
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of amphiphilic diblock copolymers of isotactic polystyrene-block-isotactic poly(p-hydroxystyrene) using a titanium complex with an [OSSO]-type bis(phenolate) ligand and sequential monomer addition†
QiHua Zhoua,
HuaQing Lianga,
WanChu Weia,
ChunFeng Mengb,
YongJiang Longa and
FangMing Zhu *ac
*ac
aKey Lab for Polymer Composite and Functional Materials of Ministry of Education, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: ceszfm@mail.sysu.edu.cn; Fax: +86-20-84114033; Tel: +86-20-84113250
bSchool of Material Science and Engineering, Jiangsu University, Zhenjiang, Jiangsu 212003, China
cGDHPRC Lab, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 510275, China
First published on 4th April 2017
Abstract
A series of isotactic diblock copolymers of polystyrene-block-poly(p-tert-butyldimethylsilyloxystyrene) (iPS-b-iP(p-TBDMSOS)) were successfully synthesized using living coordination polymerization techniques with a kind of titanium dichloro complex containing a 1,4-dithiabutandiyl-linked [OSSO]-type bis(6-cumyl-4-methyl phenolato) ligand (complex 1) activated by methylaluminoxane (MAO) as a catalyst. Nuclear magnetic resonance (NMR), gel permeation chromatography (GPC), infrared spectroscopy (IR), and differential scanning calorimetry (DSC) were used to characterize the copolymers. iPS-b-iP(p-TBDMSOS) had a higher molecular weight than that of the iPS prepolymer, and a narrow molecular weight distribution below 1.35. The diblock copolymers displayed two glass transition temperatures (Tg) at ∼97 and ∼117 °C, originating from iPS and iP(p-TBDMSOS) blocks, respectively. Furthermore, a novel well-defined amphiphilic diblock copolymer consisting of iPS (nonpolar block) and isotactic poly(p-hydroxystyrene) (iP(p-HOS), polar block), was achieved through hydrolysis of iP(p-TBDMSOS) block of iPS-b-iP(p-TBDMSOS) in the presence of hydrochloric acid. The obtained amphiphilic diblock copolymers self-assembled into spherical micelles with size of approximately 70 nm in methanol.
Introduction
Stereospecific living diblock copolymerization of nonpolar monomers with polar monomers has attracted great interest due to its capability to form amphiphilic diblock copolymers with well-defined stereochemistry and comonomer composition, controlled molecular weight as well as narrow molecular weight distribution.1,2 The interest stems from the unique features of the copolymers synthesized to be used as compatibilizers for polymer blends and their ability to self-assemble into ordered nanostructures that can be applied in various fields including nanoreactors, nanotemplates, nanocarriers, and drug delivery.3–11It is well known that the synthesis of diblock copolymers was based on living/control polymerizations to minimize undesired termination and/or transfer reactions.12 Three methods have been developed for the synthesis of diblock copolymers: (a) sequential addition of monomers; (b) macroinitiator methods and (c) coupling of two end-functionalized chains. A widely used method is the sequential addition of monomers, living polymerization of the first monomer, followed by polymerization of the second monomer. However, this method requires that the same catalytic or initiating specifies can initiate the living polymerization of both monomers, and that the conversion of the first monomer must be quantitative.13–21 Macroinitiators were prepared from end-functionalized polymers available, which initiate or catalyze living polymerizations of desired monomers to obtain well-defined diblock copolymer.22–26 Moreover, click coupling reaction of two end-functionalized macromolecular chains has been employed to synthesize diblock copolymers in recent years.27,28 Our group has reported the synthesis of amphiphilic diblock copolymers of polyethylene-block-poly(ethylene oxide), syndiotactic polypropylene-block-poly(ethylene oxide) and isotactic polystyrene-block-poly(ethylene oxide) via click coupling reactions.29–31 Nevertheless, such coupling reaction is inefficient when long polymer blocks (Mw > 104) are used because of wrapping of the reactive ends by polymer chains. Post-modification including hydrogenation,32–39 hydrolysis,34,35 hydroboration,36 and hydrosilylation,37–39 are also used to synthesize diblock copolymers.
Group IV metal complexes containing tetradentate [OSSO]-type bis(phenolate) ligands have been recently developed by Okuda and co-workers.40–46 They demonstrated that group IV complexes with 1,4-dithiabutandiyl-linked [OSSO]-type bis(6-tert-butyl-4-tert-butyl phenolato) ligands could be used as a highly active pre-catalysts for styrene isospecific polymerizations.40 A titanium complex bearing tetradentate [OSSO] bis(phenolate) ligand activated by [PhNMe2H]-[B(C6F5)4] in the presence of Al(nOct)3 at 25 °C was first used for styrene living polymerization with perfect isotacticity (mmm > 95%).43 Subsequently, the styrene stereospecific living polymerization catalyzed by titanium dichloro complex of 1,4-dithiabutandiyl-linked [OSSO]-type bis(6-cumyl-4-methyl phenolato) ligand (complex 1, Scheme 1) activated by MAO was fulfilled to produce highly isotactic polystyrene (iPS) with narrow molecular weight distribution.46 Styrene stereospecific living polymerization with other olefins such as butadiene, 4-methyl-1,3-pentadiene (4MPD),46 4-methylstyrene (PMS),47 and 4-tert-butylstyrene (PTBS)47 was carried out by using [OSSO] titanium complexes having two cumyl bulky groups in the ortho positions.46–48 The mechanism of isospecific styrene polymerization was depicted in Scheme 2. The active species were formed from the dichloro complex as a result of methyl exchange between aluminum and titanium.40 The insertion of monomers into the Ti–CH3 bond occurred in a 2, 1- or secondary fashion, and the polymerization was highly regioselective in either the initiation step or the propagation step. The β-hydrogen reaction did not represent an irreversible termination reaction, and the unsaturated polymer chain represented a resting state.47
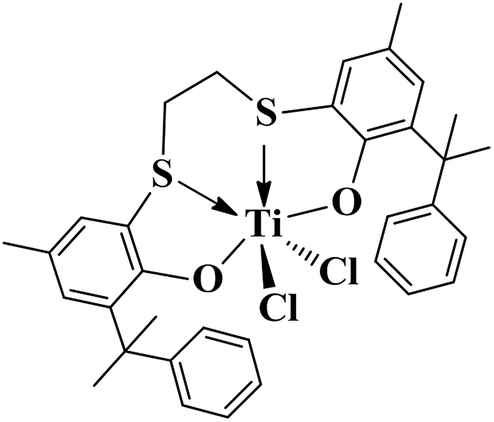 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
It is difficult to use directly transition metal catalysts for coordination polymerization of hydroxystyrene monomers, owing to the fact that hydroxyl groups coordinate with the transition metals and poison the catalysts.49 Consequently, these monomers were generally protected using bulky groups before polymerization. Syndiospecific living coordination polymerization of p-tert-butyldimethylsilyloxy styrene (p-TBDMSOS) has been succeed using transition metal catalysts.50–52 However, the stereospecific living polymerization of p-TBDMSOS base on transition metal catalysts is unprecedented. Herein we reported the first example of isospecific living polymerization of p-TBDMSOS and the synthesis of diblock copolymer of iPS-b-iP(p-TBDMSOS) via sequential monomer addition using complex 1/MAO as a catalyst. After hydrolysis of the iP(p-TBDMSOS) block by hydrochloric acid, an amphiphilic diblock copolymer of iPS-block-isotactic poly(p-hydroxystyrene) (iPS-b-iP(p-HOS)) was obtained.
Experimental
Materials
All manipulations of air- and moisture-sensitive compounds were performed under an inert atmosphere of nitrogen using a VAC glovebox or standard Schlenk-line techniques. Toluene and hexane were refluxed over sodium/benzophenone under high-pure-nitrogen. Styrene was stirred over CaH2 for about 24 h and then distilled under vacuum over CaH2. MAO (1.5 M) was purchased from Akzo-Nobel. All solvents, if not specified, were purchased from Sinopharm Chemical Reagent Co. Ltd. and used as received. Titanium tetrachloride and all other chemicals were purchased from J&K Chemicals and were used as received. Complex 1 was prepared as previously reported.40Synthesis of p-TBDMSOS
The p-TBDMSOS was prepared according to procedure reported by Kim et al.34 12.6 mL (90 mmol) of triethylamine were added to a suspension of 10.98 g (90 mmol) of p-hydroxybenzaldehyde in 150 mL of CH2Cl2, and the mixture was stirred at room temperature for 30 min. A solution of tert-butyldimethylsilyl chloride (13.56 g, 90 mmol) in 150 mL of CH2Cl2 was then added dropwise. The mixture was kept at room temperature under stirring for 3 h, and the reaction was quenched with water. The organic phase was separated, washed repeatedly with water and brine, dried over anhydrous MgSO4, filtered and concentrated to yield 20.8 g (98%) of a yellow oily product as p-tert-butyldimethylsilyloxybenzaldehyde (p-TBDMSOB).n-BuLi (1.6 M solution in hexane, 14 mL, 22.4 mmol) was added dropwise to a stirred, cold (0 °C) solution of methyltriphenylphosphonium bromide (8.19 g, 22.8 mmol) in 50 mL of THF. The suspension was stirred and kept at 0 °C for 15 min, a solution of p-TBDMSOB (5.69 g, 24 mmol) in 50 mL of THF was added dropwise via needle. The yellow suspension was stirred for 4 h and treated with NH4Cl. The solution was filtered and concentrated under vacuum. The resultant viscous liquid was purified by column chromatography using hexane as the solvent. The product was obtained as a colorless oil in a yield of 4.50 g (90%) and used immediately. 1H NMR (400 MHz, CDCl3): δ = 7.33 (d, 2H, C6H4), 6.85 (d, 2H, C6H4), 6.70 (q, 1H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.65 (d, 1H, –CHCH2), 5.17 (d, 1H, –CHCH2), 1.04 (s, 9H, C(CH3)3), 0.25 (s, 6H, Si(CH3)2).
CH2), 5.65 (d, 1H, –CHCH2), 5.17 (d, 1H, –CHCH2), 1.04 (s, 9H, C(CH3)3), 0.25 (s, 6H, Si(CH3)2).
Styrene polymerization
A 100 mL Schlenk flask equipped with a magnetic stirrer was dried more than 3 h by infrared light under vacuum and then cooled to room temperature. After replacement with high-pure-nitrogen three times, the flask was charged with high-pure-nitrogen to atmospheric pressure. Toluene, MAO solution in toluene, and complex 1 solution in toluene were charged sequentially by syringe under high-pure-nitrogen pressure. After equilibration of the solution at the polymerization temperature, the reaction was started by styrene. The run was terminated after the desired time by injection of ethanol (2 mL). The polymer was coagulated in ethanol (200 mL) acidified with aqueous HCl (ethanol/HCl, 90/10). The precipitated polymer was collected by filtration, washed several times with ethanol, and dried under vacuum at 60 °C to a constant weight.p-TBDMSOS polymerization
A 100 mL Schlenk flask equipped with a magnetic stirrer was dried more than 3 h by infrared light under vacuum and then cooled to room temperature. After replacement with high-pure-nitrogen three times, the flask was charged with high-pure-nitrogen to atmospheric pressure. Toluene, MAO solution in toluene and complex 1 solution in toluene were charged sequentially by syringe under high-pure-nitrogen pressure. Polymerization was initiated by injecting p-TBDMSOS. The reactions were kept at 25 °C under stirring for the required length of time. Polymerization was terminated by adding methanol. The polymers coagulated in methanol were recovered by filtration after washing several times with methanol, and the filtrate was dried to constant weight under vacuum at 60 °C.Block copolymerization of styrene with p-TBDMSOS via sequential monomer addition
A 100 mL Schlenk flask equipped with a magnetic stirrer was dried more than 3 h by infrared light under vacuum and then cooled to room temperature. After replacement with high-pure-nitrogen three times, the flask was charged with high-pure-nitrogen to atmospheric pressure. Toluene, MAO solution in toluene and complex 1 solution in toluene were charged sequentially by syringe under high-pure-nitrogen pressure. Polymerization was initiated by injecting styrene. After 2 h of polymerization, p-TBDMSOS was then rapidly added to this flask. The copolymerization continuously proceeded for an additional time, and then was terminated by introducing methanol. The polymers coagulated in methanol were recovered by filtration after washing several times with methanol, and the filtrate was dried to constant weight under vacuum at 60 °C.Deprotection of silylated homo- or block co-polymers
Silylated homo- or diblock co-polymers (0.2 g) were dissolved in THF (30 mL), acidified with concentrated HCl (5 mL), and the mixture was refluxed for 5 h. The resultant hydroxylated homo- or diblock co-polymers were concentrated and recovered by precipitation with cold diethyl ether and dried to constant weigh under vacuum at 50 °C.Self-assembly of iPS-b-iP(p-HOS)
Typically, 3 mg of iPS-b-iP(p-HOS) and 9.0 mL of THF were placed into a 20 mL flask. The mixture was stirred at 60 °C for 3 h to ensure that iPS-b-iP(p-HOS) dissolved completely to form a molecular solution. After the solution was added into methanol under stirring, the mixture was volatilized at room temperature for several days to remove THF entirely.Measurements
Nuclear magnetic resonance (1H NMR, 13C NMR) spectra of organic compounds and polymer were recorded on a Varian Unity Inova 400 spectrometer, operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR, respectively. Using 5 mm NMR tubes, polymer samples (30–50 mg) were dissolved in CDCl3, deuterated terahydrofuran (THF-d8) or CH3OD and analyzed at 25 °C. Chemical shifts were referenced to tetramethylsilane (TMS) or the residual solvent peak.The differential scanning calorimetry (DSC) analyses were performed using a PerkinElmer DSC-4000 instrument with the samples placed under a nitrogen atmosphere 2.0–4.0 mg of samples. Step-scan method for homopolymers: heating from 30 °C to 310 °C at a rate of 10 °C min−1; hold for 5 min at 310 °C; cooling to 30 °C at a rate of 10.0 °C min−1; hold for 5 min at 30 °C and reheating from 30 °C to 310 °C at a rate of 10 °C min−1. Step-scan method for diblock copolymers: heating from 30 °C to 310 °C at a rate of 10 °C min−1; hold for 5 min at 310 °C; cooling to 30 °C at a rate of 1.0 °C min−1; hold for 5 min at 30 °C and reheating from 30 °C to 310 °C at a rate of 10 °C min−1. Glass transition temperature (Tg) and melting temperature (Tm) were recorded on the second heating scan. The infrared (IR) spectra were obtained on a Thermo Nicolet Nexus 670 instrument with a measurement range of 400–4000 cm−1.
Gel permeation chromatography (GPC) analysis of the molecular weights and molecular weight distributions of the polymer were performed at 150 °C on a Varian PL-220 HTGPC instrument equipped with a triple-detection array, including a differential refractive-index detector, a two-angle light-scattering detector, and a four-bridge capillary viscometer that use narrow molecular weight distribution polystyrene as standards. The detection angles of the LS detector were 15 and 90°, and the laser wavelength was 658 nm. 1,2,4-Trichlorobenzene (TCB) was used as the eluent at a flow rate of 1.0 mL min−1.
The morphological observation of the self-assembled micelles in methanol was performed using scanning electron microscopy (SEM) observations (Hitachi S-4800) with an accelerating voltage of 10.0 kV. A drop from the previously prepared micelle solution was deposited onto a cover glass. The cover glass was dried at room temperature for several hours before examination by SEM.
Dynamic light scattering (DLS) measurements were conducted on a Brookhaven BI-2005M apparatus with a BI-9000AT digital correlator and a He–Ne laser at 532 nm. The angle was fixed to be 90°. The samples were placed in an index-matching decaline bath with temperature control within ±2 °C. Each solution passed through a 0.45 μm polytetrafluoroethene (PTFE) filter to remove dust. The data were analyzed by CONTIN algorithm, while the hydrodynamic diameter (Dh) and size polydispersity of the particles were obtained by a cumulant analysis of the experimental correlation function.
Results and discussion
Isospecific living coordination polymerization of styrene and p-TBDMSOS
Living coordination polymerization of styrene was carried out using complex 1 as catalyst activated by MAO at 25 °C. The number-average molecular weight (Mn) of isospecfic polystyrene synthesized (iPS) increased linearly with increasing monomer conversion (Fig. 1(a)), showing a typical living polymerization characteristics. The molecular weight distribution of iPS was below 1.30, verifying the living characteristics of the polymerization. The isospecific living coordination polymerization of styrene was quick, and reached a full conversion in 2 h.46 | ||
| Fig. 1 Plots of Mn (▲) and Mw/Mn (■) of polystyrene, poly(p-TBDMSOS) and block copolymer versus conversion of styrene (a), p-TBDMSOS (b) and comonomer (c). Polymerization conditions: styrene and p-TBDMSOS, 9.40 mmol; complex 1, 5.0 μmol; toluene, 20 mL; Al/Ti = 1500; polymerization temperature, 25 °C. | ||
The coordination polymerization of styrene derivates such as p-tert-butyldimethylsilyloxy styrene (p-TBDMSOS) was further checked using the identical catalyst system. The linear dependence of the molecular weight of poly(p-TBDMSOS) on the conversion of p-TBDMSOS was observed again (Fig. 1(b) and Table 1). The narrow molecular weight distribution of poly(p-TBDMSOS) was comparable to that of iPS. This suggested that the induction of silyloxy moiety had little influence on the coordination polymerization. Representatively, the addition of 2.2 g of p-TBDMSOS with 5.0 μmol of complex 1 and 1500 equiv. of MAO yielded 0.80 g poly(p-TBDMSOS) (Mn = 6.4 × 105 and PDI = 1.15) at 25 min. 13C NMR spectrum of the poly(p-TBDMSOS) synthesized was shown in Fig. 2 peaks at 153.43 ppm (C4), 139.51 ppm (C1 ipso-phenyl carbon), 128.17 ppm (C2), 119.57 ppm (C3), 43.25 ppm (the methylene carbons C6), and 39.78 ppm (the methine carbons C5) were observed. According to the literature data of syndiotacic poly(p-TBDMSOS) (sP(p-TBDMSOS)), the C1 singlet at 139.50 ppm in the 13C NMR was a characteristic peak of isotactic poly(p-TBDMSOS) (iP(p-TBDMSOS)) ([mmmm] > 95%).50 This showed that living isospecfic coordination polymerization of p-TBDMSOS using catalyst system of complex 1 was successful.
| Run | t (min) | Conv. (%) | Act.b | Mnc × 10−5 | Mw/Mnc | [mmmm]d (%) |
|---|---|---|---|---|---|---|
| a Polymerization conditions: complex 1, 5.0 μmol; [Al]/[Ti], 1500; toluene, 20 mL; p-TBDMSOS (2.2 g, 9.40 mmol); polymerization temperature, 25 °C.b 105 g (polymer) (mol per catalyst) per h.c Determined by GPC.d Determined by 13C NMR. | ||||||
| 1 | 5 | 10 | 4.8 | 3.08 | 1.07 | >95 |
| 2 | 15 | 28 | 4.4 | 4.84 | 1.10 | >95 |
| 3 | 25 | 40 | 3.8 | 6.43 | 1.15 | >95 |
| 4 | 35 | 48 | 3.6 | 6.96 | 1.20 | >95 |
| 5 | 45 | 55 | 3.2 | 7.56 | 1.26 | >95 |
| 6 | 60 | 62 | 2.7 | 7.84 | 1.30 | >95 |
 | ||
| Fig. 2 13C NMR spectrum of the iP(p-TBDMSOS) in CDCl3 at 25 °C. Polymerization conditions: p-TBDMSOS, 9.40 mmol; complex 1, 5.0 μmol; toluene, 20 mL; Al/Ti = 1500; polymerization temperature, 25 °C. | ||
Herein, we demonstrate first p-TBDMSOS isospecific living coordination polymerization with [OSSO]-type titanium catalyst (complex 1) activated by MAO and the formation of iP(p-TBDMSOS) with high isotacticity ([mmmm] > 95%). Consequently, isotactic poly(p-hydroxystyrene) (iP(p-HOS)) was readily acquired through hydrolyzing the iP(p-TBDMSOS) in the presence of hydrochloric acid. The deprotection of tert-butyldimethylsilyl to hydroxyl group gave rise to small chemical shift comparing to iP(p-TBDMSOS), as shown Fig. 3.
 | ||
| Fig. 3 13C NMR spectrum of the iP(p-HOS) (in CD3OD at 25 °C) obtained from hydrolysis of iP(p-TBDMSOS) with complex 1, 5.0 μmol; toluene, 20 mL; Al/Ti = 1500; polymerization temperature, 25 °C. | ||
Isospecific living block copolymerization of styrene with p-TBDMSOS via sequential monomer addition
The main results of isospecific living block copolymerization via sequential monomer addition of styrene (first monomer) and p-TBDMSOS (second monomer) using complex 1/MAO were summarized in Table 2. As in Fig. 1c, Mn of iPS with narrow molecular weight distribution (Mw/Mn < 1.35) also increased linearly as the conversion increased. This further verified that the polymerization was living. The activity decreased slightly with time.| Run | Feed (M1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M2) :M2) |
M2Conv. (%) | Act.b | XSc | Mnd × 10−5 | Mw/Mnd | Mne × 10−5 | Mw/Mne |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: complex 1, 5.0 μmol; [Al]/[Ti], 1500; toluene, 20 mL; M1, styrene (mmol); M2, p-TBDMSOS (mmol); polymerization temperature, 25 °C; first step, styrene, 120 min; second step, p-TBDMSOS, 60 min.b 105 g (polymer) (mol per catalyst) per h.c Styrene molar fraction in iPS-b-iP(p-TBDMSOS), determined by 1H NMR spectra.d Relative to the iPS block before feeding p-TBDMSOS monomer determined by GPC.e Relative to the iPS-b-iP(p-TBDMSOS), determined by GPC. | ||||||||
| 1 | 0.00:2.05 |
72 | 0.70 | 0.00 | — | — | 3.54 | 1.26 |
| 2 | 4.35:4.27 |
38 | 0.37 | 0.73 | 3.71 | 1.24 | 5.30 | 1.29 |
| 3 | 4.35:2.05 |
60 | 0.37 | 0.78 | 3.71 | 1.24 | 4.82 | 1.33 |
| 4 | 4.35:1.03 |
82 | 0.38 | 0.82 | 3.71 | 1.24 | 4.54 | 1.32 |
| 5 | 2.17:4.27 |
55 | 0.26 | 0.52 | 2.20 | 1.23 | 5.65 | 1.32 |
| 6 | 0.87:4.27 |
70 | 0.27 | 0.23 | 1.00 | 1.23 | 7.60 | 1.35 |
The obtained diblock copolymers were characterized by GPC, NMR and DSC. The results displayed that the relative length of the blocks in the diblock copolymer of iPS-b-iP(p-TBDMSOS) can be fine-tuned by changing the feed ratio of monomers. The representative GPC profiles of iPS prepolymer that was removed from the same polymerization mixture before feeding p-TBDMSOS, the second step diblock copolymer (Table 2, Run 2) and homopolymer of iP(p-TBDMSOS) under identical conditions were shown in Fig. 4. A unimodal and quite symmetric peak for iPS-b-iP(p-TBDMSOS) showed the molecular weight (Mn = 5.30 × 105) was higher than that of the iPS prepolymer (Mn = 3.71 × 105). The conversion of styrene reached 100% before adding p-TBDMSOS, and molecular weight distribution of the diblock copolymer was narrow, Mw/Mn = 1.29. This indicated the formation of iPS-b-iP(p-TBDMSOS). The 13C NMR spectrum of iPS-b-iP(p-TBDMSOS) showed characteristic peak of 146.52 ppm (C1 ipso-phenyl carbon) of iPS and characteristic peak of 153.43 ppm (C4), 139.51 ppm (C1 ipso-phenyl carbon) of iP(p-TBDMSOS) as shown in Fig. 5, showing the existence of iPS and iP(p-TBDMSOS) blocks. As the existence of chain transfer (β-hydrogen reaction) effect, the Mn of the block copolymer was not a simple sum of the Mn of the two homopolymers.
 | ||
| Fig. 4 GPC profiles of iPS (Table 2, Run 2), iPS-b-iP(p-TBDMSOS) (Table 2, Run 2) and iP(p-TBDMSOS) in THF at 25 °C. | ||
 | ||
| Fig. 5 13C NMR spectra of iPS-b-iP(p-TBDMSOS) (Table 2, Run 2), iPS and iP(p-TBDMSOS) in CDCl3 at 25 °C. | ||
In addition, IR spectrometry was used to characterize the homopolymers and block copolymers, as shown in Fig. 6. The peak at ∼1450 cm−1 which was attributed to CH2 bending vibration, and the peaks at 2840–3060 cm−1 ascribing to stretching vibration of C–H bonds indicated that the polymerizations were successful. The peak at ∼1601 cm−1 for iPS can be attributed to CC stretching vibration of benzene rings, and peaks at ∼699 cm−1 and ∼759 cm−1 were characteristic absorption peaks of mono-substituted benzene rings. For iP(p-TBDMSOS), peak at ∼1257 cm−1 derived from the stretching vibration of Si–CH3 band, ∼1170 cm−1 derived from the stretching vibration of C–O band, and ∼839 cm−1 was characteristic absorption peak of 1,4-substituted benzene rings. In Fig. 6c, the peaks at 2840–3060 cm−1, ∼1460 cm−1,∼1170 cm−1, ∼839 cm−1, ∼759 cm−1, ∼699 cm−1 indicated that iPS-b-iP(p-TBDMSOS) block copolymer was successfully synthesized.
 | ||
| Fig. 6 IR spectra of iPS (a), iP(p-TBDMSOS) (b), iPS-b-iP(p-TBDMSOS) (c), and iPS-b-iP(p-HOS) (d) samples. | ||
The thermal properties of the iPS-b-iP(p-TBDMSOS) obtained by complex 1/MAO were investigated by DSC, and compared with those of iPS and iP(p-TBDMSOS). As the complicated crystallization of the block copolymer, cooling cycle was conducted at 1.0 °C min−1. Typical DSC curves of the synthesized iPS, iP(p-TBDMSOS) and iPS-b-iP(p-TBDMSOS) of Run 6 in Table 2 were displayed in Fig. 7 iP(p-TBDMSOS) showed higher Tg at 117 °C and Tm at 294 °C than those of iPS (Tg = 97 °C, Tm = 223 °C). A bulky group in the para position of styrene restricted segmental dynamics and lead to an increase in Tg.53 iPS-b-iP(p-TBDMSOS) displayed two glass transition temperatures (∼97 and ∼117 °C) and a wide overlapped melting temperature from 267 to 275 °C originating from the iPS and iP(p-TBDMSOS) blocks. These results demonstrated that the iPS-b-iP(p-TBDMSOS) was double crystalline diblock copolymer.
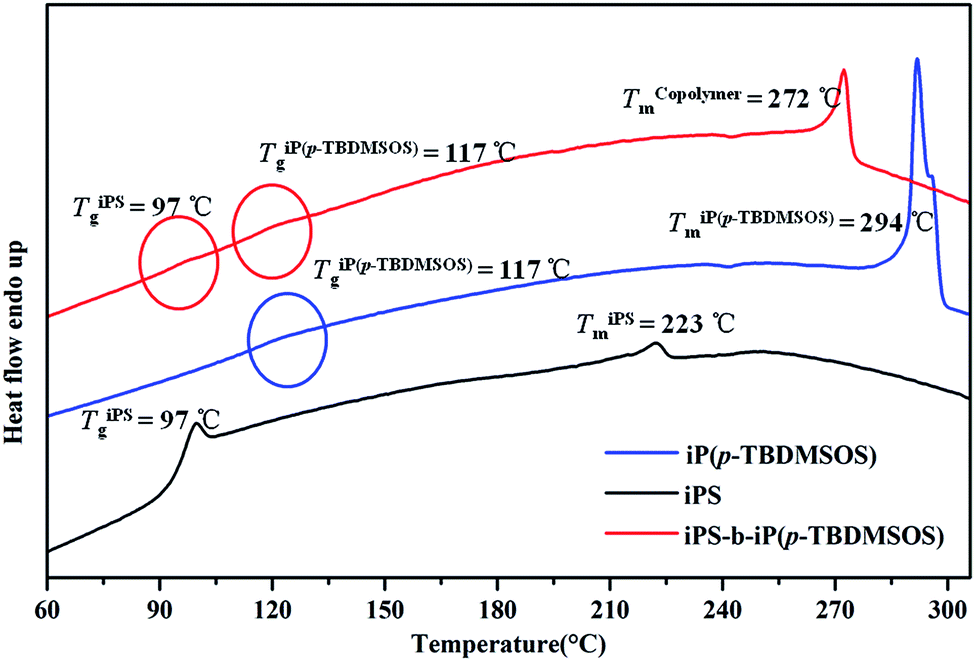 | ||
| Fig. 7 DSC curves of the second heating scan of iPS, iP(p-TBDMSOS) and iPS-b-iP(p-TBDMSOS) samples obtained in Table 2, Run 6. | ||
Preparation and solution self-assembly of iPS-b-iP(p-HOS)
iPS-b-iP(p-TBDMSOS) was hydroxylated to produce amphiphilic diblock copolymer of iPS-b-iP(p-HOS) in the presence of hydrochloric acid. iPS-b-iP(p-HOS) was characterized by 13C NMR in THF-d8 at 25 °C, as depicted in Fig. 8. The characteristic peaks at 146.57 ppm (C1 ipso-phenyl carbon) for iPS, and at 155.40 ppm (C4), 137.53 ppm (C1 ipso-phenyl carbon) for iP(p-HOS), confirmed the formation of iP(p-HOS) block. IR spectrum of iPS-b-iP(p-HOS) in Fig. 6d showed no peak at ∼1257 cm−1 and a broad peak at 3200–3500 cm−1, which also affirmed the formation of iP(p-HOS) block.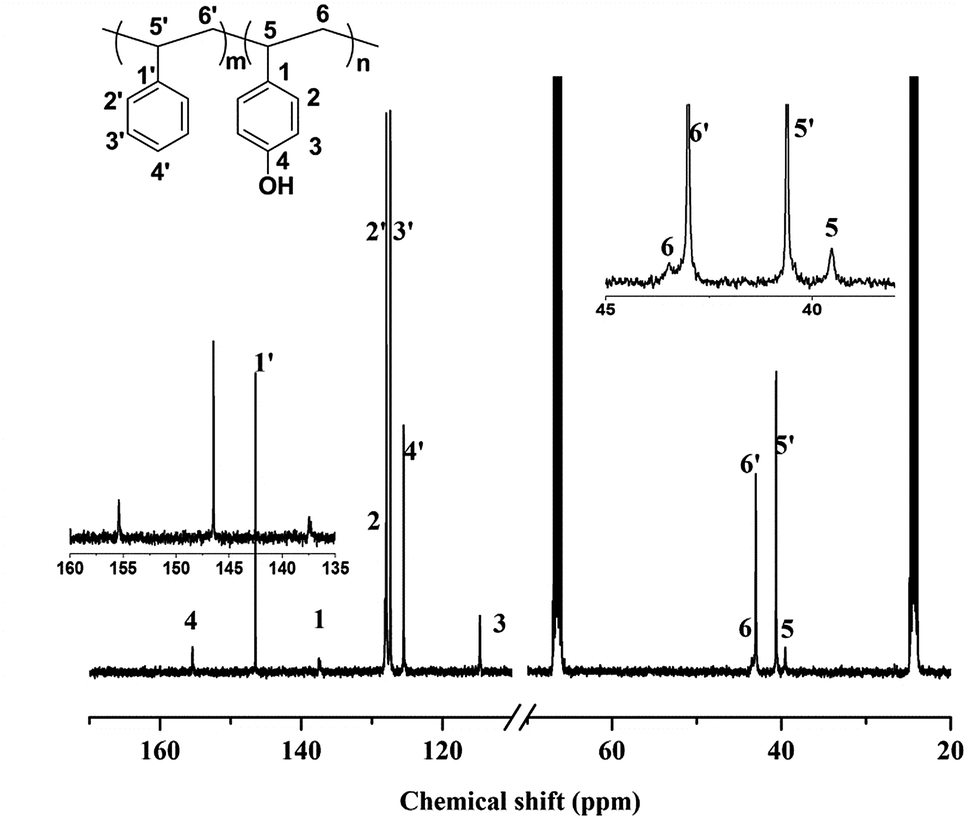 | ||
| Fig. 8 13C NMR spectrum (in THF-d8 at 25 °C) of iPS-b-iP(p-HOS) obtained from hydrolysis of iPS-b-iP(p-TBDMSOS) (Table 2, Run 2). | ||
THF is a common solvent for both iPS and iP(p-HOS) blocks, while methanol is a selective solvent for the iP(p-HOS) block but poor solvent for the iPS block. A THF solution of the iPS-b-iP(p-HOS) was added dropwise to methanol to form a light blue solution, implying the self-assembly process of the iPS-b-iP(p-HOS). After the mixture solution was volatilized at room temperature for several days to remove THF, the self-assembled micelles were observed using SEM, as displayed as in Fig. 9. The SEM images of a self-assembled iPS-b-iP(p-HOS) showed spherical nanoparticles with a diameter of approximately 70 nm. Hydrodynamic diameter of iPS-b-iP(p-HOS) micelles in methanol were further measured using DLS, as shown in Fig. 10. The hydrodynamic diameter of iPS-b-iP(p-HOS) micelles was about 74 nm, constant with SEM results.
 | ||
| Fig. 9 SEM image of the self-assembled micelles of iPS-b-iP(p-HOS) obtained from the hydrolysis of iPS-b-iP(p-TBDMSOS) (Table 2, Run 2) in methanol solution (0.03 mg mL−1). | ||
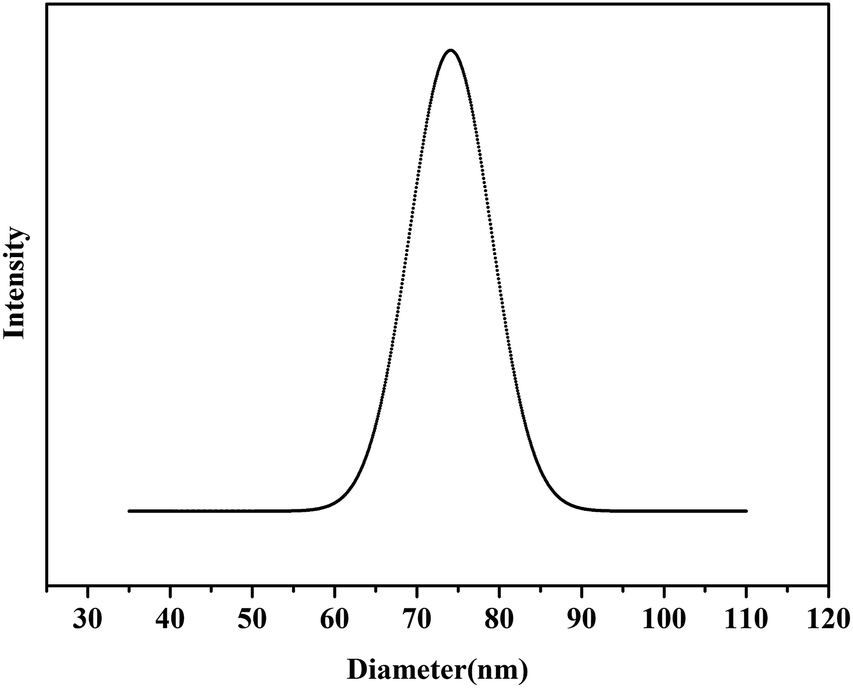 | ||
| Fig. 10 The Dh distribution of the self-assembled micelles of iPS-b-iP(p-HOS) obtained from the hydrolysis of iPS-b-iP(p-TBDMSOS) (Table 2, Run 2) in methanol solution (0.03 mg mL−1) at 25 °C. | ||
Conclusions
A series of isospecific diblock copolymers of iPS-b-iP(p-TBDMSOS) with well-controlled molecular weights, narrow molecular weight distribution, and tunable block ratios, have been successfully synthesized using sequent living coordination polymerization of styrene and p-TBDMSOS monomers with complex 1/MAO as a catalyst. iPS-b-iP(p-TBDMSOS) displays two glass transition temperatures (∼97 and ∼117 °C), originating from the iPS and iP(p-TBDMSOS) blocks, respectively. Furthermore, a novel amphiphilic diblock copolymer of iPS-b-iP(p-HOS) is prepared through the hydrolysis of the p(p-TBDMSOS) block in the presence of hydrochloric acid. iPS-b-iP(p-HOS) self assembles into spherical micelles with size of approximately 70 nm in methanol.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21174167, 51573212) and the NSF of Guangdong Province (S2013030013474, 2014A030313178).Notes and references
- L. Sung, J. F. Douglas, C. C. Han and A. Karim, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 1697–1700 CrossRef CAS
.
- H. K. Jeon, B. J. Feist, S. B. Koh, K. Chang, C. W. Macosko and R. P. Dion, Polymer, 2004, 45, 197–206 CrossRef CAS
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 237–266 CrossRef PubMed
- G. Pandav and V. Ganesan, Macromolecules, 2015, 73, 16–22 Search PubMed
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS
- B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CrossRef CAS
- N. Tomczak, D. Jancsewski, M. Han and G. J. Vancso, Prog. Polym. Sci., 2009, 34, 393–430 CrossRef CAS
- A. Meristoudi, G. Mountrichas and S. Pispas, Encyclopedia of nanoscience and nanotechnology, American Scientific Publishers, 2010 Search PubMed
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS PubMed
- N. Nishiyama and K. Kataoka, Adv. Polym. Sci., 2006, 193, 67–101 CrossRef CAS
- J. H. Park, S. Lee, J.-H. Kim, K. Park, K. Kim and I. C. Kwon, Prog. Polym. Sci., 2008, 33, 113–137 CrossRef
- N. Hadjichristidis, M. Pitsikalis and H. Iatrou, Adv. Polym. Sci., 2005, 189, 1–124 CrossRef CAS
- G. Leone, Polym. Chem., 2012, 3, 1987–1990 RSC
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS
- S. Tanaka, R. Goseki, T. Ishizone and A. Hirao, Macromolecules, 2014, 47, 2333–2339 CrossRef CAS
- Z. Q. Wu, Y. Chen, Y. Wang, X. Y. He, Y. S. Ding and N. Liu, Chem. Commun., 2013, 49, 8069–8071 RSC
- J. X. Yang, Y. Y. Long, L. Pan, Y. F. Men and Y. S. Li, ACS Appl. Mater. Interfaces, 2016, 8, 12445–12455 CAS
- J. Wu, H. Jiang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 2486–2491 RSC
- Y. Ren, Z. Wei, X. Leng, Y. Wang and Y. Li, Polymer, 2015, 78, 51–58 CrossRef CAS
- A. D. Todd and C. W. Bielawski, ACS Macro Lett., 2015, 4, 1254–1258 CrossRef CAS
- R. Goseki, S. Onuki, S. Tanaka, T. Ishizone and A. Hirao, Macromolecules, 2015, 48, 3230–3238 CrossRef CAS
- S. J. Buwalda, A. Amgoune and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 1222–1227 CrossRef
- J. F. Reuther, M. P. Bhatt, G. Tian, B. L. Batchelor, R. Campos and B. M. Novak, Macromolecules, 2014, 47, 4587–4595 CrossRef CAS
- T. C. Chung, Prog. Polym. Sci., 2002, 27, 39–85 CrossRef CAS
- T. Shiono, Y. Akino and K. Soga, Macromolecules, 1994, 27, 6229–6231 CrossRef CAS
- Y. J. Chen, B. J. Wu, F. S. Wang, M. H. Chi, J. T. Chen and C. H. Peng, Macromolecules, 2015, 48, 6832–6838 CrossRef CAS
- H. C. Kolb, M. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS
- T. Li, W. J. Wang, R. Liu, W. H. Liang, G. F. Zhao, Z. Y. Li, Q. Wu and F. M. Zhu, Macromolecules, 2009, 42, 3804–3810 CrossRef CAS
- Z. Y. Li, R. Liu, B. Y. Mai, S. Feng, Q. Wu, G. D. Liang, H. Y. Gao and F. M. Zhu, Polym. Chem., 2013, 4, 954–960 RSC
- R. Liu, Z. Y. Li, B. Y. Mai, Q. Wu, G. D. Liang, H. Y. Gao and F. M. Zhu, J. Polym. Res., 2013, 20, 1–11 CrossRef CAS
- Q. H. Zhou, Z. Y. Li, H. Q. Liang, Y. J. Long, Q. Wu, H. Y. Gao, G. D. Liang and F. M. Zhu, Chin. J. Polym. Sci., 2015, 33, 646–651 CrossRef CAS
- X. L. He, W. Li, A. M. Liang, J. J. Chen, A. D. Wang, B. L. Hu, J. W. Shi and A. M. Chen, China Synth. Rubber Ind., 2011, 34, 8–12 CAS
- Y. Kim and Y. Do, Macromol. Rapid Commun., 2000, 21, 1148–1155 CrossRef CAS
- M. Franco, F. J. Rabagliati, A. A. Rodríguez, M. I. Antxon and M.-G. Sebastián, Polymer, 2007, 48, 4646–4652 CrossRef
- C. Bae, US Pat., US7671157, 2010
- S. Andre, F. Guida-Pietrasanta, A. Rousseau and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2414–2425 CrossRef CAS
- B. C. Auman, V. Percec, H. A. Schneider and H. J. Cantow, Polymer, 1987, 28, 1407–1417 CrossRef CAS
- B. N. Gacal, V. Filiz, S. Shishatskiy, S. Rangou, S. Neumann and V. Abetz, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1252–1261 CrossRef CAS
- C. Capacchione, A. Proto, H. Ebeling, R. Mulhaupt, K. Moller, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2003, 125, 4964–4965 CrossRef CAS PubMed
- C. Capacchione, R. Manivannan, M. Barone, K. Beckerle, R. Centore, L. Oliva, A. Proto, A. Tuzi, T. P. Spaniol and J. Okuda, Organometallics, 2005, 24, 2971–2982 CrossRef CAS
- K. Beckerle, C. Capacchione, H. Ebeling, R. Manivannan, R. M€ulhaupt, A. Proto, T. P. Spaniol and J. Okuda, J. Organomet. Chem., 2004, 689, 4636–4641 CrossRef CAS
- K. Beckerle, R. Manivannan, T. P. Spaniol and J. Okuda, Organometallics, 2006, 25, 3019–3026 CrossRef CAS
- K. Beckerle, R. Manivannan, B. Lian, G.-J. M. Meppelder, G. Raabe, T. P. Spaniol, H. Ebeling, F. Pelascini, R. Mülhaupt and J. Okuda, Angew. Chem., Int. Ed., 2007, 46, 4790–4793 CrossRef CAS PubMed
- G.-J. M. Meppelder, K. Beckerle, R. Manivannan, B. Lian, G. Raabe, T. P. Spaniol and J. Okuda, Chem.–Asian J., 2008, 3, 1312–1323 CrossRef CAS PubMed
- A. Proto, A. Avagliano, D. Saviello, R. Ricciardi and C. Capacchione, Macromolecules, 2010, 43, 5919–5921 CrossRef CAS
- C. Capacchione, D. Saviello, R. Ricciardi and A. Proto, Macromolecules, 2011, 44, 7940–7947 CrossRef CAS
- M. Loria, A. Proto and C. Capacchione, Polym. Int., 2017, 66, 144–150 CrossRef CAS
- A. Grassi, P. Longo, A. Proto and A. Zambelli, Macromolecules, 1989, 22, 104–108 CrossRef CAS
- Y. J. Kim and Y. K. Do, Macromol. Rapid Commun., 2000, 21, 1148–1155 CrossRef CAS
- M. Kawabe and M. Murata, Macromol. Chem. Phys., 2002, 203, 24–30 CrossRef CAS
- M. Kawabe and M. Murata, Macromol. Chem. Phys., 2001, 202, 3157–3164 CrossRef CAS
- Y. S. Xu and H. B. Li, Chem. Ind. Eng. Prog., 2000, 19, 35–37 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01450c |
| This journal is © The Royal Society of Chemistry 2017 |