
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Twisted configuration pyrene derivative: exhibiting pure blue monomer photoluminescence and electrogenerated chemiluminescence emissions in non-aqueous media†
Ying Zhao,
Dong Xue*,
Honglan Qi and
Chengxiao Zhang *
*
Key Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710062, P. R. China. E-mail: cxzhang@snnu.edu.cn; xuedong_welcome@snnu.edu.cn
First published on 26th April 2017
Abstract
A blue light emitting pyrene derivative, 1-methyl benzoate-pyrene (MB-Py), was synthesized by employing a pyrene moiety as the parent linked with a small electron-donating group, methyl benzoate. Owing to the introduction of methyl benzoate, there is an evident twisted configuration between the methyl benzoate moiety and the central pyrene core with a dihedral angle of 56°, confirmed by density functional theory (DFT) calculations. The basic photophysical, electrochemical and electrogenerated chemiluminescence (ECL) properties of MB-Py together with those of the parent pyrene (Py) and the analogue benzo[α]pyrene (Bz-Py) in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bz (v/v = 1:1) were extensively characterized for the first time. The electrochemical behavior of MB-Py showed a quasi-reversible one-electron oxidation (E1/2 = +1.11 V vs. SCE) and two reversible closely spaced consecutive one-electron reductions (E1/2,1 = −2.12 V and E1/2,2 = −2.27 V vs. SCE). This redox behavior was confirmed by CV digital simulations and was consistent with the frontier molecular orbitals calculated. It was found that, unlike its parent Py and analogue Bz-Py, MB-Py showed a single-peaked absorption (343 nm) and a blue monomer photoluminescence (PL) emission (428 nm) with a large Stokes shift (85 nm). The electrogenerated radical ions of MB-Py are stable enough to produce an efficient ECL emission (439 nm) through ion annihilation with the S-route. The ECL spectra of MB-Py obtained in both the ion annihilation and coreactant processes are similar to the monomer PL spectrum. This nonplanar twisted conformation pyrene derivative imparts steric hindrance, prevents excimer formation and results in a high PL efficiency and efficient, stable blue ECL monomer emission. Owing to their rigid planar structures, Py and Bz-Py exhibited monomer and excimer blue-violet PL emissions and cyan/green ECL emissions, and could not be used as appropriate blue luminophores.
:Bz (v/v = 1:1) were extensively characterized for the first time. The electrochemical behavior of MB-Py showed a quasi-reversible one-electron oxidation (E1/2 = +1.11 V vs. SCE) and two reversible closely spaced consecutive one-electron reductions (E1/2,1 = −2.12 V and E1/2,2 = −2.27 V vs. SCE). This redox behavior was confirmed by CV digital simulations and was consistent with the frontier molecular orbitals calculated. It was found that, unlike its parent Py and analogue Bz-Py, MB-Py showed a single-peaked absorption (343 nm) and a blue monomer photoluminescence (PL) emission (428 nm) with a large Stokes shift (85 nm). The electrogenerated radical ions of MB-Py are stable enough to produce an efficient ECL emission (439 nm) through ion annihilation with the S-route. The ECL spectra of MB-Py obtained in both the ion annihilation and coreactant processes are similar to the monomer PL spectrum. This nonplanar twisted conformation pyrene derivative imparts steric hindrance, prevents excimer formation and results in a high PL efficiency and efficient, stable blue ECL monomer emission. Owing to their rigid planar structures, Py and Bz-Py exhibited monomer and excimer blue-violet PL emissions and cyan/green ECL emissions, and could not be used as appropriate blue luminophores.
Introduction
Development of new radical candidates for electrogenerated chemiluminescence (ECL) has become an active area of research, since the use of ECL has become commercially viable in areas such as life science research, environmental monitoring, full-color displays and so on.1 Three basic colors – red, green, and blue luminophores – are required for the improvement of ECL techniques. Green and red luminophores with good stability and efficiency have been widely used, such as Ru(bpy)32+ and its derivatives.2 However, the intrinsically large optical and transport gaps required for blue luminophores make them difficult to optimize in terms of their efficiency, color purity, and stability.3 Thus, tailor-made blue luminophores that are suitable for ECL still need to be further improved. The investigation of the relationships between the structures and ECL behaviors has been a continuous theme in ECL.Polycyclic aromatic hydrocarbons (PAHs), such as 9,10-diphenylanthracene (DPA),4 have been studied as potential ECL blue luminophores in both fundamental and application-oriented research.5 Pyrene, an important type of PAH, consists of four fused benzene rings, resulting in a large conjugated planar aromatic system. Pyrene is a possible ECL radical candidate because of its prominent photophysical properties, such as its remarkably long fluorescence lifetime and high photoluminescence (PL) efficiency.6 However, pyrene itself, with its intrinsic planar rigid structure and significant π-orbital overlap,7 has serious disadvantages for use as a blue luminophore due to its tendency to crystallize, conjugate or form extensive excimers,8 resulting in unstable radical ions and a red-shift of the emission peak to the green light-emitting region in an ECL process.9 Therefore, searching for and designing modified pyrene derivatives to overcome these disadvantages is key to improving both the efficiency and color purity of blue ECL materials.
Accordingly, much research has been done on designing highly efficient blue-emitting pyrene derivatives to suppress the formation of crystals and excimers. Mostly, approaches to such systems have focused on adopting a non-planar geometry by the introduction of other functionalized moieties10 or incorporation of an appropriate bridge linkage group.11 Bard’s group reported that capping pyrene with two fluorene derivatives produced new non-planar blue ECL materials and prevented excimer formation, resulting in enhanced ECL efficiency and stability of the cation radical.12 Kaafarani et al. combined carbazoles with pyrene and synthesized tortile tert-bis(carbazolyl) derivatives of pyrene and tetrahydropyrene, in a search for blue materials for organic electroluminescent devices.13 However, not all non-planar pyrene derivatives designed by the above strategies could completely suppress the tendency of the emission peak to undergo a red-shift in the PL/ECL process. The emission behaviors of these molecules were inevitably influenced by the attached fluorophores. For instance, Sun’s group designed and synthesized two twisted pyrene derivatives, 1,10-dipyrene and 1,4-dipyrenyl benzene, which prevented the close packing/luminescence quenching effect in pyrene-type materials, and suppressed the problematic recrystallization.14 However, due to the large conjugated system, the emission peaks were red-shifted and exhibited greenish-blue emissions. Bard and co-workers carried out photophysical, electrochemical and ECL characterization of bowl-shaped pyrene derivatives, pyrenophanes, which incorporate non-six membered rings as bridges.15 The research showed that blue monomer emission was evident in the fluorescence spectra. The ECL spectra of all the pyrenophanes showed intensive “excimer-like” emission in addition to the monomer emission observed at shorter wavelengths, which was explained as excimers could be easily formed in the electron-transfer reaction without an unexcited molecule. Therefore, we proposed that tailor-made twisted configurations of pyrene derivatives could impart steric hindrance and prevent the formation of crystals and excimers. Furthermore, we wondered if a small conjugated system could effectively decrease the effect of π-stacking interactions and suppress the formation of excimers. Overall, we came up with an idea to utilize pyrene derivatives with a simple twisted configuration to develop highly efficient blue luminophores.
The aim of this work is to study the electrochemical, photophysical and ECL properties of a pyrene derivative with a simple non-planar configuration, especially its steric hindrance, which prevents the formation of an excimer, that exhibits efficient blue monomer ECL emission in non-aqueous media. Herein, we report the properties of 1-methyl benzoate-pyrene (MB-Py) in mixed acetonitrile:benzene (v/v = 1:1) solvent for the first time. The relationships between the electrochemical and ECL properties and the molecule structures were investigated together with those of the parent pyrene (Py) and analogue benzo[α]pyrene (Bz-Py). The electrochemical and ECL mechanisms were also discussed. This study will provide insight for the future design of pyrene derivatives as blue light luminophores in ECL.
Experimental
Materials
Benzo[α]pyrene (Bz-Py), pyrene (Py), 9,10-diphenylanthracene (DPA), benzoyl peroxide (BPO), tetrabutylammonium hexafluorophosphate (TBAPF6), anhydrous acetonitrile (MeCN, 99.8%), and anhydrous benzene (Bz, 99.8%) were purchased from Sigma-Aldrich Co., Ltd (St. Louis, MO, USA). Ferrocene (98%) was obtained from Aladdin Chemical Co., Ltd. (Shanghai, China). 1-Methyl benzoate-pyrene (MB-Py) was synthesized through cross-coupling reactions, which have been described previously.16 The chemical structures of MB-Py, Bz-Py and Py are depicted in Fig. 1. All solvents were transferred directly into an argon atmosphere glove box (Mikrouna, China) and the solutions were prepared in the glove box. An airtight vessel was used for the electrochemical and ECL measurements outside of the glove box. Because of the poor solubility of pyrene derivatives in MeCN and the insolubility of TPAPF6 in pure benzene, an MeCN:Bz mixture (v/v = 1:1) was used as the solvent to obtain moderate polarity and an appropriate potential window, unless specifically stated.
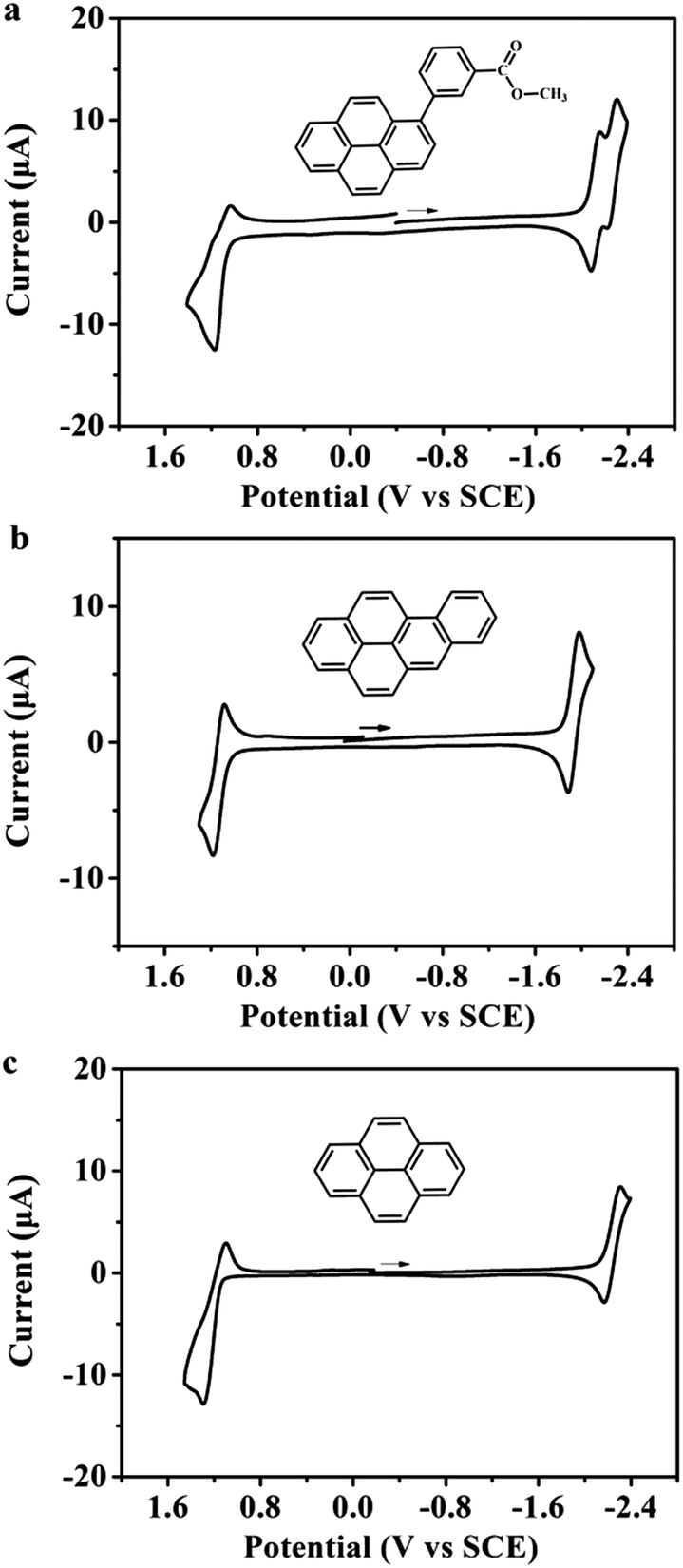 | ||
| Fig. 1 Cyclic voltammograms of (a) 1-methyl benzoate-pyrene (MB-Py, 1 mM), (b) benzo[α]pyrene (Bz-Py, 1 mM) and (c) pyrene (Py, 1 mM) in MeCN:Bz (v/v = 1:1) containing 0.1 M TBAPF6. Scan rate: 0.1 V s−1; platinum electrode area: 0.027 cm2. | ||
Electrochemical experiments
All electrochemical experiments were performed using a three-electrode setup with a 2 mm diameter platinum disk working electrode, a platinum wire auxiliary electrode, and a silver wire quasi-reference electrode. The working electrode was polished before each measurement experiment with 0.3 μm alumina (Buehler, Ltd., Lake Bluff, IL) for several minutes, sonicated in water and then ethanol (each for 2 min), and dried in an oven at 100 °C. All the glassware was oven-dried for 1 h at 100 °C before transferring into an argon atmosphere glove box. All the solvents were prepared inside the glove box and sealed in a vacuum-tight electrochemical cell with a Teflon screw cap containing three metal rods for electrode connections.The electrochemical measurements were carried out with a CHI 440a electrochemical workstation (Chenhua Instruments Co., Shanghai, China). MeCN:Bz (v/v = 1:1) was used as the solvent and 0.1 M TBAPF6 as the supporting electrolyte for all electrochemical investigations. Cyclic voltammetry potentials were calibrated with ferrocene as a standard, taking E0 = 0.424 V vs. SCE.17 DigiSim 3.03 software (Bioanalytical Systems, Inc., West Lafayette, IN, USA) was used to simulate experimental cyclic voltammograms.
Photophysical experiments
Absorption spectra and photoluminescence (PL) spectra in a quartz cell (1.0 cm path length) were obtained under air-saturated conditions in MeCN:Bz (v/v = 1:1) solvent at room temperature on a UV-vis spectrophotometer (UV-2450, Shimadzu Corporation, Japan) and a Cary Eclipse fluorescence spectrophotometer (Varian, USA), respectively. The relative photoluminescence efficiencies of the compounds were calculated using DPA as a standard (λex = 431 nm, ΦPL = 0.804 in MeCN:Bz (v/v = 2:3)).18 PL photographs were taken using a Canon 770D EOS (MP-E 65 mm Len) camera. Images were used directly from the camera.
ECL experiments
The ECL intensity–CV and ECL intensity–time simultaneous measurements were recorded on an MPI-E ECL detector (Remax Electronics, Xi’an, China). The relative ECL efficiencies were calculated with respect to DPA as a standard (λex = 430 nm, ΦECL = 0.0108 in MeCN:Bz (v/v = 2:3)).18 The ECL spectra were generated by annihilation by pulsing the potential with a pulse width of 1 s from about 80 mV past the peak potentials, or by stepping from 0 to 80 mV from the reduction peak with a step time of 1 s using benzoyl peroxide as a coreactant. The ECL spectra were generated using a CHI 440a and were recorded on a Princeton Instruments PyLon 100BX CCD camera (Trenton, USA) coupled with an Acton SP2300 monochromator. ECL images were obtained in a darkroom using the integrated system including an Olympus IX-51 inverted microscope (Olympus Corporation, Tokyo, Japan), an Olympus DP80 camera (Olympus Corporation, Tokyo, Japan) and a CHI 660a electrochemical workstation.
Computations
Geometry optimizations, frequency calculations, and molecular orbital calculations were performed in Gaussian 09 using the B3LYP/6-31G(d,p) basis set.19 Only positive frequencies were found for the optimized structures. Molecular orbitals were visualized using Gaussview 5.0 software. All calculations were performed in the gas phase. Structures and electronic properties were estimated by density functional theory (DFT) calculations at the B3LYP/6-31G level of theory due to the relatively simple structure of pyrene derivatives of this type.Results and discussion
Electrochemical properties
Cyclic voltammetry was used to obtain information about the energetic formation and stability of the radical ions generated from the studied organic compounds. Fig. 1 shows the cyclic voltammogram of MB-Py together with those of Bz-Py and Py in MeCN:Bz (v/v = 1:1) containing 0.1 M TBAPF6. Details of the electrochemical data are summarized in Table 1. MB-Py at a scan rate of 0.1 V s−1 exhibits a quasi-reversible one-electron oxidation wave with E1/2 = +1.11 V vs. SCE and two reversible closely spaced consecutive one-electron reduction waves with E1/2,1 = −2.12 V and E1/2,2 = −2.27 V vs. SCE. Bz-Py exhibits a reversible one-electron oxidation wave with E1/2 = +1.13 V vs. SCE and a reversible one-electron reduction wave with E1/2 = −1.94 V vs. SCE, while Py displays a quasi-reversible one-electron oxidation wave with E1/2 = +1.18 V vs. SCE and a quasi-reversible one-electron reduction wave with E1/2 = −2.22 V vs. SCE. These results are consistent with the previous reports for Py15 and Bz-Py.20 Compared with Py, MB-Py shows a negative shift (0.07 V) of the first oxidation potential and a positive shift (0.10 V) of the first reduction potential, indicating that MB-Py is more easily oxidized and reduced than Py. In addition, the second reduction of MB-Py at E1/2,2 = −2.27 V vs. SCE is reversible even with a high scan rate (see scan rate studies in Fig. S-1†). These shift potentials and the reversibility of the reduction process indicated that compared with Py, MB-Py could generate the radical ions and doubly charged moieties more easily, and the generated species from MB-Py are more stable. These findings can be attributed to the fact that MB-Py has an enhanced conjugation effect and electronic interactions, causing the delocalization of charges throughout the parent pyrene core and methyl benzoate substituent. DFT computation details are discussed below. Compared with Bz-Py, MB-Py shows a negligible negative shift (0.02 V) of the first oxidation potential but an obvious negative shift (0.18 V) of the first reduction potential. These observations indicate that the reduction of MB-Py is harder than that of Bz-Py, and are attributed to the electron-donating effect of the ester group linked with the benzene ring in MB-Py. Moreover, the perturbation of the reduction potential after capping with methyl benzoate is more pronounced than that of the oxidation potential in the anodic region, implying that more remarkable electronic interactions occur when MB-Py is reduced. The quasi-reversibility of the oxidation in MB-Py can be attributed to a strong contribution from the C–O σ-bond orbitals.21 Overall, it is reasonable to anticipate that the new molecule, MB-Py, should provide efficient ECL with higher stability due to the formation of the oppositely charged radicals, which subsequently annihilate to form the excited states.
:Bz (v/v = 1:1)
| E1/2(A2+/A)a (V) | E1/2(A+/A)a (V) | E1/2(A/A−)a (V) | E1/2(A/A2−)a (V) | 106D (cm2 s−1) | −ΔGannb (eV) | −ΔHannb (eV) | Egc (eV) | |
|---|---|---|---|---|---|---|---|---|
| a E1/2 values are obtained either by digital simulation or averaging the redox peak potentials and are reported versus SCE.b −ΔGann = Epa,ox − Epc,red, −ΔHann = −ΔGann − 0.1.c Eg = Epa,ox − Epc,red. | ||||||||
| MB-Py | +1.72 | +1.11 | −2.12 | −2.27 | 13.6 | 3.23 | 3.13 | 3.23 |
| Bz-Py | +1.59 | +1.13 | −1.94 | — | 9.80 | 3.07 | 2.97 | 3.07 |
| Py | — | +1.18 | −2.22 | — | — | 3.40 | 3.30 | 3.40 |
The CV scans at more extreme potentials can provide information about the energies needed to form the divalent or trivalent radical ions.22 The cyclic voltammograms of MB-Py and Bz-Py at more positive potentials (Fig. S-2†) showed that both the derivatives produced a second irreversible oxidation wave at +1.72 V and +1.59 V vs. SCE, respectively, which are frequently observed for the dications.23 This indicates that it is more difficult for MB-Py to lose the second electron to form the dication than it is for Bz-Py. The irreversible oxidation waves indicate that the dications of these two pyrene derivatives are generally unstable, and can be attributed to a subsequent homogeneous reaction. In the cathodic region, the more negative waves of these two derivatives could not be detected within the potential window of the solvent used in this experiment (data not shown). Overall, the introduction of methyl benzoate to the pyrene core not only stabilizes the radical ions, but also blocks the active positions subject to decomposition, giving rise to extra stabilization on the highly charged species (dianion). As discussed below, this important observation is consistent with the theoretically determined structures of the compounds.
Density functional theory (DFT) calculations for MB-Py and Bz-Py were done in computations at the B3LYP/6-31G(d,p) level of theory as described. The calculation results of the HOMO and LUMO energies and the HOMO–LUMO gaps for MB-Py and Bz-Py are sketched in Fig. 2. The orbital diagrams of MB-Py show that the HOMO is mainly delocalized to the pyrene core and the LUMO is localized on the pyrene core and the methyl benzoate substituent. This indicates that the oxidation of MB-Py mostly occurs on pyrene and the reduction occurs on the whole molecule. Importantly, the calculations revealed that there is a twisted conformation between the methyl benzoate moiety and the central pyrene core with a dihedral angle of 56°, indicating that the geometric configuration of MB-Py is nonplanar.24 A small amount of charge delocalization between the pyrene core and methyl benzoate group is observed on the HOMO of MB-Py. This indicates that the HOMO is affected by the methyl benzoate substituent group, and also provides a reasonable explanation for the quasi-reversibility of the oxidation in MB-Py. The HOMO energies of MB-Py and Bz-Py were estimated at −5.34 eV and −5.11 eV, while the LUMO energies of MB-Py and Bz-Py were estimated at −1.66 eV and −1.75 eV, respectively. The HOMO energy of MB-Py (−5.34 eV) is lower (by 0.23 eV) than that of Bz-Py (−5.11 eV), while the LUMO energy of MB-Py (−1.66 eV) is higher (by 0.09 eV) than that of Bz-Py (−1.75 eV). These calculated data suggest that MB-Py is more easily oxidized and more difficult to reduce than Bz-Py, which is consistent with the potential shifts of MB-Py relative to Bz-Py described above. The HOMO–LUMO energy gap of MB-Py (3.68 eV) is larger than that of Bz-Py (3.36 eV), owing to the p-orbital formed by the lone-pair electrons of the O atom in the ester group, which decreases the electron density and recedes the π conjugation effect of the molecule.25 Additionally, the HOMO−1 energies of MB-Py and Bz-Py were estimated at −6.07 eV and −6.27 eV, while the LUMO+1 energies of MB-Py and Bz-Py were estimated at −1.26 eV and −0.85 eV, respectively. The second oxidation of MB-Py occurred at a more positive potential than that of Bz-Py, which is consistent with the DFT data of HOMO−1. The slight difference between the energies of the LUMO and LUMO+1 is consistent with two closely spaced consecutive one-electron transfer reductions of MB-Py. As can be seen, the LUMO+1 of MB-Py is localized on the methyl benzoate substituent and is much lower (0.41 eV) than that of Bz-Py. In this circumstance, methyl benzoate serves as an electron donor and attaches to the electron-deficient pyrene core as an electron acceptor. In the electrochemical reaction, charges are transferred from the donor to the directly attached acceptor with some intramolecular rearrangement.26 This profound electron effect is responsible for the easier removal of a second electron from MB-Py, which requires a lower energy relative to Bz-Py. In contrast to MB-Py, the HOMO energies (HOMO, HOMO−1) and LUMO (LUMO, LUMO+1) energies of Bz-Py are all localized on the whole planar molecule. On account of the strong coulombic repulsive forces in the whole rigid structure,27 we deduced that the second reduction wave of Bz-Py possibly shifted to a more negative potential which could not be detected within the present potential window of the solvent used in this experiment.
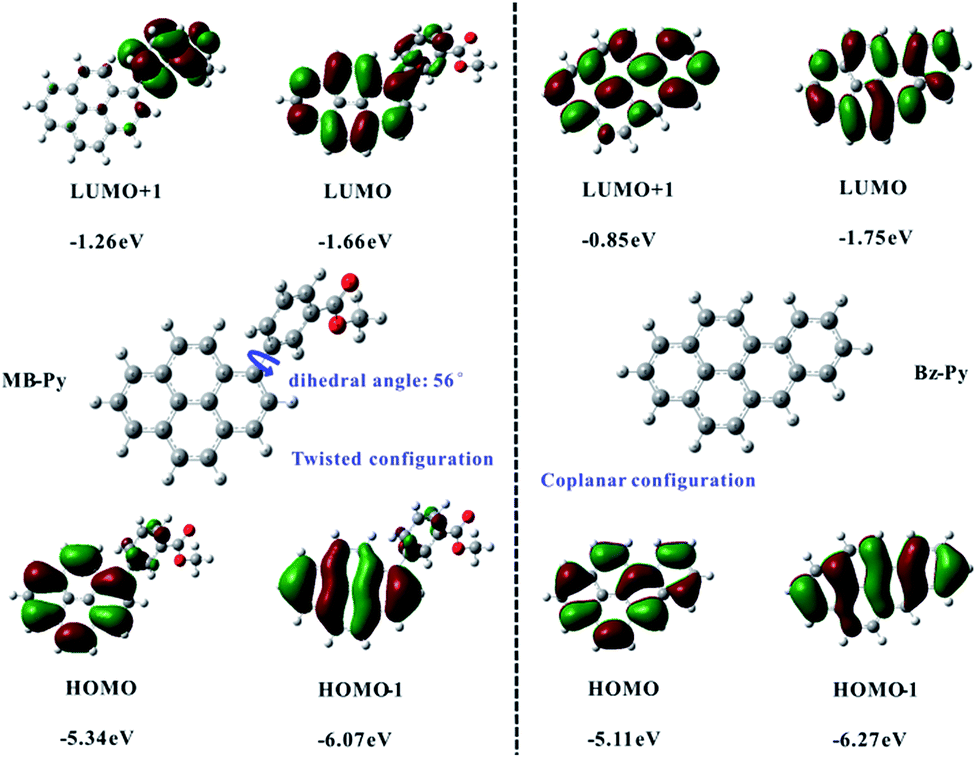 | ||
| Fig. 2 The ball-stick structures and frontier molecular orbitals for MB-Py and Bz-Py calculated by DFT (B3LYP/6-31G(d,p)). | ||
The cyclic voltammograms of MB-Py and Bz-Py obtained at different scan rates showed that the peak currents of the reduction and oxidation wave changed linearly with the square root of the scan rate (v1/2, Fig. S-1†) for both the reduction (ip,r) and the oxidation (ip,o), indicating diffusion control for both oxidation and reduction processes. The diffusion coefficients, D, were found to be 1.36 × 10−5 cm2 s−1 for MB-Py and 9.80 × 10−6 cm2 s−1 for Bz-Py, by plotting the peak current versus v1/2, and are listed in Table 1. The electron-donating effect of the ester group in the MB-Py molecule might be the reason for the relatively larger diffusion coefficient of the electrochemical processes than that of Bz-Py.28 Additionally, the peak current ratio (ipa/ipc) was approximately beyond unity down to a scan rate of 50 mV s−1, indicating that a subsequent chemical reaction followed the electrochemical reaction. The cyclic voltammogram (CV) of MB-Py obtained at a Pt ultramicroelectrode (UME) (Fig. S-3†) showed one oxidation wave and two reduction waves, consistent with the CV results at the macroelectrode. The value of the steady-state current for the reduction is twice the value of that for the oxidation wave, indicating that the number of electron transfers for reduction is twice the number of electron transfers for oxidation.
In order to obtain better insight into the mechanism and kinetics of both anodic and cathodic processes, the experimental CVs of MB-Py and Bz-Py were digitally simulated with different scan rates using DigiSim 3.03. The oxidation wave for MB-Py could be fit to an EC mechanism (Fig. S-4†), a chemical reaction following an electrochemical electron transfer, with a heterogeneous rate constant k0 = 1.0 × 104 cm s−1 and a homogeneous forward rate constant kf = 20 s−1, keq = 0.8. An ECE mechanism with two Nernstian reduction waves (Fig. S-5†) was used in the digital simulation for the reduction of MB-Py, with a heterogeneous rate constant, k0, of approximately k01 = 0.012 cm s−1, k02 = 0.015 cm s−1. Both the cathodic and anodic waves of Bz-Py were characterized by an EC mechanism with a heterogeneous rate constant, k0 = 1.0 × 104 cm s−1 with coupled homogeneous chemical reactions, kf = 18 s−1, keq = 0.8 (Fig. S-6 and S-7†). The CV simulations for MB-Py and Bz-Py are consistent with the experimental data of scan rates from 0.05 V s−1 to 2 V s−1. The occurrence of the chemical reaction after the electrochemical electron transfer process could explain the disappearance or consumption of the electrogenerated product. A clear explanation for the electrogenerated product is still a challenge. The slight differences between the simulated and experimental CV curves may be attributed to the weak adsorption of the pyrene derivatives to the surface of the working electrode.
Photophysical properties
We studied the absorption and PL properties of MB-Py together with those of Bz-Py and Py in MeCN:Bz (v/v = 1:1). The basic photophysical data obtained are listed in Table 2. Fig. 3 shows the normalized UV-vis absorption spectra and PL spectra of MB-Py with those of Bz-Py and Py in MeCN:Bz (v/v = 1:1) under air-saturated conditions. In the normalized UV-vis absorption spectrum, the absorption wavelength maximum (λmax) of MB-Py appears at 343 nm with an extinction coefficient of 2.39 × 104 L mol−1 cm−1, and is dominated by a strong π–π* transition.29 Bz-Py shows four absorption bands with the absorption wavelength maximum at 387 nm and Py shows three absorption bands with the absorption wavelength maximum at 336 nm in the scan range. Since both are unsaturated aromatic compounds, the defined vibrational modes generated from π–π* transitions are clearly observed. Owing to the extended π-conjugation, the absorption of Bz-Py shows an evident shift (51 nm relative to Py, 44 nm relative to MB-Py) to the lowest energy in comparison to that of Py and MB-Py, which is attributed to it having the largest π-conjugated system of the three compounds. As a result of the attachment of the methyl benzoate substituent to the parent pyrene, the defined vibrational mode of MB-Py disappears. The absorption spectrum of the new compound, MB-Py, shows a slight shift (7 nm) to a lower energy as compared to the parent pyrene, which is attributed to the moderate contribution of p–π conjugation in the attached chromophores, and agrees with our observations in electrochemical studies.30
:Bz (v/v = 1:1)
| λabs (nm) | 104ε (M−1 cm−1) | λem (nm) | Stokes shift | ΦPLa | Esb (eV) | λECL (nm) | ΦECLc | λECL/BPO (nm) | |
|---|---|---|---|---|---|---|---|---|---|
| a Using DPA as the standard, for which ΦPL = 0.804 in MeCN:Bz (v/v = 2:3).18b Es: approximate energy of the first singlet excited state taken as the photoluminescence wavelength maximum; Es (eV) = 1239.81/λem (nm).c ECL efficiencies were determined using DPA as the standard, for which ΦECL = 0.0108 in MeCN:Bz (v/v = 2:3).18d ECL efficiency of pyrene is quoted from a previous report (λex = 334 nm, in Bz).31 |
|||||||||
| MB-Py | 343 | 2.39 | 428 | 85 | 0.96 | 2.90 | 439 | 0.26 | 446 |
| Bz-Py | 349 | 0.83 | 407 | 433 | 435 | ||||
| 367 | 1.67 | 430 | 20 | 0.72 | 3.05 | 498 | 0.18 | 499 | |
| 387 | 1.95 | 457 | |||||||
| Py | 306 | 0.76 | 374 | 426 | |||||
| 320 | 1.84 | 393 | 38 | 0.60d | 3.32 | 475 | — | — | |
| 336 | 2.96 | 412 | |||||||
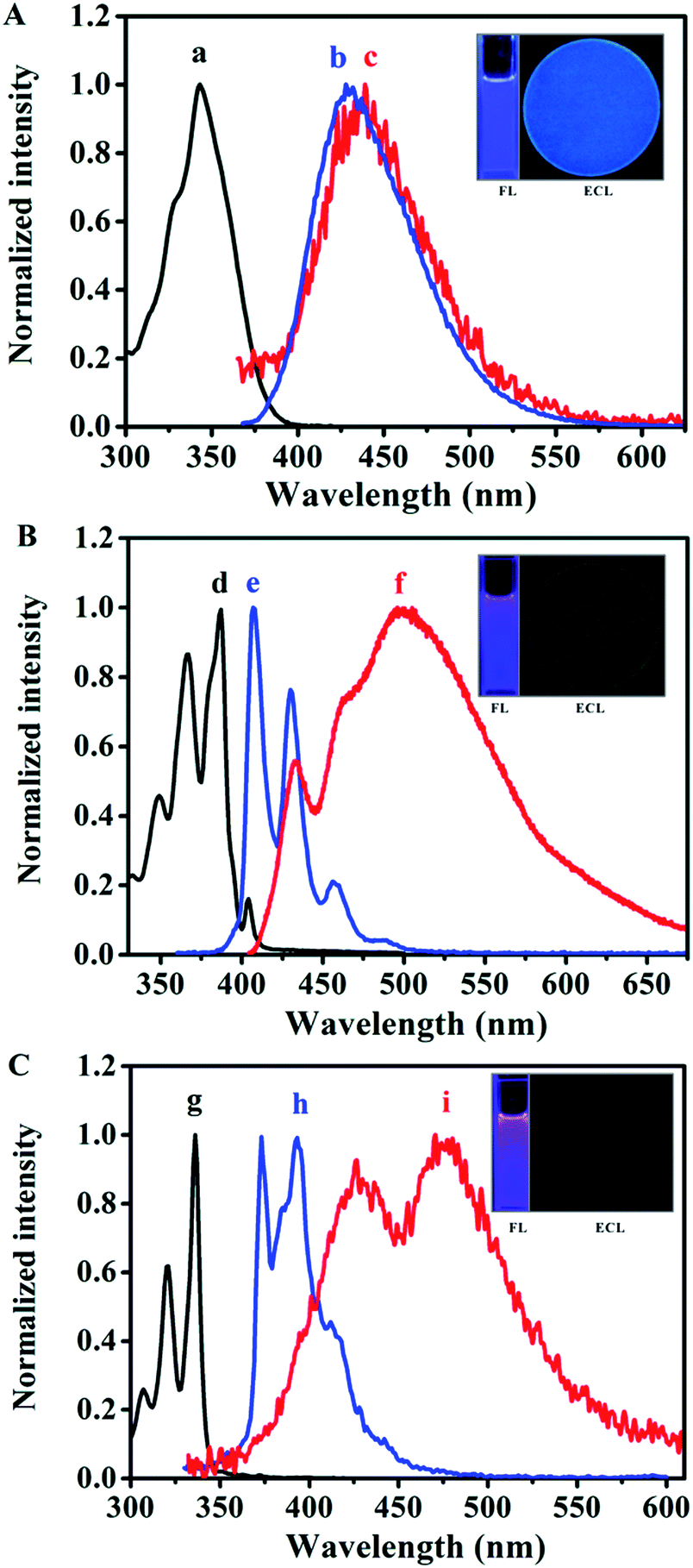 | ||
| Fig. 3 Normalized absorbance (black line), PL (blue line) and ECL (red line) spectra of (A) MB-Py, (B) Bz-Py and (C) Py in MeCN:Bz (v/v = 1:1). PL spectra were excited at the absorption maximum. Normalized ECL spectra were generated through ion radical annihilation by pulsing the potential with a pulse width of 1 s from approximately 80 mV past the peak potentials, slit width: 20 nm. Inset photographs: PL and annihilation ECL emission images. The concentrations of the luminophores were 10 μM for absorbance and PL, and 1 mM for annihilation ECL. | ||
As can be seen from the insets of Fig. 3, Py and Bz-Py show blue-violet PL emissions at the concentration of 10 μM in MeCN:Bz (v/v = 1:1). Specifically, Py exhibits two evident vibrational structure emissions around 374 and 393 nm in addition to a smaller emission band with the observed peak of 412 nm. Moreover, broad tailing above 430 nm can be observed, which is ascribed to the excimer of pyrene formed in MeCN:Bz (v/v = 1:1).12 It is clear that the maximum emission wavelength of Bz-Py is red-shifted (by 33 nm) from Py, and is characterized by three defined vibrational structures around 407, 430, and 457 nm, which are similar to the vibrational structures observed in the PL spectrum of pyrene. The bump in the spectral line of Bz-Py around 480 nm is attributed to the Bz-Py excimer emission, which was confirmed in a previous report.32 Importantly, unlike Py or Bz-Py, MB-Py shows a single-peaked structureless, broad, pure blue emission centered around 428 nm under the same experimental conditions. It is attributed to the increased conjugation system and the effect of the methyl benzoate chromophore of MB-Py, which shifts the maximum emission wavelength to the lowest energy band. The red-shifted PL without excimer emission of MB-Py indicates that the introduction of the methyl benzoate substituent not only increases the conjugation, but also imparts steric hindrance that prevents excimer formation. On the basis of the DFT computations for MB-Py (Fig. 2), the severe distortion in this new compound probably minimizes intermolecular interactions and, therefore, impedes excimer formation within the lifetime of the molecular excited state.15 A relatively large Stokes shift of 85 nm was observed between the absorption and emission maxima of MB-Py, whereas smaller Stokes shifts of 38 nm and 20 nm were observed in Py and Bz-Py, respectively. The largest Stokes shift is observed in the twisted configuration of MB-Py, probably owing to the changes in the molecular geometrical structure of the compound upon excitation. Since MB-Py could be considered as a molecule with donor and acceptor constituents (see DFT computations above), the increase in the Stokes shift could be due to the intramolecular charge transfer in the excited state for the coupling of donor and acceptor.33 In addition, the relative PL efficiencies were calculated to be 0.96 for MB-Py and 0.72 for Bz-Py, using diphenylanthracene (DPA) as a standard (λex = 431 nm, ΦPL = 0.804 in MeCN:Bz (v/v = 2:3)).18 As is expected, the π-conjugation extent of the rigid features in Bz-Py is larger than that in MB-Py, which is beneficial for generating more electronic excited state molecules and contributes to a higher PL efficiency. However, the PL efficiency of MB-Py is superior to that of Bz-Py, which may be attributed to the dimerization of coplanar Bz-Py and the electron-donating effect of the methyl benzoate substituent in MB-Py.
To further explore the differences in PL emission behaviors between MB-Py, the parent Py and analogue Bz-Py, we investigated the PL spectra of these three compounds in different concentrations (1 × 10−5 M, 5 × 10−5 M, 1 × 10−4 M and 1 × 10−3 M) in MeCN:Bz (v/v = 1:1). (Fig. S-8†) As can be seen in Fig. S-8A,† the emission wavelength maxima of MB-Py are all centered on 428 nm, and no obvious peak-shape changes occur with the increase of MB-Py concentration. This indicates that MB-Py only emits monomer photoluminescence with blue color and no excimer appears even at high concentrations. In contrast, the spectra of both Bz-Py and Py change obviously with the increase in concentration. Briefly, with the increase in Py and Bz-Py concentration in the range from 1 × 10−5 M to 1 × 10−3 M, the intensity of the photoluminescence emission at the S1–S0 band (the emission maximum) gradually decreases until it disappears, while a green photoluminescence emission around 500 nm appears and its intensity gradually increases (Fig. S-8B and C†). This indicates that the emission at the lower energy band is attributed to the excimer formation and the excimer emission gradually increases along with the increase in concentration.
The absorbance and PL spectra of 10 μM MB-Py, Bz-Py and Py in different ratios of benzene/MeCN solvents (Bz/MeCN 1:0, 3:1, 0:1) are shown in Fig. S-9.† As can be seen, both the absorption and PL emission intensities of all three luminophores decrease along with the increase in solvent polarity, while there is no evident wavelength change in the absorption spectra in different ratios of the above-mentioned solvents. This is attributed to the fact that no interactions in the molecules occur in the ground state.34 Meanwhile, no solvatochromism effect was observed for Bz-Py and Py, which is attributed to the planar configurations of the two analogues. However, the excimer emissions of Bz-Py and Py were observed in all the different ratios of solvents and increased with the decrease in solvent polarity. The PL emission of MB-Py was slightly red-shifted along with the increase in solvent polarity. Since the twisted MB-Py could be considered as a molecule with donor and acceptor constituents (see DFT computations), the positive solvatochromism effect is attributed to the typical twisted intramolecular charge transfer (TICT) which occurs in donor–acceptor PAHs.35 Although the PL emission of MB-Py was slightly shifted due to TICT, the maximum of the wavelength was still observed around 430 nm with blue-light emission and no excimer emissions were observed.
Electrogenerated chemiluminescence properties
The ECL properties of MB-Py along with those of Py and Bz-Py were investigated in a radical ion annihilation process and coreactant process, respectively. Fig. 4 shows the ECL intensity–potential profile of MB-Py in MeCN:Bz (v/v = 1:1) containing 0.1 M TBAPF6. For MB-Py, a moderately intense ECL emission (I = 2746) in the cathodic region during dynamic forward potential scanning (Fig. 4a) and a more intense ECL emission (I = 3975) in the anodic region during dynamic backward potential scanning are observed (Fig. 4c). Under the same experimental conditions, Bz-Py shows a weak ECL emission (I = 727) in the cathodic region during dynamic forward potential scanning (Fig. 4b) and a slightly more intense ECL emission (I = 1463) in the anodic region during dynamic backward potential scanning (Fig. 4d). These observations are attributed to the fact that both the radical cations and radical anions generated from MB-Py are much more stable than those from Bz-Py.
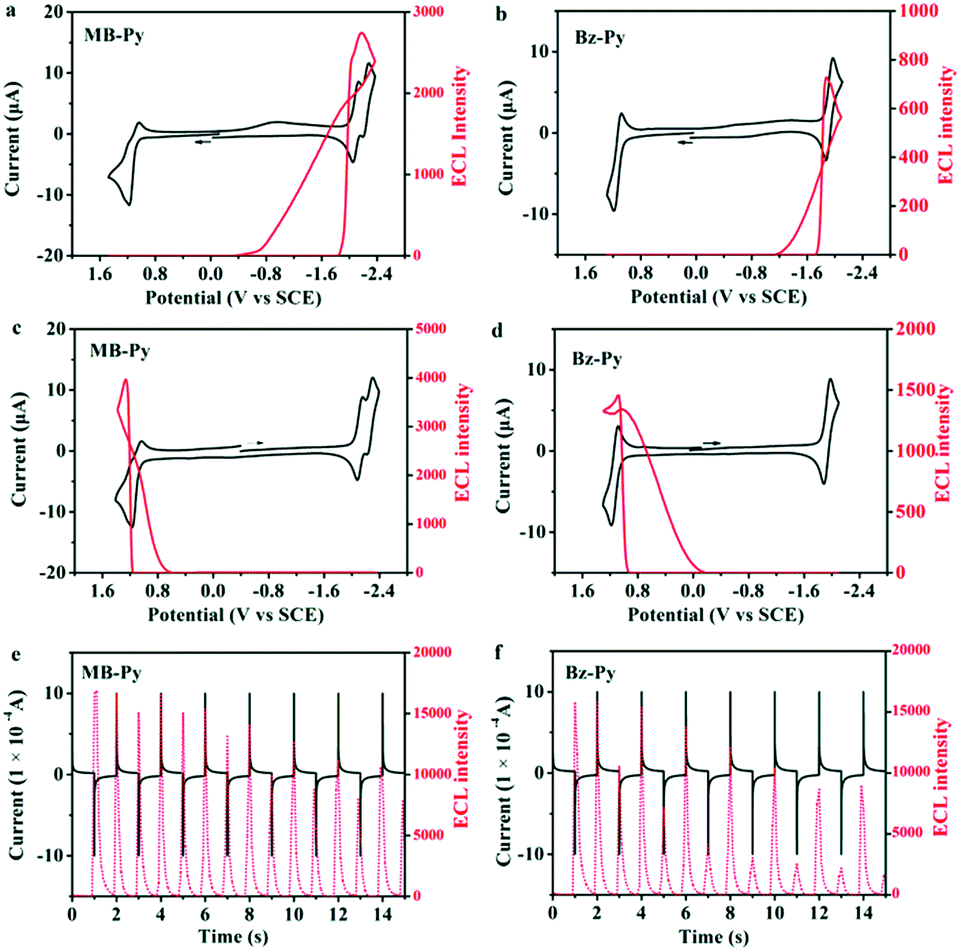 | ||
| Fig. 4 ECL (red) and CV (black) simultaneous measurements for 1 mM (a) MB-Py and (b) Bz-Py during dynamic forward potential scanning and 1 mM (c) MB-Py and (d) Bz-Py during dynamic backward potential scanning with a scan rate of 0.1 V s−1; initial current (black) and ECL light (red) transients for 1 mM (e) MB-Py and (f) Bz-Py pulsed between 80 mV past the first reduction peak and 80 mV past the first oxidation potential, respectively. Pulse width is 1 s. Experimental conditions, solvent: MeCN:Bz (v/v = 1:1), supporting electrolyte: 0.1 M TBAPF6, negative high-voltage: 600 V, Pt electrode area: 0.027 cm2. | ||
To further estimate the stability of the radical ion species and calculate the ECL efficiencies, potential pulsing ECL transients were collected. When the potential was stepped from the reduction wave at Epc −80 mV to the oxidation wave at Epa +80 mV for MB-Py and Bz-Py in this study, quick emissions and different ECL intensities were observed for the cathodic and anodic pulses. Radical anion–cation annihilations of both MB-Py and Bz-Py produced by potential steps showed asymmetric ECL transients in which the cathodic pulses were always smaller than the anodic pulses (Fig. 4e and f). This kind of ECL behavior suggests that the reduced form is stable while the oxidized form is unstable and is depleted in the solution when the anodic pulse occurs. Only a fraction of the oxidized form is available for ECL, even though the current for its oxidation on a subsequent pulse is essentially the same. As shown in Fig. 4e, the transient ECL of MB-Py retains a relatively constant intensity for the cathodic pulse while the ECL emission of Bz-Py at the cathodic pulse is too weak and significantly decreases (Fig. 4f). This indicates that the radical cations of MB-Py exhibit relatively superior stability to those of Bz-Py, and can generate more constant ECL emission in radical ion processes. The relative ECL efficiencies of MB-Py and Bz-Py were determined by comparing the number of photons emitted per electron with that for the standard ECL luminophore, DPA (ΦECL = 0.0108 in MeCN:Bz (v/v = 2:3)18), based on the data from the annihilation and chronoamperometric experiments (Fig. 4e and f). The relative ECL efficiencies in the radical ion annihilation were calculated to be 0.26 for MB-Py and 0.18 for Bz-Py. The higher ΦECL of MB-Py is attributed to the more stable radical cations produced in the annihilation process. This indicates that the change of the configuration also facilitates the charge delocalization, permitting the formation of stable radical ions. Owing to its long-term stability upon pulse cycling, which has always been desirable for ECL applications,36,37 this new pyrene derivative, MB-Py, can be utilized as an efficient luminescent material.
As shown in Fig. 3A(c) and the inset ECL image, in the radical ion annihilation process MB-Py exhibits intense blue light ECL emission with a maximum at the wavelength of 439 nm in MeCN:Bz (v/v = 1:1), and the ECL spectrum of MB-Py is nearly the same as its PL spectrum. This indicates that the emission pathways of PL and ECL both originate from the monomer without an excimer emission process. This is attributed to the twisted configuration of MB-Py, which increases steric hindrance that prevents π-stacking for excimer formation during the electron transfer reaction, leading to high FL and ECL efficiencies. A slight red-shift in the ECL spectrum was observed as compared to the PL spectrum. This is probably due to an inner filter effect as a result of the high concentrations of the compound needed to obtain measurable ECL, as well as the difference in resolution between the two instruments with which the PL and ECL spectra were measured.38,39 As shown in Fig. 3B(f) and C(i), the ECL spectra of both Bz-Py and Py in Bz:MeCN (v/v = 1:1) can be divided into two main components: the emission maximum peaks at around 498 nm and 475 nm, and the smaller peaks at 433 nm and 426 nm, respectively. The two peaks with broad shapes in the ECL spectra of Bz-Py and Py correspond to the simultaneous monomer and excimer emissions, which are attributed to the fact that these two strongly π-conjugated planar molecules favor the formation of a large amount of the excimer in the excited state.7 As observed in Fig. 3 and S-8,† more excimer emissions of Bz-Py and Py are evident in the ECL spectra when compared to the PL spectra obtained at the same concentration. This is attributed to the fact that the ECL excimer emission is generally much more intense than the PL excimer emission at the same concentration, due to the different modes of excimer formation.15 Briefly, the excited molecule requires an encounter with an unexcited molecule to generate an excimer in photoluminescence spectroscopy, while the excimers could be formed directly in the electron transfer reaction by the annihilation or coreactant process in ECL. Additionally, these ECL emission peaks were red-shifted from the PL emission wavelengths of both the monomer and excimer and most likely represent the emission from byproducts of annihilation, owing to the instability of the cation.40 Overall, on account of the red-shift of the spectra and the intense excimer ECL emissions for Py and Bz-Py, the blue monomer emissions are covered up and these two molecules mainly show cyan, or even green, excimer emissions, and these compounds are obviously not appropriate to be used as blue-emitting ECL materials (Fig. 3B and C, ECL images).
In order to clarify the ECL route in radical ion annihilation, the energy of the excited singlet state was compared with the energy of the annihilation reaction. The energy of the annihilation reaction, −ΔHann (eV), was estimated by the equation −ΔHann = E1/2(A+/A) − E1/2(A/A−) − 0.1 eV. The values for MB-Py, Bz-Py and Py are listed in Table 1. The energy of the excited singlet state Es (eV) was estimated by the equation Es (eV) = 1239.81/λem (nm); the values for the three compounds are listed in Table 2. The value of −ΔHann is 3.13 eV for MB-Py, 2.97 eV for Bz-Py, and 3.30 eV for Py; Es is 2.90 for MB-Py, 3.05 for Bz-Py, and 3.32 eV for Py. For MB-Py, −ΔHann is larger than Es, which indicates that the electron-transfer process is energy-sufficient and the direct production of 1MB-Py* is possible – that is, the ECL route is the S-route.41 However, for Bz-Py and Py, −ΔHann is smaller than Es, which further indicates that these two planar molecules have significant π-orbital overlap and ion annihilation reactions lead to the formation of excimers. The ECL routes for Bz-Py and Py are said to follow the E-route.41
Also, we investigated the ECL properties of MB-Py and Bz-Py, separately, in coreactant processes. BPO was chosen as a coreactant since it forms a strong oxidizing agent (+1.5 V vs. SCE) after being reduced.42 This strong oxidizing agent can react directly with the electrogenerated radical anion, generating light. As shown in Fig. 5A, a strong ECL signal (I = 9166) was observed upon the reduction of MB-Py in the presence of the coreactant BPO. Under these conditions, an ECL spectrum (λECL/BPO = 446 nm) was obtained, as shown in Fig. 5B. This ECL spectrum is similar to that of MB-Py in the ion annihilation ECL process, indicating that the ECL emission of the coreactant process is also a monomer emission. We deduced that the byproducts generated upon BPO reduction contributed to the slight red-shift observed in the coreactant spectrum. In addition, a lower ECL signal (I = 8580) was observed upon the reduction of Bz-Py in the presence of the coreactant BPO, which is attributed to the lower ECL efficiency (Fig. S-10a†). Compared with the ECL spectrum of MB-Py in the same coreactant process, the ECL spectrum of Bz-Py showed a broader shape with two peaks (λECL/BPO = 435 nm, 499 nm) (Fig. S-10b†), which each correspond to the monomer and excimer emissions of Bz-Py. The red-shift and the peak broadening of the two peaks in the spectrum may be attributed to the byproducts generated in the reactions of the reduced BPO with the monomer and excimer, respectively. These results indicate that the ECL emitters in the coreactant process are the same as that in ion radical annihilation for both MB-Py and Bz-Py.
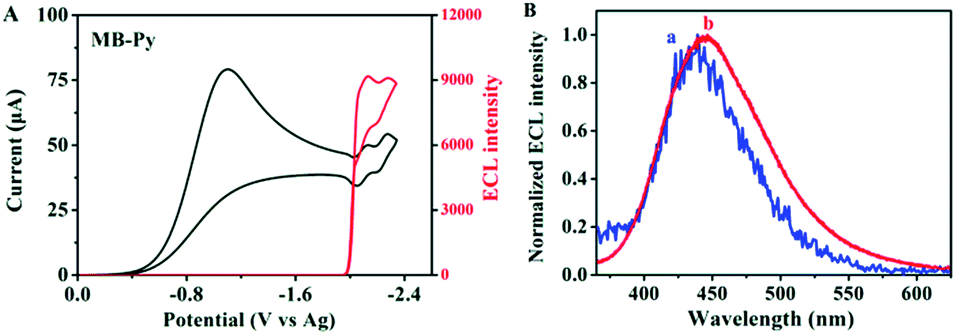 | ||
| Fig. 5 (A) ECL (red) and CV (black) simultaneous measurements of 1 mM MB-Py in the presence of 10 mM BPO with a scan rate of 0.1 V s−1; (B) ECL spectra for 1 mM MB-Py in the absence (blue, a) and presence (red, b) of 10 mM BPO; spectra were generated by pulsing the potential from 0 to −2.3 V vs. Ag in MeCN:Bz (v/v = 1:1) containing 0.1 M TBAPF6. Pulse width is 1 s, negative high-voltage: 600 V, Pt electrode area: 0.027 cm2. | ||
Compared with the ECL spectra of MB-Py, the ECL emission wavelength maxima of Bz-Py and Py were not only red-shifted to lower energy bands, but also showed intense excimer emissions, which resulted in a large amount of green ECL emissions. In contrast, the new pyrene derivative, MB-Py, only exhibited monomer ECL centered at about 440 nm with efficient pure blue ECL in both the annihilation and coreactant process. Alongside previous approaches for improving the PL and ECL properties of pyrene derivatives through capping pyrene with other sophisticated molecules,10 introducing a small substituent group linked to pyrene is a promising simple way to form a twisted configuration pyrene derivative that also exhibits excellent blue emission behavior.
Conclusions
In summary, a comparison study of the electrochemical, photophysical and ECL properties of a twisted pyrene derivative (MB-Py) with those of the parent Py and analogue Bz-Py was performed. The introduction of methyl benzoate evidently changes the molecular geometric configuration, leading to intramolecular electron rearrangement. This profound electron effect between the electron-deficient pyrene core and electron-donating methyl benzoate prompts the molecule to exhibit two closely spaced consecutive one-electron reductions. Owing to the rigid planar structures of Bz-Py and Py, these two compounds exhibited monomer and excimer blue-violet PL emissions and cyan/green ECL emissions. Unlike Bz-Py and Py, the twisted conformation of MB-Py imparts steric hindrance and prevents excimer formation, giving the molecule pure blue monomer PL emission and high PL efficiency. Moreover, the change in the configuration also facilitates charge delocalization, permitting the formation of stable radical ions, allowing MB-Py to show highly efficient blue monomer ECL spectra similar to the corresponding PL emission in both the ion annihilation process (assigned as the S-route) and coreactant process. In this work, we simply tailored the classic planar PAH molecular structure by introducing a small substituent group to tune the electronic and spectroscopic properties, effectively overcoming the excimer formation to obtain a material with a high PL efficiency and intense, pure blue ECL monomer emission. This study provides significant electrochemical and ECL information on pyrene derivatives as well as on structure–property relationships for future research. It will guide the future design of pyrene derivatives for various potential applications as ECL materials.Acknowledgements
We gratefully acknowledge the financial support from The National Science Foundation of China (No. 21475082, 21375084, 51373092).Notes and references
- L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275 RSC; C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- J. K. Leland and M. J. Powell, J. Electrochem. Soc., 1990, 137, 3127 CrossRef CAS; W. Miao, J. P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478 CrossRef PubMed.
- C. Tang, F. Liu, Y. J. Xia, L. H. Xie, A. Wei, S. B. Li, Q. L. Fan and W. Huang, J. Mater. Chem., 2006, 16, 4074 RSC.
- J. H. Huang, J. H. Su, X. Li, M. K. Lam, K. M. Fung, H. H. Fan, K. W. Cheah, C. H. Chen and H. Tian, J. Mater. Chem., 2011, 21, 2957 RSC.
- A. W. Knight and G. M. Greenway, Analyst, 1994, 119, 879 RSC; L. R. Faulkner and A. J. Bard, Electroanalytical Chemistry, Marcel Dekker, New York, 1977 Search PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley Interscience, London, 1970 Search PubMed.
- B. Fleet, G. F. Kirkbright and C. J. Pickford, J. Electroanal. Chem. Interfacial Electrochem., 1971, 30, 115 CrossRef CAS; A. Weller and K. Zachariasse, Chem. Phys. Lett., 1971, 10, 197 CrossRef.
- J. Shinar, Organic Light-Emitting Devices, Springer-Verlag, New York, 2003 Search PubMed.
- Y. J. Zhang, Y. X. Jin, R. Bai, Z. W. Yua, B. Hua, M. Ouyang, J. W. Sun, C. H. Yu, J. L. Liu and C. Zhang, J. Photochem. Photobiol., A, 2012, 227, 59 CrossRef CAS.
- M. Teresa, F. Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef PubMed.
- H. Hopf, Classics in hydrocarbon chemistry, Wiley-VCH, Weinheim, 2000 CrossRef CAS; G. J. Bodwell, J. J. Fleming and D. O. Miller, Tetrahedron, 2001, 57, 3577 CrossRef CAS.
- K. M. Omer, S. Y. Ku, K. T. Wong and A. J. Bard, Angew. Chem., Int. Ed., 2009, 48, 9300 CrossRef CAS PubMed.
- B. R. Kaafarani, A. O. El-Ballouli, R. Trattnig, A. Fonari, S. Sax, B. Wex, C. Risko, R. S. Khnayzer, S. Barlow, D. Patra, T. V. Timofeeva, E. J. W. List, J. Bredas and S. R. Marder, J. Mater. Chem. C, 2013, 1, 1638 RSC.
- C. H. Yang, T. F. Guo and I. W. Sun, J. Lumin., 2007, 124, 93 CrossRef CAS.
- R. Y. Lai, J. J. Fleming, B. L. Merner, R. J. Vermeij, G. J. Bodwell and A. J. Bard, J. Phys. Chem. A, 2004, 108, 376 CrossRef CAS.
- A. H. Sato, M. Maeda, S. Mihara and T. Iwasawa, Tetrahedron Lett., 2011, 52, 6284 CrossRef CAS.
- S. Rashidnadimi, T. H. Hung, K. T. Wong and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 634 CrossRef CAS PubMed.
- D. J. Vinyard, S. Su and M. M. Richter, J. Phys. Chem. A, 2008, 112, 8529 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. TIyengar, J. omasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- L. Jeftif and R. N. Adams, J. Am. Chem. Soc., 1970, 92, 1332 CrossRef.
- J. Jayabharathi, R. Sathishkumar, V. Thanikachalam and K. Jayamoorthy, J. Fluoresc., 2014, 24, 445 CrossRef CAS PubMed.
- H. L. Qi, J. J. Teesdale, R. C. Pupillo, J. Rosenthal and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 13558 CrossRef CAS PubMed.
- R. Y. Lai, E. F. Fabrizio, L. D. Lu, S. A. Jenekhe and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 9112 CrossRef CAS PubMed.
- H. Y. Chen and I. Chao, Chem. Phys. Lett., 2005, 401, 539 CrossRef CAS.
- R. J. Chen, F. Wu, L. Li, Y. B. Guan, X. P. Qiu, S. Chen, Y. J. Li and S. X. Wu, J. Power Sources, 2007, 172, 395 CrossRef CAS.
- A. L. Sobolewskia and W. Domcke, Chem. Phys. Lett., 1996, 259, 119 CrossRef.
- H. L. Qi, J. Chang, S. H. Abdelwahed, K. Thakur, R. Rathore and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 16265 CrossRef CAS PubMed.
- K. M. Kadish, M. M. Morrison, L. A. Constant, L. Dickens and D. G. Davis, J. Am. Chem. Soc., 1976, 98, 8387 CrossRef CAS PubMed.
- S. K. Lee, Y. B. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513 CrossRef CAS.
- S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816 CrossRef CAS PubMed.
- W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251 CrossRef CAS.
- J. R. Lakowicz and D. R. Bevan, Biochemistry, 1979, 18, 5170 CrossRef CAS PubMed.
- R. R. Hu, C. F. A. Gómez-Durán, J. W. Y. Lam, J. L. Belmonte-Vázquez, C. Deng, S. J. Chen, R. Q. Ye, E. Peña-Cabrera, Y. C. Zhong, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 10099 RSC.
- Y. B. Jiang, Spectrochim. Acta, Part A, 1999, 55, 833 CrossRef.
- M. Shigeta, M. Morita and G. Konishi, Molecules, 2012, 17, 4452 CrossRef CAS PubMed.
- L. R. Faulkner and A. J. Bard, J. Am. Chem. Soc., 1968, 90, 6284 CrossRef CAS.
- L. R. Faulkner and A. J. Bard, J. Am. Chem. Soc., 1969, 91, 209 CrossRef CAS.
- R. Y. Lai and A. J. Bard, J. Phys. Chem. A, 2003, 107, 3335 CrossRef CAS.
- M. Bandini, M. Bianchi, G. Valenti, F. Piccinelli, F. Paolucci, M. Monari, A. Umani-Ronchi and M. Marcaccio, Inorg. Chem., 2010, 49, 1439 CrossRef CAS PubMed.
- J. B. Birks and L. G. Christopherou, Spectrochim. Acta, 1963, 19, 401 CrossRef.
- W. J. Miao, Chem. Rev., 2008, 108, 2506 CrossRef CAS PubMed.
- E. A. Chandros and F. I. Sonntag, J. Am. Chem. Soc., 1966, 88, 1089 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemistry and spectroscopic data. See DOI: 10.1039/c7ra01586k |
| This journal is © The Royal Society of Chemistry 2017 |