
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN,N-Dimethylformamide-stabilized copper nanoparticles as a catalyst precursor for Sonogashira–Hagihara cross coupling†
Hideo Okaa,
Katsuya Kitaia,
Takeyuki Suzukib and
Yasushi Obora *a
*a
aDepartment of Chemistry and Materials Engineering, Faculty of Chemistry, Materials and Bioengineering, Kansai University, Suita, Osaka 564-8680, Japan. E-mail: obora@kansai-u.ac.jp
bComprehensive Analysis Center, The Institute of Scientific and Industrial Research (ISIR), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka 567-0057, Japan
First published on 26th April 2017
Abstract
We report the catalysis of a Sonogashira–Hagihara cross-coupling reaction using a DMF-stabilized copper nanoparticle catalyst. The reaction proceeded with low catalyst loadings, and a turnover number of 4.0 × 103 was recorded for 0.01 mol% catalyst loading. DMF-stabilized copper nanoparticles thus showed high catalytic activity.
Introduction
Transition metal catalysis is a very useful strategy for the synthesis of a wide variety of compounds with carbon structures. Multiple catalytic reactions that use transition metal catalysts have been reported; well-known examples include Suzuki–Miyaura cross coupling, the Mizoroki–Heck reaction, Negishi coupling, Migita–Kosugi–Stille coupling, and Sonogashira coupling.1–3 Sonogashira coupling is a remarkably valuable approach to the synthesis of acetylene compounds with unsaturated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, which can be used as intermediates, bioactive substances, pharmaceutical compounds, and electronic materials.4–6 However, the strategy originally reported by Sonogashira and co-workers used expensive palladium and copper as co-catalysts.7 In recent years, copper- or palladium-free catalysis has been studied.8–13 Low-cost metals such as iron and nickel have also been used as catalysts, but these metals require copper to be used as a co-catalyst, complex ligands, zinc additives, and harsh reaction conditions.14–21 Sonogashira coupling using a palladium-based catalyst system proceeds with a low catalyst loading (0.07 mol%) and under mild conditions. Copper, nickel, or iron catalysts with a cost advantage have not yet been developed.22,23
C bonds, which can be used as intermediates, bioactive substances, pharmaceutical compounds, and electronic materials.4–6 However, the strategy originally reported by Sonogashira and co-workers used expensive palladium and copper as co-catalysts.7 In recent years, copper- or palladium-free catalysis has been studied.8–13 Low-cost metals such as iron and nickel have also been used as catalysts, but these metals require copper to be used as a co-catalyst, complex ligands, zinc additives, and harsh reaction conditions.14–21 Sonogashira coupling using a palladium-based catalyst system proceeds with a low catalyst loading (0.07 mol%) and under mild conditions. Copper, nickel, or iron catalysts with a cost advantage have not yet been developed.22,23
Transition metal nanoparticles (M NPs) have been extensively studied with regard to their application in catalytic reactions, with the aim of identifying the specific advantages of their high surface area and unique properties.24,25 The synthesis of metal NPs commonly requires a metal precursor, reductant, protectant, and solvent. M NPs stabilized by dendrimers and supported on metal oxides have also been reported.26–29 Copper NPs (Cu NPs) with various nanostructures have also been developed, and research into their applicability as catalysts has been carried out.26,27,30 There are many reports of Sonogashira coupling catalyzed by M NPs.31–37 However, in almost all cases, these reports describe Pd NP catalyst systems.31–33,38–41 Cu NPs are superior in terms of cost, but there are a limited number of examples in which Cu NPs are used as catalysts for the Sonogashira reaction.34–37 In addition, a greater amount of catalyst is required for Cu NP-catalyzed reactions, and multi-step procedures are required for the synthesis of these catalysts.28,29,34–37 Rothenberg and co-workers were the first to report a copper-catalyzed palladium-free and ligand-free Sonogashira cross coupling with various aryl iodides and bromides.35 For this they used Cu nanoclusters prepared from chloride salt precursors by using tetra-n-octylammonium formate (TOAF) as a stabilizer in DMF at 65 °C. However, a multi-step operation was required for the synthesis of the Cu nanoparticles and the Sonogashira reaction was carried out with 5 mol% of the Cu NP catalyst.35 Tang, Zhang, and co-workers reported that octahedral Cu2O NPs could be used in a reusable Cu2O/PPh3/n-Bu4NBr (TBAB) system to catalyze Sonogashira coupling.37 However, a multistep synthesis is required to access Cu NPs in this system; this includes the use of hazardous reductants, such as hydrazine Cu NPs. Furthermore, this Cu NP system required a relatively large amount of the Cu NP catalyst (10 mol%), and TBAB surfactant was also needed.37 Therefore, the development of more effective Cu NPs that require smaller catalytic amounts and less demanding synthetic procedures is desired.
We have reported the synthesis of Cu NPs using the DMF-reduction method without the need for specialized reductants or surfactants, as well as catalytic reactions using these Cu NPs.42 DMF-stabilized M NPs synthesized with this method have very small (<10 nm) particle sizes. The specific surface area of the M NPs increases as the particle diameter decreases, and this leads to an improvement in the catalytic activity. We have previously reported that DMF-stabilized Pd NCs catalyze Suzuki–Miyaura cross coupling,43 the Mizoroki–Heck reaction,43 and Migita–Kosugi–Stille coupling,44 and that DMF-stabilized Cu NPs catalyze the Ullmann coupling reaction.45 For these catalytic reactions, we recorded higher turnover numbers (TONs), and thus concluded that DMF-stabilized M NPs have high catalytic activity. Herein, we report that DMF-stabilized Cu NPs can catalyze the Sonogashira coupling reaction. DMF-stabilized Cu NPs prepared by a simple methodology from CuCl2 and DMF (as solvent, reductant, and stabilizer) promoted the reaction of terminal alkynes with aryl halides at low catalyst loadings (0.01 mol%) with a TON of 4.0 × 103. This is an improvement over previously reported TBAB- and TOAF-protected Cu NPs, which require 5–10 mol% catalyst loadings.35,37
Results and discussion
First, we prepared Cu NPs by the DMF-reduction method, according to the procedure reported in the literature.45 Characterization of the Cu NPs was conducted by various instrumental analytical techniques.The transmission electron microscopy (TEM) image and particle size distribution of the prepared Cu NPs revealed that their diameters mainly ranged from 2 to 7 nm (Fig. 1). In addition, X-ray diffraction (XRD) spectra showed that the NPs had an amorphous nature (Fig. S1†).
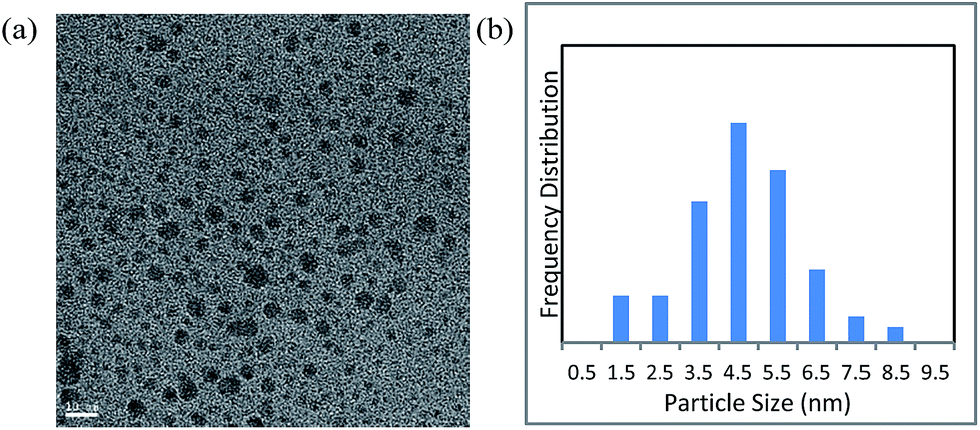 | ||
| Fig. 1 (a) Transmission electron microscopy image of DMF-stabilized copper nanoparticles (scale bar = 10 nm); (b) particle size distribution of the nanoparticles. | ||
The FT-IR spectrum of the DMF-stabilized Cu NPs showed a strong absorption at around 1670 cm−1 (Fig. S2(a)†), which corresponds to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration of the DMF molecules. These results suggest that the DMF molecules interact with the Cu NPs.46,47 Thermogravimetric analysis-differential thermal analysis (TG-DTA) of the Cu NPs confirmed that the nanoparticles are thermally stable up to around 150 °C (Fig. S3†).
O) vibration of the DMF molecules. These results suggest that the DMF molecules interact with the Cu NPs.46,47 Thermogravimetric analysis-differential thermal analysis (TG-DTA) of the Cu NPs confirmed that the nanoparticles are thermally stable up to around 150 °C (Fig. S3†).
The synthesis of acetylene by the coupling of aryl halides with terminal alkynes has a wide scope, and is utilized in areas such as the pharmaceutical and electronic materials fields. In terms of cost advantage, it would be more useful to develop an M NP-catalyzed version of this reaction than of any other reaction. Furthermore, a wide variety of additives and solvents are tolerated in this reaction (Table 1). First, the Sonogashira reaction of the model compounds iodobenzene (1a, 0.5 mmol) and phenylacetylene (2a) was catalyzed by Cu NPs in DMF at 135 °C for 48 hours, using K2CO3 as the base. However, this reaction showed low conversion and we only obtained a trace of the desired product (entry 1). Addition of PPh3 promoted the reaction and improved conversion; we obtained desired product 3a in quantitative yield (entry 2). We then examined other phosphine, nitrogen, and diketone compounds because of the great improvement observed when using the PPh3 additive. Phosphine compounds other than PPh3, for example P(o-tolyl)3, PCy3, and dppe did not promote the reaction (entries 3–5). In contrast, nitrogen compounds such as bipyridine (bpy) and 4,4′-di-tert-butyl-2,2′-bipyridine (dbbpy) were very effective and the reaction proceeded quantitatively when these additives were used (entries 6 and 7). 1,10-Phenanthroline (1,10-phen) and N,N,N′,N′-tetramethylethylenediamine (TMEDA) improved the yield slightly (entries 8 and 9), and using 2,2,6,6-tetramethyl-3,5-heptadione (TMHD) gave the product in moderate yields (entry 10). However, when acetylacetone (acac) was used instead of PPh3 the product yield was 19% (entry 11).
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Base | Solvent | Conv. (%) | Yieldb (%) |
| a Conditions: 1a (0.5 mmol), 2a (0.75 mmol), Cu NPs (2 × 10−1 mol%), additive (10 mol%), base (1.0 mmol), solvent (1 mL), 135 °C, 48 h, under Ar.b GC yield. The number in parentheses shows the isolated yield.c With 0.01 mol% Cu NPs.d At 80 °C.e At 120 °C.f Without catalyst. | |||||
| 1 | — | K2CO3 | DMF | 9 | 7 |
| 2 | PPh3 | K2CO3 | DMF | >99 | >99 (96) |
| 3 | P(o-tol)3 | K2CO3 | DMF | 73 | 14 |
| 4 | PCy3 | K2CO3 | DMF | 47 | Trace |
| 5 | dppe | K2CO3 | DMF | 65 | Trace |
| 6 | bpy | K2CO3 | DMF | >99 | >99 |
| 7 | dbbpy | K2CO3 | DMF | >99 | >99 |
| 8 | 1,10-Phen | K2CO3 | DMF | 82 | 40 |
| 9 | TMEDA | K2CO3 | DMF | 63 | 25 |
| 10 | TMHD | K2CO3 | DMF | 84 | 45 |
| 11 | acac | K2CO3 | DMF | 60 | 19 |
| 12 | PPh3 | Na2CO3 | DMF | 66 | 55 |
| 13 | PPh3 | Cs2CO3 | DMF | 65 | Trace |
| 14 | PPh3 | KOH | DMF | 27 | 18 |
| 15 | PPh3 | NEt3 | DMF | <1 | Trace |
| 16 | PPh3 | — | DMF | 59 | Trace |
| 17 | PPh3 | K2CO3 | NMP | 89 | 73 |
| 18 | PPh3 | K2CO3 | DMSO | >99 | >99 |
| 19 | PPh3 | K2CO3 | H2O | >99 | 88 |
| 20 | PPh3 | K2CO3 | Toluene | >99 | >99 |
| 21c | PPh3 | K2CO3 | DMF | 84 | 41 |
| 22d | PPh3 | K2CO3 | DMF | 3 | Trace |
| 23e | PPh3 | K2CO3 | DMF | 64 | 51 |
| 24f | PPh3 | K2CO3 | DMF | <1 | n.d. |
To investigate the effect of the DMF molecules on the surface of the Cu NPs during the catalytic reaction, FT-IR measurements featuring the DMF ν(CO) vibration peak at around 1670 cm−1 were conducted on Cu NP samples that had undergone complete removal of the DMF solvent using an NaCl plate method (Fig. S2†). These analyses indicated that DMF was liberated from the Cu NPs after catalytic reactions with PPh3 (entry 2 and Fig. S2(b) and (c)†) and bipyridine (entry 6 and Fig. S2(d)†). However, the ν(CO) vibration peak remained for a mixture of Cu NPs with PCy3 after the reaction had finished. This result indicates that addition of PCy3 did not induce the liberation of DMF, probably owing to steric constraints (entry 4 and Fig. S2(e)†).
We next measured the particle size distribution of the Cu NPs by TEM and dynamic light scattering under the conditions shown in entry 2 of Table 1 (Fig. 2 and S4†). A slight growth in the particle size was observed after the reaction. However, by inhibiting Ostwald ripening42,48,49 during the course of the catalytic reaction, the nanoparticles retained their original size of <10 nm.
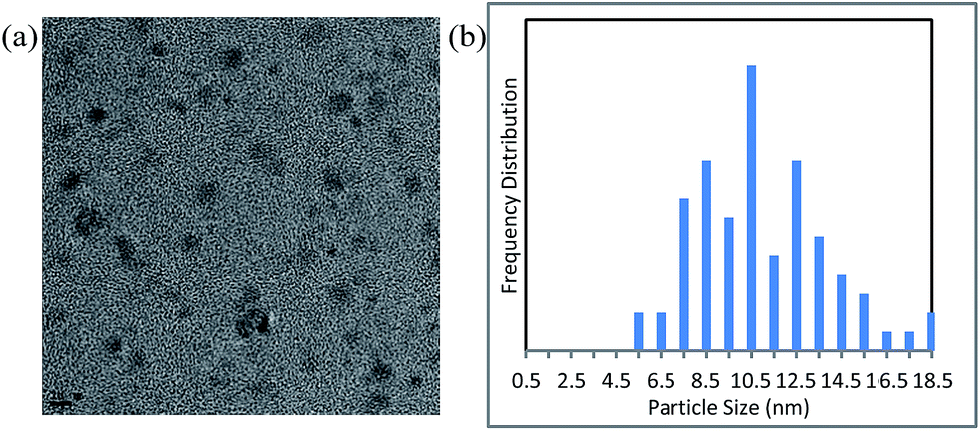 | ||
| Fig. 2 (a) Transmission electron microscopy image (scale bar = 10 nm) and (b) particle size distribution of the nanocatalyst after reaction under the conditions shown in entry 2, Table 1. | ||
The Cu NPs are themselves inactive as catalysts in this reaction owing to the strong coverage of DMF molecules on their surfaces (Fig. S5† and entry 1, Table 1). The liberation of the DMF molecules during the course of the reaction, which is assisted by additives such as PPh3, generates the active NP catalyst. PPh3 (or bipyridine) presumably serves to stabilize the uncoordinated sites on the Cu nanoparticles during the course of the reaction (Fig. 2 and S4†).
As shown in entry 2 of Table 1, K2CO3 was a good choice of base and gave excellent yields when used with the PPh3 additive. We also investigated other carbonate salts under the same conditions. Using a weak base, such as Na2CO3, gave the desired product in moderate yield (entry 12), but strong bases, such as Cs2CO3 or KOH, were not suitable for this reaction (entries 13 and 14). The substrates did not convert at all when using triethylamine or base-free conditions (entries 15 and 16). We next investigated the effect of changing the solvent and found that aprotic polar solvents, such as N-methyl-2-pyrrolidone (NMP) and dimethyl sulfoxide (DMSO), were most appropriate for this catalytic system (entries 17 and 18). Using H2O as the solvent gave good yields (entry 19), and quantitative yields of the product were also obtained with toluene (entry 20), which is less toxic than NMP and DMSO. Thus the reaction proceeded smoothly with various solvents under these conditions (entries 2, 18–20).
For this reaction, TONs of up to 4.0 × 103 were obtained by reducing the amount of the catalyst to 0.01 mol% (entry 21), although a small amount of a dehalogenated byproduct was observed. The time–yield curves for the formation of 3a with the Cu NP catalyst under the conditions in entry 2 of Table 1 (Fig. S6†) show that the reaction was mostly complete after about 24 h, and that the turnover frequency at the initial stage of the reaction was 29 h−1.
The reaction temperature is another important factor in this catalytic system. The reaction substrates did not convert at 80 °C (entry 22, Table 1) and only moderate yields were obtained at 120 °C (entry 23). The reaction did not proceed without the catalyst (entry 24).
We next attempted Cu NP-catalyzed Sonogashira cross-coupling reactions using various reaction substrates (Table 2), and found that DMF-stabilized Cu NPs had a broad scope in Sonogashira coupling reactions when the optimized reaction conditions were used. First, the reactivity of iodobenzene derivatives in reactions with phenylacetylene was studied (entries 1–9). Sterically hindered substrates and iodobenzenes bearing electron-donating or electron-withdrawing substituents gave the corresponding internal alkyne in moderate-to-good yields (3b–f, entries 1–5). Heteroaromatic iodides, such as 3-iodopyridine or 2-iodothiophene, provided the corresponding heteroaromatic internal alkynes (3g and 3h) in good yields (95% and 75%, respectively, entries 6 and 7).
|
||||
|---|---|---|---|---|
| Entry | Ar-X (1) | Alkyne (2) | Product (3) | Yielda (%) |
| a Conditions: same as entry 2, Table 1. Yields of the isolated product after purification.b Bromobenzene (2.0 mmol) and phenylacetylene (0.5 mmol) were used.c Not detected by GC. | ||||
| 1 | 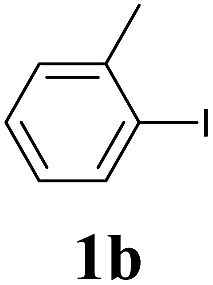 |
 |
 |
57 |
| 2 |  |
2a |  |
90 |
| 3 |  |
2a |  |
97 |
| 4 |  |
2a |  |
84 |
| 5 | 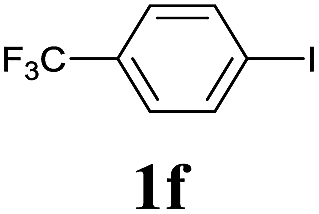 |
2a | 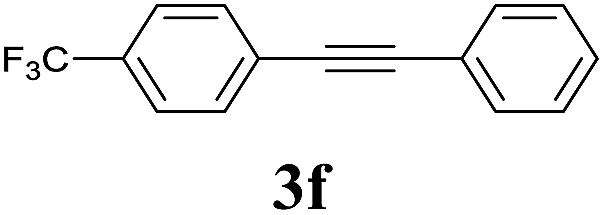 |
93 |
| 6 | 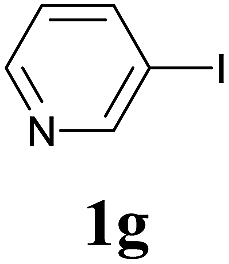 |
2a | 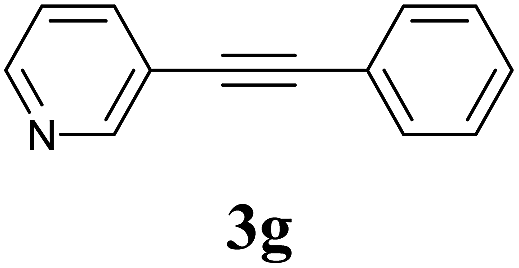 |
95 |
| 7 | 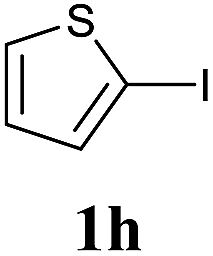 |
2a | 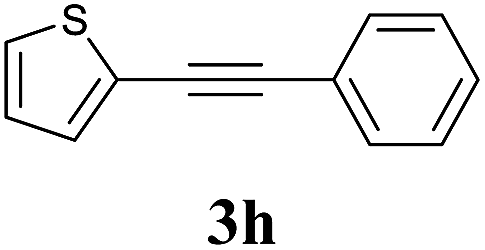 |
75 |
| 8b |  |
2a | 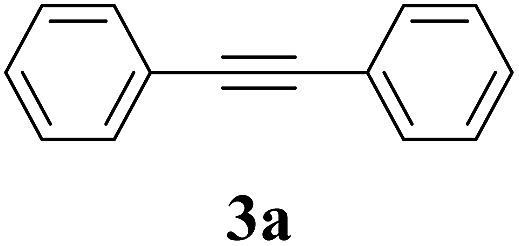 |
90 |
| 9 | 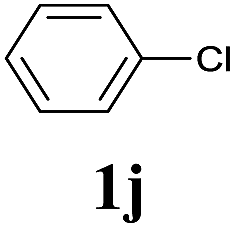 |
2a | 3a | n.d.c |
| 10 | 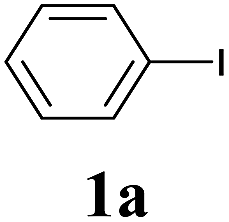 |
 |
3e | 84 |
| 11 | 1a |  |
 |
92 |
| 12 | 1a | 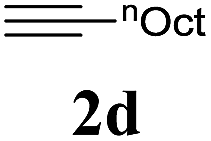 |
 |
57 |
| 13 | 1a | 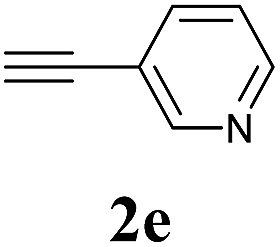 |
3g | 87 |
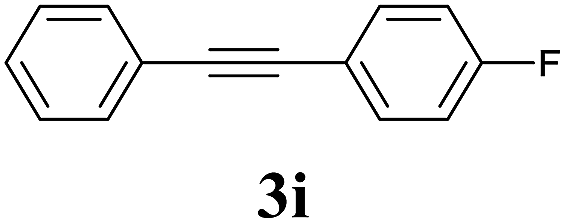
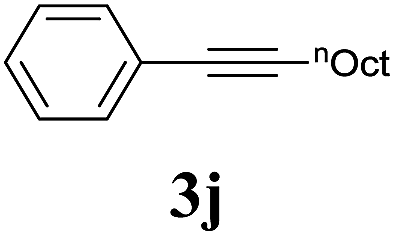
We also applied this catalytic system to the less reactive substrates bromobenzene and chlorobenzene in an attempt to extend the scope of the halide leaving group. Sonogashira coupling products were obtained from bromobenzene by using excess aryl halide reagent. However, no coupling products were obtained from chlorobenzene. Both electron-donating and electron-withdrawing groups were tolerated on the terminal alkyne and gave desired products 3e and 3i in good yields (entries 10 and 11). This is a result of the high acidity of the C–H bond attached to the electron-withdrawing alkyne group. In contrast, because aliphatic terminal alkynes lower the acidity of the C–H position, we obtained the internal alkyne with an aliphatic moiety (3j) in only moderate yield (entry 12). In addition, heteroaromatic substrate 3-ethynylpyridine gave corresponding heteroaromatic internal alkyne 3-(phenylethynyl) pyridine (3g) in good yield (87%, entry 13).
To gain further insight into the function of PPh3 on the catalytic activity of the Cu NPs, we carried out a poisoning test in the presence of mercury (5 equiv.) under the conditions shown in entry 2 of Table 1. This reaction gave coupling product 3a in only 3% yield and with low conversion (20%). This result indicates that PPh3-stabilized Cu NPs have more uncoordinated active sites available on their Cu surfaces, whereas the DMF seems to passivate the surface of the nanoparticles.
The reusability of the Cu NP catalyst was examined in toluene, and the results showed that the catalyst could be reused at least five times without loss of activity (Fig. 3).
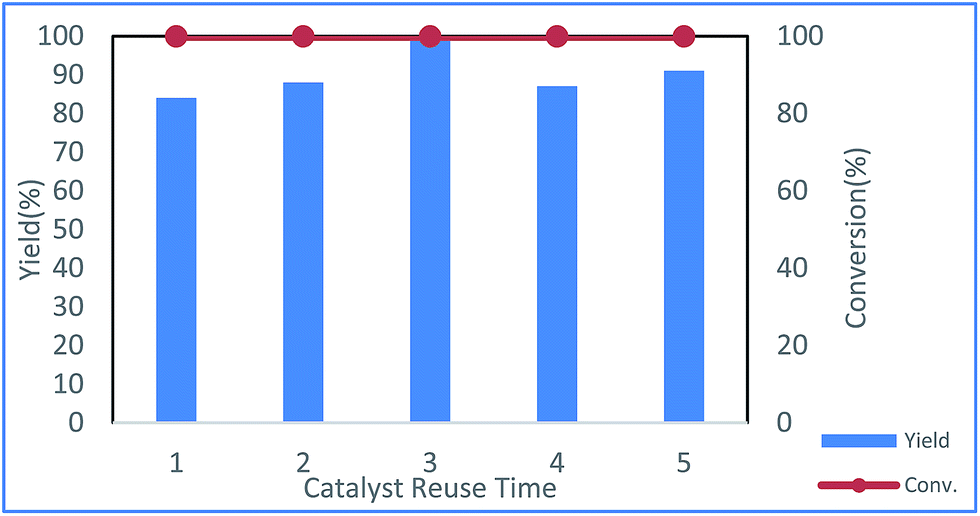 | ||
| Fig. 3 Repeated runs of the same catalyst under the reaction conditions shown in entry 20 of Table 1 (red: conversion of 1a; blue: yield of 3a). | ||
Conclusions
We found that DMF-stabilized Cu NPs have high catalytic activity in the Sonogashira cross-coupling reaction and can be applied in the reactions of a wide range of substrates, including aryl halides and alkynes bearing electron-donating and electron-withdrawing substituents, as well as sterically hindered and heterocyclic substrates. The reactions were successful with low catalyst loadings and it was possible to reuse the catalyst multiple times.Experimental section
General
Transmission electron microscopy (TEM) images were recorded by using a JEOL JEM-ARM200F at an acceleration voltage of 200 kV. 1H and 13C NMR were measured by using a JEOL JNM-ECA 400 spectrometer at 400 and 100 MHz, respectively, in CDCl3 with Me4Si as the internal standard. Thermogravimetric analysis (TG) was performed by using a Thremo plus EVO device (Rigaku, Japan) at a heating rate of 10 °C min−1 under nitrogen flow.Typical reaction procedure for Cu NP-catalyzed Sonogashira cross coupling of 1a with 2a (Table 1, entry 2)
A mixture of iodobenzene 1a (102 mg, 0.5 mmol), phenylacetylene 2a (77 mg, 0.75 mmol), K2CO3 (138 mg, 1.0 mmol), and PPh3 (13 mg, 10 mol%) were placed in a pressure container and a suspension of Cu NPs in DMF (1 mM, 1.0 mL) was added as the catalyst. The reaction mixture was stirred at 135 °C for 48 h under argon. The conversion of substrates and yields of the products were calculated from their peak areas in the GC based on an internal standard (n-tridecane). The reaction mixture was extracted with water and n-hexane to separate the product from the Cu NPs and K2CO3. Product 3a was isolated by column chromatography (silica gel (230–400 mesh), n-hexane as eluent) in 96% yield (86 mg).Mercury poisoning test for Cu NP-catalyzed Sonogashira cross coupling of 1a with 2a (Table 1, entry 2)
A mixture of iodobenzene 1a (102 mg, 0.5 mmol), phenylacetylene 2a (77 mg, 0.75 mmol), K2CO3 (138 mg, 1.0 mmol), PPh3 (13 mg, 10 mol%) and Hg (0.5 g, 2.5 mmol) were placed in a pressure container and a suspension of Cu NPs in DMF (1 mM, 1.0 mL) was added as the catalyst. The reaction mixture was stirred at 135 °C for 48 h under argon. The conversion of substrates and yields of the products were calculated from their peak areas in the GC based on an internal standard (n-tridecane).Reusability of the catalyst in Cu NP-catalyzed Sonogashira cross coupling of 1a with 2a in toluene (Table 1, entry 20)
A mixture of iodobenzene 1a (102 mg, 0.5 mmol), phenylacetylene 2a (77 mg, 0.75 mmol), K2CO3 (138 mg, 1.0 mmol) and PPh3 (13 mg, 10 mol%) were placed in a pressure container and a suspension of Cu NPs in toluene (1 mM, 1.0 mL) was added as the catalyst. The reaction mixture was stirred at 135 °C for 48 h under argon. The conversion of the substrate and yield of the product were calculated from their peak areas in the gas chromatogram based on an internal standard (n-tridecane). Then, 1a (102 mg, 0.5 mmol), phenylacetylene 2a (77 mg, 0.75 mmol), K2CO3 (138 mg, 1.0 mmol) and PPh3 (13 mg, 10 mol%) were added to the resulting mixture for the next reaction cycle.Acknowledgements
This work was performed under the Research Program of “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” in “Network Joint Research Center for Materials and Devices”. We thank the members of the Comprehensive Analysis Center, SANKEN (ISIR), Osaka University for TEM analysis.Notes and references
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 Search PubMed.
- V. Mehta, M. Panchal, A. Kongor, U. Panchal and V. K. Jain, Catal. Lett., 2016, 146, 1581–1590 Search PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- Y. N. Kotovshchikov, G. V. Latyshev, N. V. Lukashev and I. P. Beletskaya, Org. Biomol. Chem., 2015, 13, 5542–5555 CAS.
- C. W. D. Gallop, M.-T. Chen and O. Navarro, Org. Lett., 2014, 16, 3724–3727 CrossRef CAS PubMed.
- C.-X. Lin, J.-F. Zhu, Q.-S. Li, L.-H. Ao, Y.-J. Jin, F.-B. Xu, F.-Z. Hu and Y.-F. Yuan, Appl. Organomet. Chem., 2014, 28, 298–303 CrossRef CAS.
- R. Zhou, W. Wang, Z.-J. Jiang, H.-Y. Fu, X.-L. Zheng, C.-C. Zhang, H. Chena and R.-X. Li, Catal. Sci. Technol., 2014, 4, 746–751 CAS.
- H. Zhong, J. Wang, L. Li and R. Wang, Dalton Trans., 2014, 43, 2098–2103 RSC.
- K. Okuro, M. Furuune, M. Miura and M. Nomura, Tetrahedron Lett., 1992, 33, 5363–5364 CrossRef CAS.
- F. Monnier, F. Turtaut, L. Duroure and M. Taillefer, Org. Lett., 2008, 10, 3203–3206 CrossRef CAS PubMed.
- A. R. Hajipour, S. H. Nazemzadeh and F. Mohammadsaleh, Tetrahedron Lett., 2014, 55, 654–656 CrossRef CAS.
- N. T. Tran, C. S. Cho, H.-S. Sohn and S. C. Shim, Bull. Korean Chem. Soc., 2011, 32, 1080–1082 CrossRef CAS.
- D. N. Sawant, P. J. Tambade, Y. S. Wagh and B. M. Bhanage, Tetrahedron Lett., 2010, 51, 2758–2761 CrossRef CAS.
- T.-T. Hung, C.-M. Huang and F.-Y. Tsai, ChemCatChem, 2012, 4, 540–545 CrossRef CAS.
- M. Bakherad, A. Keivanloo and S. Mihanparast, Synth. Commun., 2010, 40, 179–185 CrossRef CAS.
- O. Vechorkin, D. Barmaz, V. Proust and X. Hu, J. Am. Chem. Soc., 2009, 131, 12078–12079 CrossRef CAS PubMed.
- I. P. Beletskaya, G. V. Latyshev, A. V. Tsvetkov and N. V. Lukashev, Tetrahedron Lett., 2003, 44, 5011–5013 CrossRef CAS.
- L. Wang, P. Li and Y. Zhang, Chem. Commun., 2004, 514–515 RSC.
- L.-H. Zou, A. J. Johansson, E. Zuidema and C. Bolm, Chem.–Eur. J., 2013, 19, 8144–8152 CrossRef CAS PubMed.
- B. J. Borah and D. K. Dutta, J. Mol. Catal. A: Chem., 2013, 366, 202–209 CrossRef CAS.
- G. Chen, X. Zhu, J. Cai and Y. Wan, Synth. Commun., 2007, 37, 1355–1361 CrossRef CAS.
- S. Sun, Nanoscale, 2015, 7, 10850–10882 RSC.
- H. Xu, W. Wang and W. Zhu, J. Phys. Chem. B, 2006, 110, 13829–13834 CrossRef CAS PubMed.
- M. B. Thathagar, J. Beckers and G. Rothenberg, J. Am. Chem. Soc., 2002, 124, 11858–11859 CrossRef CAS PubMed.
- B. White, M. Yin, A. Hall, D. Le, S. Stolbov, T. Rahman, N. Turro and S. O'Brien, Nano Lett., 2006, 6, 2095–2098 CrossRef CAS PubMed.
- M. Zhang, L. Wang, B. Tang, X. Shen and R. Hu, Chin. J. Chem., 2010, 28, 1963–1966 CrossRef CAS.
- M. A. Bhosale, T. Sasaki and B. M. Bhanage, Catal. Sci. Technol., 2014, 4, 4274–4280 CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- P. K. Mandali and D. K. Chand, Catal. Commun., 2014, 47, 40–44 CrossRef CAS.
- D. Sengupta, J. Saha, G. De and B. Basu, J. Mater. Chem. A, 2014, 2, 3986–3992 CAS.
- G. Li, D.-E. Jiang, C. Liu, C. Yu and R. Jin, J. Catal., 2013, 306, 177–183 CrossRef CAS.
- N. Panda, A. K. Jena and S. Mohapatra, Chem. Lett., 2011, 40, 956–958 CrossRef CAS.
- M. B. Thathagar, J. Beckers and G. Rothenberg, Green Chem., 2004, 6, 215–218 RSC.
- Y. Yuan, H. Zhu, D. Zhao and L. Zhang, Synthesis, 2011, 1792–1798 CrossRef CAS.
- B.-X. Tang, F. Wang, J.-H. Li, Y.-X. Xie and M.-B. Zhang, J. Org. Chem., 2007, 72, 6294–6297 CrossRef CAS PubMed.
- M. Shunmughanathan, P. Puthiaraj and K. Pitchumani, ChemCatChem, 2015, 7, 666–673 CrossRef CAS.
- A. R. Hajipour, Z. Shirdashtzade and G. Azizi, Appl. Organomet. Chem., 2014, 28, 696–698 CrossRef CAS.
- L. N. Parshina, A. P. Tantsyrev, L. A. Grishchenko and B. A. Trofimov, Russ. J. Org. Chem., 2013, 49, 412–416 CrossRef CAS.
- A. Khazaei, S. Rahmati and S. Saednia, Catal. Commun., 2013, 37, 9–13 CrossRef CAS.
- H. Yamamoto, H. Yano, H. Kouchi, Y. Obora, R. Arakawa and H. Kawasaki, Nanoscale, 2012, 4, 4148–4154 RSC.
- M. Hyotanishi, Y. Isomura, H. Yamamoto, H. Kawasaki and Y. Obora, Chem. Commun., 2011, 47, 5750–5752 RSC.
- H. Yano, Y. Nakajima and Y. Obora, J. Organomet. Chem., 2013, 745, 258–261 CrossRef.
- Y. Isomura, T. Narushima, H. Kawasaki, T. Yonezawa and Y. Obora, Chem. Commun., 2012, 48, 3784–3786 RSC.
- Y. Sugii, M. Inada, H. Yano, Y. Obora, Y. Iwasaki, R. Arakawa and H. Kawasaki, J. Nanopart. Res., 2013, 15, 1379–1385 CrossRef.
- K. Oikawa, S. Itoh, H. Yano, H. Kawasaki and Y. Obora, Chem. Commun., 2017, 53, 1080–1083 RSC.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS PubMed.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- M. B. Thathagar and G. Rothenberg, Org. Biomol. Chem., 2006, 4, 111–115 CAS.
- T. Suzuka, Y. Okada, K. Ooshiro and Y. Uozumi, Tetrahedron, 2010, 66, 1064–1069 CrossRef CAS.
- A. R. Katritzky, A. A. A. Abdel-Fattah and M. Wang, J. Org. Chem., 2002, 67, 7526–7529 CrossRef CAS PubMed.
- C.-H. Lin, Y.-J. Wang and C.-F. Lee, Eur. J. Org. Chem., 2010, 23, 4368–4371 Search PubMed.
- R. Najman, J. K. Cho, A. F. Coffey, J. W. Devies and M. Bradley, Chem. Commun., 2007, 5031–5033 RSC.
- U. S. Sørensen and E. Pombo-Villar, Tetrahedron, 2005, 61, 2697–2703 CrossRef.
- C. W. D. Gallop, M.-T. Chen and O. Navarro, Org. Lett., 2014, 16, 3724–3727 CrossRef CAS PubMed.
- M. Bandini, R. Luque, V. Budarin and D. J. Macquarrie, Tetrahedron, 2005, 61, 9860–9868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27910d |
| This journal is © The Royal Society of Chemistry 2017 |