao, A. Van Der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- S. Jeon, H. Park, J. Yeo, S. Yang, C. H. Cho, M. H. Han and D. K. Kim, Energy Environ. Sci., 2013, 6, 1471–1475 CAS.
- J. Ma, D. He, W. Tang, P. Kovalsky, C. He, C. Zhang and T. D. Waite, Environ. Sci. Technol., 2016, 50, 13495–13501 CrossRef CAS PubMed.
- G. J. Doornbusch, J. E. Dykstra, P. M. Biesheuvel and M. E. Suss, J. Mater. Chem. A, 2016, 4, 3642–3647 CAS.
- H. Cohen, S. E. Eli, M. Jõgi and M. E. Suss, ChemSusChem, 2016, 9, 3045–3048 CrossRef CAS PubMed.
- Y. Gendel, A. K. E. Rommerskirchen, O. David and M. Wessling, Electrochem. Commun., 2014, 46, 152–156 CrossRef CAS.
- S. Porada, D. Weingarth, H. V. M. Hamelers, M. Bryjak, V. Presser and P. M. Biesheuvel, J. Mater. Chem. A, 2014, 2, 9313–9321 CAS.
- J. Lee, S. Kim, C. Kim and J. Yoon, Energy Environ. Sci., 2014, 7, 3683–3689 CAS.
- K. C. Smith and R. Dmello, J. Electrochem. Soc., 2016, 163, A530–A539 CrossRef CAS.
- F. Xing, T. Li, J. Li, H. Zhu, N. Wang and X. Cao, Nano Energy, 2017, 31, 590–595 CrossRef CAS.
- P. Srimuk, F. Kaasik, B. Krüner, A. Tolosa, S. Fleischmann, N. Jäckel, M. C. Tekeli, M. E. Suss, M. Aslan and V. Presser, J. Mater. Chem. A, 2016, 4, 18265–18271 CAS.
- A. M. Johnson and J. Newman, J. Electrochem. Soc., 1971, 118, 510–517 CrossRef CAS.
- I. Cohen, E. Avraham, M. Noked, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2011, 115, 19856–19863 CAS.
- Y. Bouhadana, E. Avraham, M. Noked, M. Ben-Tzion, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2011, 115, 16567–16573 CAS.
- I. Cohen, E. Avraham, Y. Bouhadana, A. Soffer and D. Aurbach, Electrochim. Acta, 2015, 153, 106–114 CrossRef CAS.
- Y. Qu, P. G. Campbell, L. Gu, J. M. Knipe, E. Dzenitis, J. G. Santiago and M. Stadermann, Desalination, 2016, 400, 18–24 CrossRef CAS.
- M. E. Suss, T. F. Baumann, W. L. Bourcier, C. M. Spadaccini, K. A. Rose, J. G. Santiago and M. Stadermann, Energy Environ. Sci., 2012, 5, 9511–9519 CAS.
- E. N. Guyes, A. N. Shocron, A. Simanovski, P. M. Biesheuvel and M. E. Suss, Desalination, 2017 DOI:10.1016/j.desal.2017.03.013.
- D. Gerteisen, T. Heilmann and C. Ziegler, J. Power Sources, 2008, 177, 348–354 CrossRef CAS.
- D. Gerteisen and C. Sadeler, J. Power Sources, 2010, 195, 5252–5257 CrossRef CAS.
- M. P. Manahan, M. C. Hatzell, E. C. Kumbur and M. M. Mench, J. Power Sources, 2011, 196, 5573–5582 CrossRef CAS.
- M. P. Manahan and M. M. Mench, J. Electrochem. Soc., 2012, 159, F322–F330 CrossRef CAS.
- I. Mayrhuber, C. R. Dennison, V. Kalra and E. C. Kumbur, J. Power Sources, 2014, 260, 251–258 CrossRef CAS.
- V. P. Nemani, S. J. Harris and K. C. Smith, J. Electrochem. Soc., 2015, 162, A1415–A1423 CrossRef CAS.
- R. Zhao, O. Satpradit, H. H. M. Rijnaarts, P. M. Biesheuvel and A. van der Wal, Water Res., 2013, 47, 1941–1952 CrossRef CAS PubMed.
- J. E. Dykstra, R. Zhao, P. M. Biesheuvel and A. Van der Wal, Water Res., 2016, 88, 358–370 CrossRef CAS PubMed.
- A. Hemmatifar, J. W. Palko, M. Stadermann and J. G. Santiago, Water Res., 2016, 104, 303–311 CrossRef CAS PubMed.
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 CrossRef CAS PubMed.
- S. Yao and J. G. Santiago, J. Colloid Interface Sci., 2003, 268, 133–142 CrossRef CAS PubMed.
- Z. Dagan, S. Weinbaum and R. Pfeffer, J. Fluid Mech., 1982, 115, 505–523 CrossRef CAS.
- S. Porada, L. Borchardt, M. Oschatz, M. Bryjak, J. S. Atchison, K. J. Keesman, S. Kaskel, P. M. Biesheuvel and V. Presser, Energy Environ. Sci., 2013, 6, 3700–3712 CAS.
- E. A. Guggenheim, Trans. Faraday Soc., 1954, 50, 1048 RSC.
- P. M. Biesheuvel, H. V. M. Hamelers and M. E. Suss, J. Colloid Interface Sci., 2015, 9, 1–5 CrossRef CAS.
- X. Gao, A. Omosebi, J. Landon and K. Liu, J. Electrochem. Soc., 2014, 161, E159–E166 CrossRef.
- I. Cohen, E. Avraham, Y. Bouhadana, A. Soffer and D. Aurbach, Electrochim. Acta, 2013, 106, 91–100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00459a |
| This journal is © The Royal Society of Chemistry 2017 |