
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomically thin SiC nanoparticles obtained via ultrasonic treatment to realize enhanced catalytic activity for the oxygen reduction reaction in both alkaline and acidic media†
Jiahao Guo *a,
Kaiyue Songa,
Bingbing Wua,
Xiang Zhua,
Beilin Zhanga and
Yantao Shib
*a,
Kaiyue Songa,
Bingbing Wua,
Xiang Zhua,
Beilin Zhanga and
Yantao Shib
aCollege of Chemistry and Materials Engineering, Anhui Science and Technology University, Fengyang, Anhui 233100, P. R. China. E-mail: guojiahao1974@163.com
bState Key Laboratory of Fine Chemicals, School of Chemistry, Dalian University of Technology, Dalian, 116024, P. R. China
First published on 26th April 2017
Abstract
Inorganic graphene analogues (IGAs) are promising catalysts for the oxygen reduction reaction (ORR) in fuel cells; however, their catalytic activities need to be improved. Herein, we report atomically thin SiC obtained via a simple ultrasonic treatment of commercial SiC as an efficient electrocatalyst for the ORR in alkaline and acidic media. Due to the advantages of its unique surface composition and structure, the atomically thin SiC shows remarkable catalytic activity for the ORR and the number of electrons transferred per O2 is close to four in both alkaline and acidic media. Compared with commercial Pt/C, the atomically thin SiC possesses prominent durability and tolerance to methanol crossover, and thus can be developed into a low-cost and efficient alternative as a cathodic electrode in fuel cells.
1. Introduction
The aggravation of environmental pollution requires new energy resources coupled with storage techniques to substitute for fossil fuels such as coal, petroleum, and natural gas.1,2 Fuel cells and batteries are considered to be very promising energy storage and conversion devices.3 One of the most important problems encountered by these new energy storage and conversion devices is the sluggish kinetic processes of the oxygen reduction reaction (ORR).4 Great efforts have been made in catalysis research to overcome the intrinsic ORR disadvantages.5–8 In recent years, despite the tremendous progress in the development of Pt-based catalytic materials, it is still a major challenge to develop highly active catalysts. Even if Pt-based catalysts are considered to be the best catalysts for the ORR in fuel cells, they still suffer from multiple disadvantages, such as high cost, limited reserves, and the crossover effect of fuel molecules.9–11 In order to promote the commercialization of fuel cells, the development of nonprecious metal or metal-free ORR catalysts has attracted great interest.12–16Graphene, which has a two-dimensional layer structure, has attracted considerable attention because of its unique physical and chemical properties that exhibit significant advances in energy storage and conversion devices.17 In the area of electrocatalysis, graphene has been recognized as a promising material and has been modified via various methods for use in ORR catalysts.16–23 Inorganic graphene analogues (IGAs) such as MoS2, MoSe2, and WSe2 also belong to two-dimensional layered materials, which have similar properties with graphene. These materials generally possess large surface areas and large amounts of coordination-unsaturated surface atoms, which provide outstanding catalytic properties and are conducive for the adsorption of reactants.24 Xie et al. showed that defect-rich MoS2 ultrathin nanosheets exhibit excellent hydrogen evolution reaction catalytic activity with a small onset potential, large current density, small Tafel slope, and prominent electrochemical durability.25 Our group confirmed the excellent ORR activity of atomically thin MoSe2/reduced graphene oxide and WSe2/reduced graphene oxide nanosheets associated with the number of exchanged electrons close to four.26 As a typical IGA, silicon carbide (SiC) possesses low density, high thermal conductivity, good chemical stability, and high saturated carrier mobility, which result in wide applications in various fields.27–30 SiC with an atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 has an imbalance of charge distribution on different atoms caused by the different electronegativities of Si (1.90) and C (2.55), which leads to more active sites than those by doped carbon materials that serve as ORR catalysts. Based on density functional theory (DFT), Zhang et al. exploited the potential of layered SiC as an ORR catalyst in alkaline media.31 However, no research on the electrocatalytic activity of layered SiC for the ORR and its preparation has been reported to date.
:1 has an imbalance of charge distribution on different atoms caused by the different electronegativities of Si (1.90) and C (2.55), which leads to more active sites than those by doped carbon materials that serve as ORR catalysts. Based on density functional theory (DFT), Zhang et al. exploited the potential of layered SiC as an ORR catalyst in alkaline media.31 However, no research on the electrocatalytic activity of layered SiC for the ORR and its preparation has been reported to date.
In this study, we highlight a simple pathway to prepare atomically thin SiC nanosheets derived from commercial SiC through ultrasonic exfoliation. Systematic characterization of its morphology, structure, composition and catalytic activity for the ORR in alkaline and acidic media is conducted. Compared with commercial SiC, the atomically thin SiC presents excellent catalytic activity for the ORR in both alkaline and acidic media, which is comparative to that of Pt/C catalysts.
2. Experimental
2.1 Preparation of atomically thin SiC nanomaterial
Commercial SiC (300 mg) was added in a mixture of deionized water (125 mL) and anhydrous ethanol (125 mL) in a 500 mL flask. The mixed system was sealed and placed in a low-power ultrasonic bath (KQ3200DA) for 12 h. The resultant dispersion was centrifuged at 2000 rpm for 20 min, and the supernatant was collected. The as-obtained product was collected after the supernatant, centrifuged at 12000 rpm for 20 min, washed with deionized water and anhydrous ethanol successively multiple times, and finally dried at 50 °C under vacuum overnight for further characterization.
2.2 Materials characterization
X-ray diffraction (XRD) patterns were collected using a Rigaku D/max-rA X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). Field emission scanning electron microscopy (FESEM) images were obtained using an FEI HITACHI S-4800 FESEM. Transmission electron microscopy (TEM) images, high-resolution TEM (HRTEM) images, and selected-area electron diffraction (SAED) patterns were obtained using a JEOL-2010 TEM with an acceleration voltage of 200 kV. Atomic force microscopy (AFM) was performed using a Veeco DI NanoScope MultiMode V system. The Brunauer–Emmett–Teller (BET) surface area was determined via nitrogen adsorption on a Tristar II 3020 Micromeritics adsorption analyzer at 77 K. Prior to the adsorption measurements, the samples were degassed at 200 °C for a minimum of 10 h. X-ray photoelectron spectra (XPS) were acquired on an ESCALAB MKII with Mg Kα (hν = 1253.6 eV) as the excitation source. The binding energies obtained in the XPS spectral analysis were corrected for specimen charging by referencing C 1s to 284.5 eV.2.3 Electrochemical measurements
The ORR activity of the atomically thin SiC was evaluated using a standard three-electrode electrochemical cell on an electrochemical workstation (CHI 660C, Chenhua, Shanghai) at room temperature. To prepare the working electrode, a homogeneous catalyst ink was made by mixture of 4 mg catalyst, deionized water, isopropanol, and Nafion ionomer solution (5 wt%) (v/v/v = 2.5:1:0.06) to form a 4 mg mL−1 suspension, which was ultrasonicated for approximately 1 h.
Furthermore, 5 μL of catalyst slurry was spread onto the surface of a glassy carbon (GC) disk electrode (0.2 cm2) to form a catalyst loading of ∼100 μg cm−2 and was dried under ambient conditions. The electrochemical properties of the catalyst were measured in 0.1 M KOH or 0.1 M HClO4 aqueous solution. The modified GC electrode, a Pt plate, and an Ag/AgCl electrode were used as the working, counter, and reference electrodes, respectively. The Ag/AgCl reference electrode was calibrated against the reversible hydrogen electrode (RHE). Before testing, an O2/N2 flow was passed through the electrolyte in the cell for 30 min to saturate it with O2/N2. The ORR catalytic activity was determined using the rotating disk electrode (RDE) and rotating ring disk electrode (RRDE) techniques with linear sweep voltammetry (LSV) from 0 to 1.1 V (vs. RHE) at a scan rate of 10 mV s−1 in an O2-saturated 0.1 M KOH or 0.1 M HClO4 solution. Cyclic voltammetry curves (CV) were collected in the potential range of 0 to 1.2 V (vs. RHE) at a sweep rate of 10 mV s−1. In this study, all the potentials were normalized to the RHE, all current densities were normalized to the BET surface area of the catalyst and all electrochemical experiments were carried out at room temperature and ambient pressure. Steady-state polarization and methanol crossover measurements were performed on the atomically thin SiC and Pt/C using the same setup as CV in O2-saturated 0.1 M KOH or 0.1 M HClO4.
3. Results and discussions
3.1 Material characterization
The XRD patterns of the atomically thin SiC and commercial SiC are shown in Fig. 1a. There are four main diffraction peaks at 34.90°, 40.52°, 59.35°, and 71.12°, which can be indexed to the lattice planes (111), (200), (220), and (311) of cubic SiC (JCPDS card no. 29-1129), respectively. There is a small peak at 33.12° (marked with an asterisk), which is likely due to the stacking faults within the crystal.32 The diffraction peaks are strong and sharp, which indicate that the atomically thin SiC possesses good crystallinity.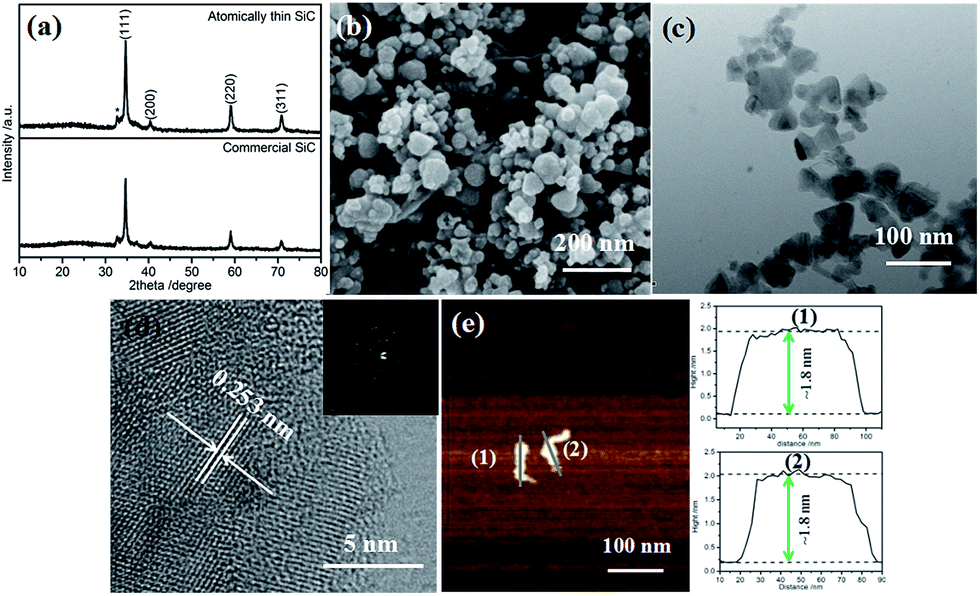 | ||
| Fig. 1 (a) XRD, (b) SEM, (c) TEM, (d) HRTEM, and (e) AFM images and height profiles from the sections indicated by gray lines of the atomically thin SiC. The inset in (d) shows the corresponding SAED pattern. | ||
The FESEM images of the atomically thin SiC and commercial SiC are shown in Fig. 1b and S1a,† respectively. As shown in Fig. S1a,† the commercial SiC is composed of both dispersed nanosized particles and some aggregates. The sizes of the nanoparticles range from 50 nm to 170 nm. After ultrasonic treatment, the nanoparticles of the atomically thin SiC are significantly reduced and no aggregates are observed, and its particle size is between 20 and 80 nm. The decrease in particle size can cause an increase in the number of catalytic activity sites in SiC and thus improve its catalytic activity. Fig. 1c and S1b† depict the TEM images of the atomically thin SiC and commercial SiC. Compared to the commercial SiC, the irregular nanoparticles of the atomically thin SiC are much smaller and their sizes range from 23 nm to 75 nm. As can be seen from Fig. 1d, the lattice fringe spacing of the atomically thin SiC is measured to be ∼0.253 nm, which is in good agreement with the value obtained using the Bragg diffraction from the (111) planes of β-SiC (Fig. 1c).33 The SAED patterns in Fig. 1d display diffraction points rather than diffraction circles, which further reveals that the atomically thin SiC exhibits an exfoliated thin layer. To confirm the thickness of the as-prepared sample, AFM imaging was carried out and the corresponding height profiles are shown in Fig. 1e. As can be seen in Fig. 1e, the atomically thin SiC possesses a relatively irregular, smooth 2D sheet structure with an average height of approximately 1.8 nm. These results confirm that the commercial SiC was effectively exfoliated via ultrasonic treatment and an ultrathin nanomaterial was successfully obtained. This atomically thin structure can effectively improve the electrical conductivity, conducive to the charge transfer. Furthermore, it also facilitates reactant adsorption and increases the reactive sites, resulting in excellent catalytic activity.
The specific surface area and porous nature of the atomically thin SiC were measured via nitrogen adsorption/desorption measurements conducted at 77 K. Compared with commercial SiC, the isotherms of the atomically thin SiC can be classified as type-III with a distinct hysteresis loop at relative pressures (P/P0) from 0.45 to 1.0 (Fig. 2a), suggesting the presence of mesopores that can facilitate oxygen adsorption and molecular transportation during the ORR. The atomically thin SiC has a huge Brunauer–Emmett–Teller surface area of 53.85 m2 g−1 with an average pore diameter of 34 nm and pore volume of 0.365 cm3 g−1, which are higher than the commercial SiC. These results clearly illustrate that with a simple ultrasonic treatment, commercial SiC can be effectively exfoliated into atomically thin SiC with a thickness of only 1.8 nm, which possesses a higher surface area.
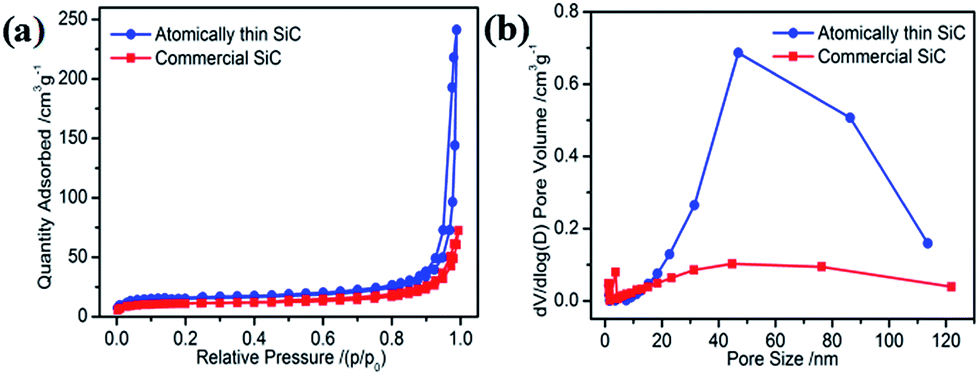 | ||
| Fig. 2 (a) Nitrogen adsorption–desorption isotherms and (b) pore size distribution curves of the atomically thin SiC and commercial SiC. | ||
The surface composition of the electrocatalysts has an important influence on their catalytic performances. The surface element composition of SiC was investigated via XPS. Fig. 3a shows the full-range XPS spectra of the atomically thin SiC and commercial SiC. As shown, the photoelectron peaks at 103.08 eV, 154.08 eV, and 284.08 eV can be indexed to Si 2p, Si 2s, and C 1s, respectively. Oxygen can be detected for SiC on the basis of its binding energy. The presence of O can be attributed to the adsorption of oxygen molecules on the surface of SiC and further result in a strong effect between Si or C and O. The unsaturated chemical bonds on the surface of the atomically thin SiC obtained by ultrasonic treatment should be responsible for this phenomenon, and resulting in the combination of C or Si and N on the surface of SiC.34 Fig. 3 and S2† show the core-level XPS spectra of C 1s, Si 2p, N 1s, and O 1s and the corresponding deconvoluted spectra for the atomically thin SiC and commercial SiC. Based on the deconvoluted XPS spectra, the percentages and binding energies of the different groups are shown in Table S1.† From Fig. 3b, the deconvoluted peaks with binding energies of 283.7, 284.6, 285.3 and 286.2 eV can be assigned to the C–Si, C–C, C–O and C–N bonds, respectively.35,36 The C–C and C–Si bonds are associated with carbon and SiC on the surface, respectively. The C–O and C–N bonds are attributed to the adsorption of O2 or N2. The N content is very low due to the low activity of N2. Compared with commercial SiC, the binding energies of the C–Si, C–C, and C–O bonds increase in different degrees. The increase in binding energy is beneficial for the adsorption and dissociation of O2 on the surface of the catalyst, which promotes the catalytic activity of the atomically thin SiC. The deconvoluted peak with the binding energy of 288.9 eV for commercial SiC can be assigned to the COO bond, which indicates that the commercial SiC surface contains more oxygen and is not conducive to oxygen adsorption. The deconvolution of the corresponding Si 2p spectrum for the atomically thin SiC (Fig. 3c) indicates that three chemical bonding states, Si–C, Si–N, and Si–O, are on its surface.37 The appearance of the Si–N bond is consistent with previous results. The binding energies of Si–C and Si–O are located at 102.1 and 104.0 eV, respectively, which are also higher than the corresponding bonds in commercial SiC. The content of Si–O bonds is significantly higher than that of Si–C bonds, which indicates that Si mainly exists in the form of Si–O on the surface of SiC, and O2 can be easily adsorbed and dissociated on the Si sites. As can be seen from Fig. S2d,† O–C and O–Si bonds are detected for the atomically thin SiC, and their binding energies are higher than that of commercial SiC. Compared with that of commercial SiC, the fraction of O–Si bonds increases from 47.4% to 63.0%, and the fraction of O–C bonds significantly decreases from 52.6% to 37.0% for the atomically thin SiC. The O–Si bond with the binding energy of 533.2 eV corresponds to amorphous SiO2.38 The XPS results show that the binding energies of the atomically thin SiC are higher than that of commercial SiC because of its surface unsaturated bonds, which are convenient for the adsorption and dissociation of O2 on the surface of the catalyst. The adsorbed oxygen mainly exists in the form of amorphous SiO2. Fig. 3d displays the high-resolution N 1s XPS spectrum of the atomically thin SiC sample as well as the corresponding deconvolution spectra of the different types of nitrogen probably existing in the sample. The asymmetric N 1s spectrum indicates the existence of three types of N: N–Si at 398.2 eV, pyridinic N at 398.9 eV, and pyrrolic N at 400.1 eV.39 The content of pyridinic N is as high as 80.9%. The pyridinic N and pyrrolic N atoms are located at the edge or defect sites,40 which indicates that the ultrasonic treatment is responsible for the existence of N. Pyridinic N atoms, as a marker of plane exposure, can provide actual active sites and improve the ORR activity of the atomically thin SiC.41,42
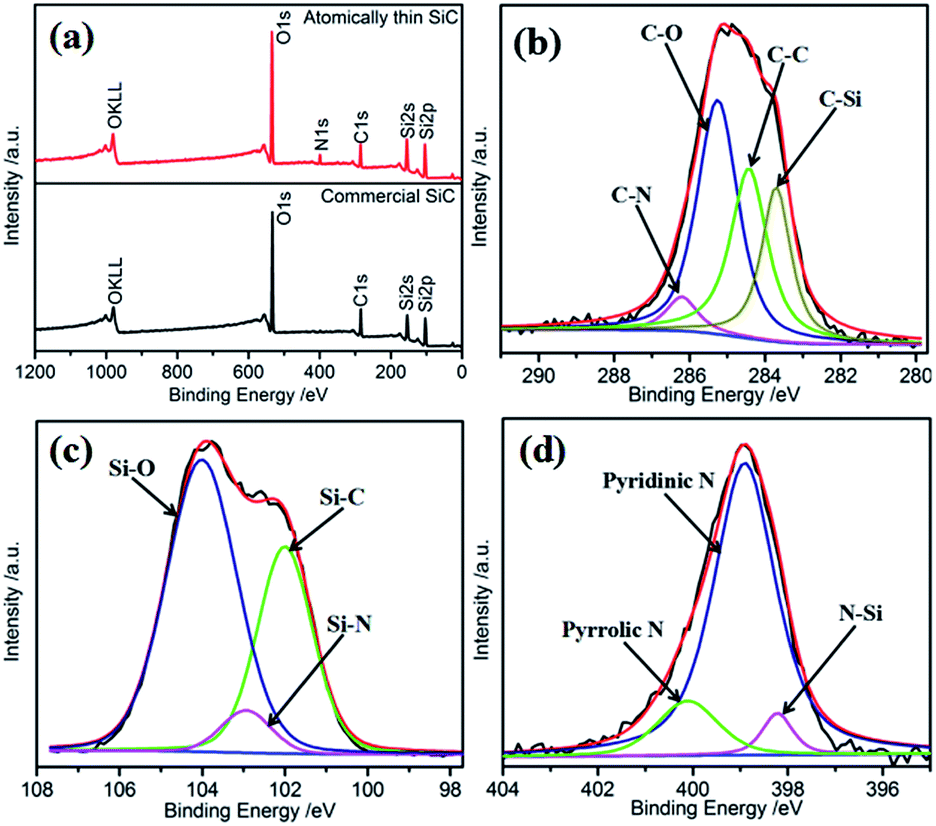 | ||
| Fig. 3 (a) Overall XPS spectra of the atomically thin SiC and commercial SiC; (b) C 1s core-level and corresponding deconvoluted spectra for the atomically thin SiC; (c) Si 2p core-level and corresponding deconvoluted spectra for the atomically thin SiC; and (d) N 1s core-level and corresponding deconvoluted spectra for the atomically thin SiC. | ||
3.2 Electrocatalytic analyses
First, the ORR catalytic activity of the atomically thin SiC in alkaline medium was investigated. Cyclic voltammetry measurements were conducted in O2- or N2-saturated 0.1 M KOH solution to evaluate the ORR activities of the atomically thin SiC (Fig. 4a). There is no visible peak in the plot of the measurements in N2 saturated KOH solution, which indicates that the oxygen reduction reaction did not occur. Nevertheless, a well-defined oxygen reduction peak at 0.76 V (vs. RHE) appeared in the presence of O2, which indicates that SiC possesses ORR catalytic activity in alkaline electrolyte. Compared with commercial SiC, the atomically thin SiC possesses a higher reduction peak potential and larger cathodic current, which suggest higher catalytic activity for the ORR.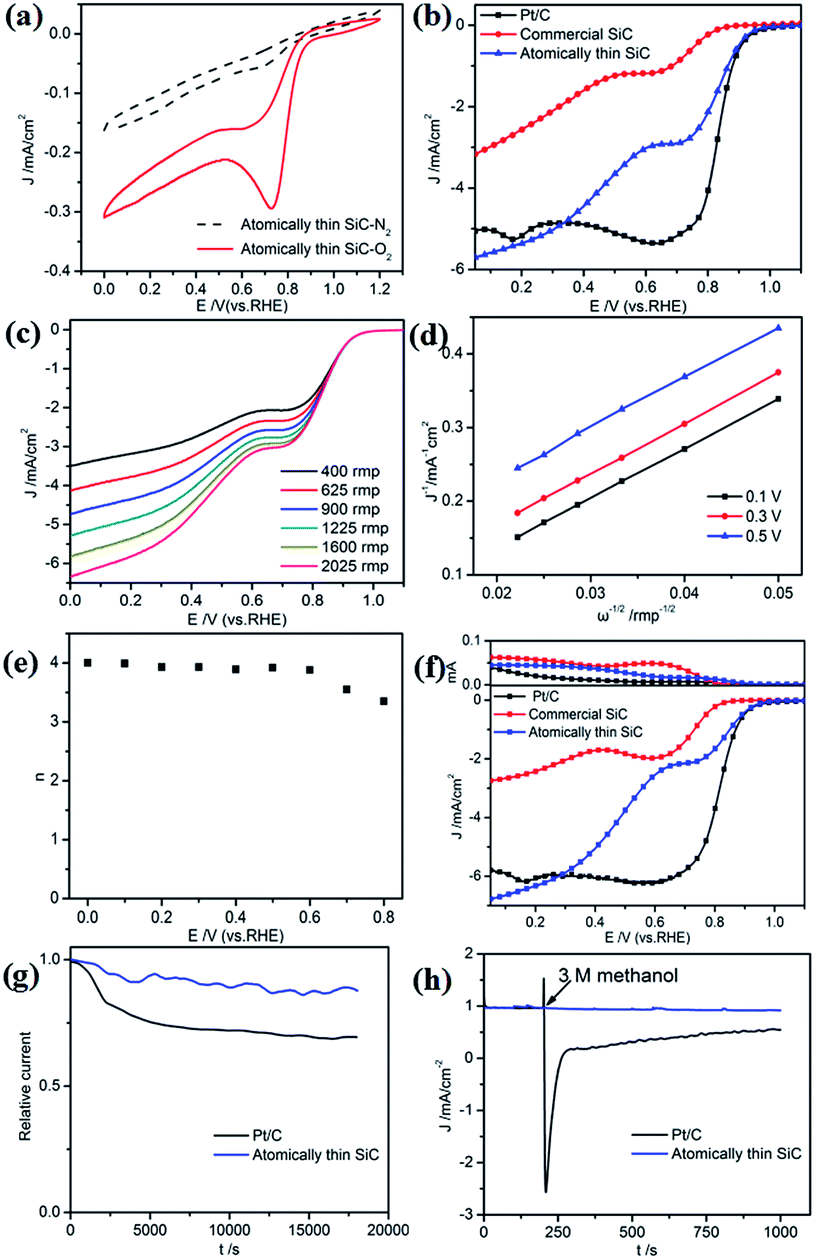 | ||
| Fig. 4 (a) CVs of the atomically thin SiC in N2- and O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1. (b) LSV of the atomically thin SiC, commercial SiC, and Pt/C in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 with an RDE rotation rate of 1600 rpm. (c) LSV of the atomically thin SiC at different RDE rotation rates. (d) Calculated K–L plots of the ORR from the atomically thin SiC. (e) Electron-transfer number n derived from the K–L plots at different potentials. (f) RRDE from the atomically thin SiC. (g) Chronoamperometric response (i–t) of the atomically thin SiC and Pt/C and (h) i–t of the atomically thin SiC and Pt/C before and after the addition of 3 M methanol. Tests were conducted in O2-saturated 0.1 M KOH solution at 0.7 V. | ||
To further investigate its ORR performance, linear sweep voltammetry (LSV) measurements on an RDE for the atomically thin SiC, commercial SiC, and Pt/C electrodes were carried out in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 and a rotation rate of 1600 rpm. As shown in Fig. 4b, the ORR based on the commercial SiC catalyst commences around 0.83 V, followed by a continuous increase in current density with no plateau. The onset potential for oxygen reduction at the atomically thin SiC electrode significantly shifts positively to 0.95 V, which is close to the onset potential for Pt/C electrodes (0.97 V). The limiting diffusion current on the atomically thin SiC electrode at 0.3 V becomes about 2.5 times stronger with a relatively wide plateau with respect to the commercial SiC electrode, which indicates a diffusion-controlled process related to an efficient 4e− dominated ORR pathway.43–45 For the atomically thin SiC electrode, its LSV curve exhibits two current plateaus at 0.8–0.6 V and below 0.6 V, respectively, corresponding to a two-electron process and four-electron process, respectively. This result may affect the catalytic activity of the atomically thin SiC. The results of CV and LSV prove that the atomically thin SiC electrode shows favorable ORR catalytic activity, which is reflected by its positive onset potential and high current density.
RDE voltammetry measurements were also carried out to gain further insight into the ORR performance of the atomically thin SiC. Fig. 4c shows the RDE current–potential curves at different rotation rates for the atomically thin SiC in an alkaline medium. In order to obtain the number of electrons transferred per oxygen molecule (n) on the atomically thin SiC, K–L plots from the LSVs at various rotation speeds were calculated from the Koutecky–Levich equations as follows:46–48
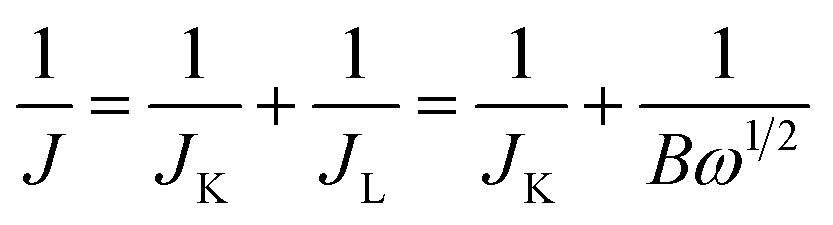 | (1) |
| B = 0.2nCO(DO)2/3ν−1/6 | (2) |
485 C mol−1), DO is the diffusion coefficient of O2, CO is the bulk concentration of O2, and ν is the kinetic viscosity of the electrolyte. In this study, the electrolyte is O2-saturated 0.1 M KOH. The DO, CO and ν values used are 1.9 × 10−5 cm2 s−1, 1.2 × 10−6 mol cm−3, and 0.01 cm2 s−1, respectively. The constant 0.2 in eqn (2) is adopted when the rotation speed is expressed in rpm. Fig. 4d shows three linear K–L plots at different potentials, which suggest that the inverse current density (1/j) is a function of the inverse of the square root of the rotation speed (ω−1/2) at different potential values and first order reaction kinetics toward the concentration of O2 on the atomically thin SiC from 0.1 V to 0.5 V. As shown in Fig. 4e, the corresponding numbers of electrons transferred were calculated to be 3.7–4.0 per O2 molecule at 0–0.6 V for the atomically thin SiC. The n value is lower at high potential. This result shows that the ORR process at the atomically thin SiC electrode is dominated by a four-electron process with water as the product at low potential. Fig. 4f shows the rotation ring-disk electrode (RRDE) curves of the three catalysts in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 and rotation rate of 1600 rpm. The atomically thin SiC exhibits a much higher diffusion-limiting current density than that of commercial SiC. The RRDE measurements show that the HO2− yield of the atomically thin SiC is less than 9%, which is significantly lower than commercial SiC (Fig. 4f and S3†). The number of electrons transferred is calculated to be 3.6–3.9 over the potential range of 0.1–0.8 V. This is well consistent with the result obtained from the K–L plots based on RDE measurements.
Regarding the potential use of a catalyst in direct methanol fuel cells, its stability and tolerance to methanol crossover are key parameters to evaluate its performance. The durability of the atomically thin SiC and commercial Pt/C for the ORR was evaluated by continuous chronoamperometric measurements at 0.7 V in O2-saturated 0.1 M KOH solution (Fig. 4g). The ORR current density of the atomically thin SiC decreases more slowly than that of commercial Pt/C, maintaining 91% of its initial current density even after 18000 s. This result indicates that the atomically thin SiC demonstrates better stability than commercial Pt/C in alkaline medium. The high stability of the atomically thin SiC is mainly derived from its thin layer structure, which ensures no significant decrease in catalytic active sites on its surface. Moreover, the catalytic selectivity of the atomically thin SiC was determined from its chronoamperometric responses at the potential of 0.7 V in 0.1 M KOH solution with 3 M methanol added (Fig. 4h). For comparison, the corresponding chronoamperometric response for the Pt/C catalyst was also tested. No significant response is observed in the ORR current for the atomically thin SiC catalyst. However, a sharp decrease in the ORR current can be observed for the Pt/C catalyst after the addition of methanol. This result indicates that the atomically thin SiC catalyst has higher selectivity toward the ORR and better methanol tolerance than the commercial Pt/C in an alkaline medium, which is relevant in practical applications.
The atomically thin SiC was also used as a catalyst for the ORR in acidic medium. The electrocatalytic activity of the atomically thin SiC in 0.1 M HClO4 was investigated via CV (Fig. 5a). Compared with N2 saturated solution, the CV curve in O2 saturated 0.1 M HClO4 solution presents a remarkable ORR current peak at 0.62 V (vs. RHE) and a larger cathodic current comparable with commercial SiC appears, which indicate high ORR catalytic activity in acidic medium.
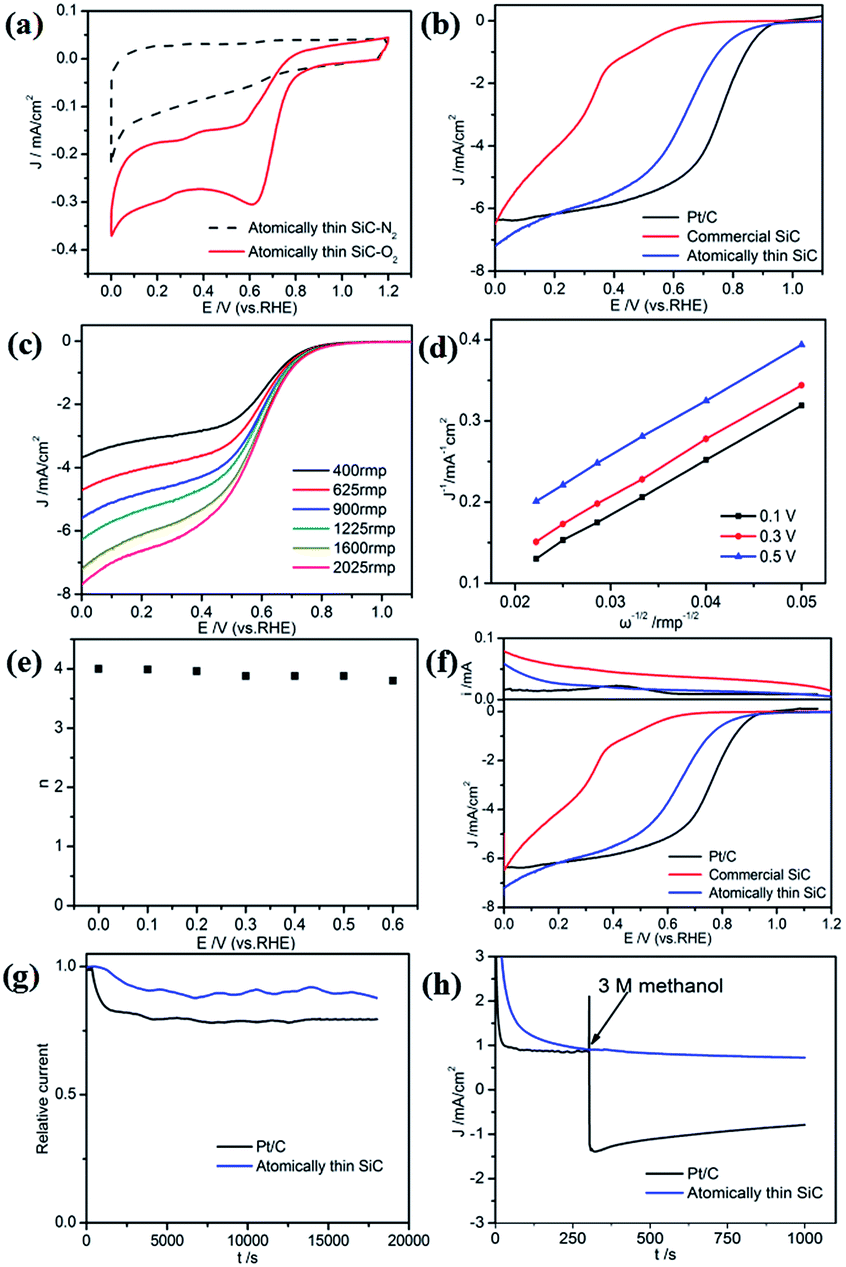 | ||
| Fig. 5 (a) CVs of the atomically thin SiC in N2- and O2-saturated 0.1 M HClO4 solution at a scan rate of 10 mV s−1. (b) LSV of the atomically thin SiC, commercial SiC, and Pt/C in O2-saturated 0.1 M HClO4 at a scan rate of 10 mV s−1 with an RDE rotation rate of 1600 rpm. (c) LSV of the atomically thin SiC at different RDE rotation rates. (d) Calculated K–L plots of the ORR on the atomically thin SiC. (e) Electron-transfer number n derived from the K–L plots at different potentials. (f) RRDE curves of the atomically thin SiC. (g) Chronoamperometric response (i–t) of the atomically thin SiC and Pt/C and (h) i–t of the atomically thin SiC and Pt/C before and after the addition of 3 M methanol. Tests were conducted in O2-saturated 0.1 M HClO4 solution at 0.7 V. | ||
The ORR performance was characterized by LSV measurements on an RDE in O2-saturated 0.1 M HClO4 solution. As seen from Fig. 5b, commercial SiC shows weaker catalytic activity and lower onset potential in an acidic medium. The ORR onset potential for the atomically thin SiC commenced at around 0.88 V, which is close to Pt/C electrodes (0.92 V). The limiting diffusion current on the atomically thin SiC electrode presents a relatively wide plateau with respect to the commercial SiC electrode.
To estimate the number of electrons transferred for the atomically thin SiC in acidic medium, LSV and K–L plots at different potentials were obtained according to the K–L equations, which are shown in Fig. 5c and d. The values of DO, CO and ν are 1.93 × 10−5 cm2 s−1, 1.26 × 10−6 mol cm−3, and 0.01 cm2 s−1, respectively in 0.1 M HClO4 solution.49 The three curves show a good linear relationship and the corresponding number of electrons transferred is 3.8–4.0 per O2 molecule at different potentials for the atomically thin SiC. According to the RRDE results obtained from 0.1 M HClO4 solution (Fig. 5e), the atomically thin SiC also exhibits a much higher diffusion-limiting current density than that of commercial SiC. The RRDE measurements show that the HO2− yield of the atomically thin SiC is less than 7%, which is significantly lower than that of commercial SiC (Fig. 5f and S3†). The number of electrons transferred is calculated to be 3.7–4.0 over the potential range of 0–0.6 V. The results of the electrochemical experiments prove that the atomically thin SiC electrode shows favorable ORR catalytic activity in acidic media. Moreover, we speculate that the catalytic activity of the atomically thin SiC is also reduced by the solubility of acid and can be further improved by changing the structure of the catalyst.
The stability and tolerance to methanol crossover of the atomically thin SiC in acidic medium were also evaluated using the same method. Compared with the performance of commercial Pt/C, the attenuation of the ORR current density for the atomically thin SiC is evidently slow (Fig. 5g). Its selectivity toward the ORR and methanol tolerance are better than that of commercial Pt/C in an acidic medium (Fig. 5h).
The ORR performance of the atomically thin SiC is mainly attributed to its ultrathin structure and the presence of a small amount of nitrogen. These microstructure and composition characteristics facilitate reactant adsorption, increase the reactive sites and improve the electrical conductivity of the atomically thin SiC, which lead to an improvement in the catalytic activity.
4. Conclusions
In summary, atomically thin SiC is obtained using an ultrasonic exfoliation method and used as an ORR catalyst in fuel cells. The particle size of SiC considerably decreases, its surface area increases, and the surface of the atomically thin SiC contains numerous unsaturated bonds and a small amount of nitrogen, which are favorable for the adsorption and dissociation of reactants. The ultrathin structure of the atomically thin SiC significantly improves the ORR catalytic activity in both alkaline and acidic media. The number of electrons transferred for the ORR on the atomically thin SiC catalyst is close to 4, which indicates that a four-electron reduction pathway is favored. The atomically thin SiC also exhibits outstanding durability and methanol crossover resistance in both alkaline and acidic media. This study provides a new idea for the design and application of non-precious ORR catalysts.Acknowledgements
Financial support was provided by the Natural Science Foundation of Anhui Province (KJ2016A171), the Stable Talent Foundation of Anhui Science and Technology University (HCWD201601), the Anhui college students innovation and entrepreneurship training program (201610879048), and Materials Science and Engineering Key Discipline foundation (AKZDXK2015 A01).Notes and references
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878 Search PubMed
.
- M. L. Xiao, J. B. Zhu, L. G. Feng, C. P. Liu and W. Xing, Adv. Mater., 2015, 27, 2521–2527 Search PubMed
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS PubMed
- E. M. Ericson, M. S. Thorum, R. Vasic, N. S. Marinkovic, A. L. Frenkel, A. A. Gewirth and R. G. Nuzzo, J. Am. Chem. Soc., 2012, 134, 197–200 CrossRef PubMed
- C. H. Choi, M. W. Chung, H. C. Kwon, S. H. Park and S. I. Woo, J. Mater. Chem. A, 2013, 1, 3694–3699 CAS
- Z. Y. Mo, H. L. Peng, H. G. Liang and S. J. Liao, Electrochim. Acta, 2013, 99, 30–37 CrossRef CAS
- S. Y. Wang, D. S. Yu, L. M. Dai, D. W. Chang and J. B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed
- S. Y. Wang, D. S. Yu and L. M. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed
- N. Hoshi, M. Nakamura and A. Hitotsuyanagi, Electrochim. Acta, 2013, 112, 899–904 CrossRef CAS
- J. Chen, B. Lim, E. P. Lee and Y. Xia, Nano Today, 2009, 4(1), 81–95 CrossRef CAS
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS PubMed
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed
- E. Proietti, F. Jaouen, M. Lefvre, N. Larouche, J. Tian, J. Herranz and J. P. Dodelet, Nat. Commun., 2011, 2, 416–424 CrossRef PubMed
- H. W. Liang, W. Wei, Z. S. Wu, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed
- L. J. Yang, S. J. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Z. Wang, Q. Wu, J. Ma, Y. W. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed
- Z. H. Sheng, H. L. Gao, W. J. Bao, F. B. Wang and X. H. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC
- L. Qu, Y. Liu, J. B. Back and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed
- Z. Y. Lin, G. Waller, Y. Liu, M. L. Liu and C. P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS
- Z. J. Jiang, Z. Q. Jiang and W. H. Chen, J. Power Sources, 2014, 251, 55–65 CrossRef CAS
- H. J. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed
- J. E. Park, Y. J. Jang, Y. J. Kim, M. S. Song, S. H. Yoon, D. H. Kim and S. J. Kim, Phys. Chem. Chem. Phys., 2014, 16, 103–109 RSC
- Y. F. Sun, F. C. Lei, S. Gao, B. C. Pan, J. F. Zhou and Y. Xie, Angew. Chem., Int. Ed., 2013, 52, 10569 CrossRef CAS PubMed
- J. F. Xie, H. Zhang, S. Li, R. X. Wang, X. Sun, M. Zhou, J. F. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed
- J. H. Guo, Y. T. Shi, X. G. Bai, X. C. Wang and T. L. Ma, J. Mater. Chem. A, 2015, 3, 24397–24404 CAS
- R. B. Wu, K. Zhou, C. Y. Yue, J. Wei and Y. Pan, Prog. Mater. Sci., 2015, 72, 1–30 CrossRef CAS
- Y. W. Kim, K. Y. Lim and W. S. Seo, J. Am. Ceram. Soc., 2014, 97, 923–928 CrossRef CAS
- M. Herrmann, K. Sempf, M. Schneider, U. Sydow, K. Kremmer and A. Michaelis, J. Eur. Ceram. Soc., 2014, 34, 229–235 CrossRef CAS
- A. M. Kueck, Q. M. Ramasse, L. C. De Jonghe and R. O. Riche, Acta Mater., 2010, 58, 2999–3005 CrossRef CAS
- P. Zhang, B. B. Xiao, X. L. Hou, Y. F. Zhu and Q. Jiang, Sci. Rep., 2014, 4, 3821–3828 CrossRef CAS PubMed
- R. B. Wu, Z. H. Yang, M. S. Fu and K. Zhou, J. Alloys Compd., 2016, 687, 833–838 CrossRef CAS
- W. Liu, L. F. Cheng, Y. G. Wang and H. J. Ma, J. Eur. Ceram. Soc., 2016, 36, 3901–3907 CrossRef CAS
- J. H. Boo, M. Kim, S. B. Lee, S. J. Park and J. G. Han, J. Vac. Sci. Technol., A, 2000, 18, 1713–1717 CAS
- E. Beche, M. Balat-Pichelin, V. Flaud, J. Esvan, T. Duguer, D. Sciti and D. Alfano, Surf. Interface Anal., 2014, 46, 817–822 CrossRef CAS
- Y. Mizokawa, K. Geib and C. Wilmsen, J. Vac. Sci. Technol., A, 1986, 4, 1696–1700 CAS
- A. Stoch, J. Stoch and A. Rakowska, Surf. Interface Anal., 1994, 22, 242–247 CrossRef CAS
- Y. S. Jin, Q. J. Yan, Z. R. Yin and Y. Chen, J. Chem. Soc., Faraday Trans., 1995, 91, 381–384 RSC
- H. T. Liu, Y. Q. Liu and D. B. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC
- Y. Nie, L. Li and Z. D. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed
- H. R. Byon, J. Suntivich and Y. S. Horn, Chem. Mater., 2011, 23, 3421–3428 CrossRef CAS
- X. D. Zhang, J. J. Zhang, J. Y. Zhao, B. C. Pan, M. G. Kong, J. Chen and Y. Xie, J. Am. Chem. Soc., 2012, 134, 11908 CrossRef CAS PubMed
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. J. Du, W. M. Zhang, Z. H. Zhu, S. C. Smith, M. Jaroniec, G. Q. (Max) Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116 CrossRef CAS PubMed
- J. J. Duan, S. Chen, S. Dai and S. Z. Qiao, Adv. Funct. Mater., 2014, 24, 2072 CrossRef CAS
- S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182 CrossRef CAS PubMed
- S. Wang, D. Yu, L. Dai, D. W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202 CrossRef CAS PubMed
- L. Y. Zhang, C. X. Guo, Z. Cui, J. Guo, Z. Dong and C. M. Li, Chem.–Eur. J., 2012, 18, 15693 CrossRef CAS PubMed
- J. N. Zheng, L. L. He, C. Chen, A. J. Wang, K. F. Ma and J. J. Feng, J. Power Sources, 2014, 268, 744–751 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: SEM, TEM, HRTEM, AFM images of commercial SiC; O 1s, C 1s, Si 2p core-level of commercial SiC; binding energy and percentages of different bindings calculated from the deconvoluted XPS spectra of C 1s, Si 2p, and O 1s peak; CVs of commercial SiC. See DOI: 10.1039/c7ra01701d |
| This journal is © The Royal Society of Chemistry 2017 |