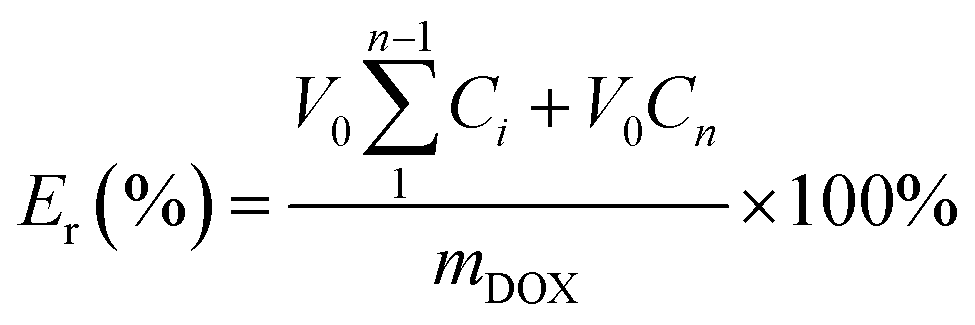DOI:
10.1039/C7RA01440F
(Paper)
RSC Adv., 2017,
7, 21093-21106
Bioreducible amphiphilic block copolymers based on PCL and glycopolypeptide as multifunctional theranostic nanocarriers for drug delivery and MR imaging†
Received
4th February 2017
, Accepted 30th March 2017
First published on 12th April 2017
Abstract
Amphiphilic diblock poly(ε-caprolactone)-b-glycopolypeptides (PCL–SS–GPPs) bearing disulfide bonds were synthesized from a clickable poly(ε-caprolactone)–SS–poly(2-azidoethyl-L-glutamate) diblock copolymer. Galactosyl and lactosyl sugar units as targeting ligands were conjugated to the polypeptide blocks via an efficient click reaction. The chemical structures of PCL–SS–GPPs were characterized by Fourier transform infrared spectroscopy and nuclear magnetic resonance analysis. Owing to their amphiphilic nature, these copolymers could self-assemble into spherical nano-sized micelles in an aqueous medium, as confirmed by fluorometry, transmission electron microscopy, and dynamic light scattering. The hydrophobic anticancer drug doxorubicin (DOX) and superparamagnetic iron oxide (SPIO) nanoparticles (NPs), as the magnetic resonance imaging (MRI) contrast agent, were simultaneously encapsulated in the hydrophobic core of the micelles via dialysis. The release profiles of encapsulated DOX from the SPIO/DOX-loaded PCL–SS–GPPs micelles were shown to be rapid in the presence of 10 mM glutathione (GSH) within 24 h, whereas in the absence of GSH, there was less than 35% DOX released from the PCL–SS–GPPs micelles in 24 h. Inverted fluorescence microscopy revealed the specific interaction between the sugars units on the PCL–SS–GPPs surface with the FITC-lectin. MTT assay demonstrated that the blank PCL–SS–GPPs micelles were nontoxic to the HepG2 cells, even for concentrations up to 500 μg mL−1, whereas the free DOX and DOX-loaded PCL–SS–GPPs micelles showed significant cytotoxicity against the HepG2 cells. The fluorescence images and flow cytometry tests revealed that DOX could be efficiently transported into HepG2 tumor cells by PCL–SS–GPPs micelles. The PCL–SS–GPPs micelles loaded with superparamagnetic iron oxide (SPIO) nanoparticles enabled excellent MRI contrast enhancement, thus confirming their effectiveness for MR imaging.
1. Introduction
Cancer remains a major threat to human health today and its treatment still faces serious challenges.1 Chemotherapy is one of the most common treatments for various human malignancies in oncology. However, the poor pharmacokinetics, lack of tumor selectivity, and dose-limiting toxicities of many anti-cancer drugs can directly cause many chemotherapeutic failures in patients with metastatic tumors.2,3 Currently, polymeric nanoparticles have been widely employed as drug delivery vehicles for cancer chemotherapy as they can effectively improve drug aqueous solubility, blood circulation time, and biodistribution.4–6 However, the lack of specific tumor targetability and inefficient drug release usually lead to a modest therapeutic effect.7,8 To overcome these limitations, it is desirable to design multifunctional delivery systems that can achieve targeted delivery and triggered release of the payload under intracellular stimuli such as mildly acidic pH, reductive agents, and enzymes,9–13 as well as the real-time monitoring of drug localization.9–13
Currently, theranostic nanocarriers have attracted significant interest due to the combination of therapeutic and diagnostic agents on a single platform. The theranostic nanocarriers can be equipped with a variety of functional agents including target ligands for precise cancer cell targeting, anticancer drugs for treatment, and diagnostic agents for the detection or even real-time monitoring of the therapeutic agents. After loading the therapeutic and diagnostic agents, the theranostic nanocarriers can achieve systemic circulation, evade host defenses, and deliver the drug and diagnostic agents at the targeted site for diagnosis and disease treatment at the cellular and molecular level.14,15 Lee et al. prepared theranostic micelles containing the photosensitizing drug Ce6 for photodynamic tumor therapeutic treatment and found that iron oxide (Fe3O4) offered high contrast magnetic resonance (MR) imaging of the tumor in vivo.16 It has been reported that glutathione (GSH), a thiol-containing tripeptide capable of cleaving disulfide bonds by a redox reaction, is abundant in the cytoplasm of cells (110 mM), whereas it is rarely present in blood plasma (2 mM). Reduction-sensitive nanocarriers have been developed for specific tumor targetability and can achieve the rapid release of encapsulated drugs inside the target cells or under reductive conditions, mimicking the intracellular compartments.17,18 As for target delivering, carbohydrates are widely used as targeting ligands owning to their special cell interaction and recognition. Galactosyl and lactosyl sugar units have the ability to recognize the asialoglycoprotein receptors (ASGP-R), which are overexpressed on hepatocellular carcinoma cells, due to their ability to bind lectins.19,20 The interaction behavior between galactosyl and lactosyl sugar units with cell surface receptors make them attractive therapeutic and diagnostic targets. However, the interaction of these proteins with a carbohydrate is quite weak, so a multivalent effect of glycopolymers are widely used to enhance the biological binding or internalization.21 This phenomenon is often referred to as “the cluster glycoside effect”, where the clustering of carbohydrates leads to high avidity receptor binding.22 As important analogs of natural glycoproteins, synthetic glycopolypeptides are regarded as new-generation drug carriers, owning to their biodegradability, well-defined secondary structures, and specific carbohydrate–protein interactions.23–25 Normally, glycopolypeptides have been widely obtained by direct ring-opening polymerization (ROP) of the glycosylated α-amino acid N-carboxyanhydride (NCA) monomers.26 However, it is still difficult to obtain the glycosylated NCA monomers with enough purity to allow their controlled polymerization.27 There has been tremendous interest and activity in recent years in the preparation of α-amino acid NCA containing reactive functional groups, such as alkyne, azido, and alkene functional groups.23,28 These functional groups can undergo highly effective “click” types reactions, such as azide–alkyne cycloadditions, thiol–ene, and thiol–yne reactions, to prepare glycopolypeptides.29,30 Chen's lab reported three different azide-functionalized monosaccharides conjugated onto alkyne functional polypeptides using azide–alkyne cycloadditions. The resulting glycopolypeptides were water soluble and could bind with Con A lectin.31 Zhang et al. reported the synthesis of glycopolypeptides by the azido functional polypeptides reaction with alkyne-functionalized D-mannose using copper catalysis in DMF.32 Schlaad et al. reported the synthesis of stimuli-responsive glycopolypeptides by the combination of the ROP with α-amino NCA monomers and “thiol–ene” click reaction with thio-functionalized sugar.33 Lecommandoux et al. synthesized amphiphilic glycopeptides by the combination of the sequential ring-opening polymerization of α-amino acid NCA monomers and a “click” reaction with azide-functionalized sugar molecules. The amphiphilic glycopeptides self-assembled in water into spherical and worm-like micelles as well as polymersomes, which showed specific recognition/binding capability to glycosidase.34,35 Many existing amphiphilic glycopeptides are homopolypeptides, where the hydrophobic block originates from the polypeptide backbone. The incorporation of a nonpeptidic hydrophobic block would widen the scope of the resultant polymer properties.36 Gupta et al. synthesized biocompatible multi-arm star copolymers comprising a hydrophilic glycopolypeptide and hydrophobic poly(ε-caprolactone) block using a ring-opening polymerization and click chemistry. The resulting noncytotoxic mannosylated rods and polymersomes could not only encapsulate hydrophobic and hydrophilic dyes within the nanostructures, but also displayed carbohydrate-specific uptake toward MDA-MB-231 cells.37 Recently, Cao et al. reported the synthesis of disulfide bonds linked to the amphiphilic glycopolypeptide analog poly(6-O-methacryloyl-D-galactopyranose)–SS–poly(γ-benzyl-L-glutamate) (PMAgala–SS–PBLG). The hydrophilic block was a polymathacrylate derivative synthesized by RAFT polymerization in a controlled manner. The poly(γ-benzyl-L-glutamate) acted as a hydrophobic block. Nanoparticles obtained from the glycopolypeptide analogs self-assembly could act as multifunctional vehicles for protein recognition, drug delivery, and hepatoma cell targeting.38
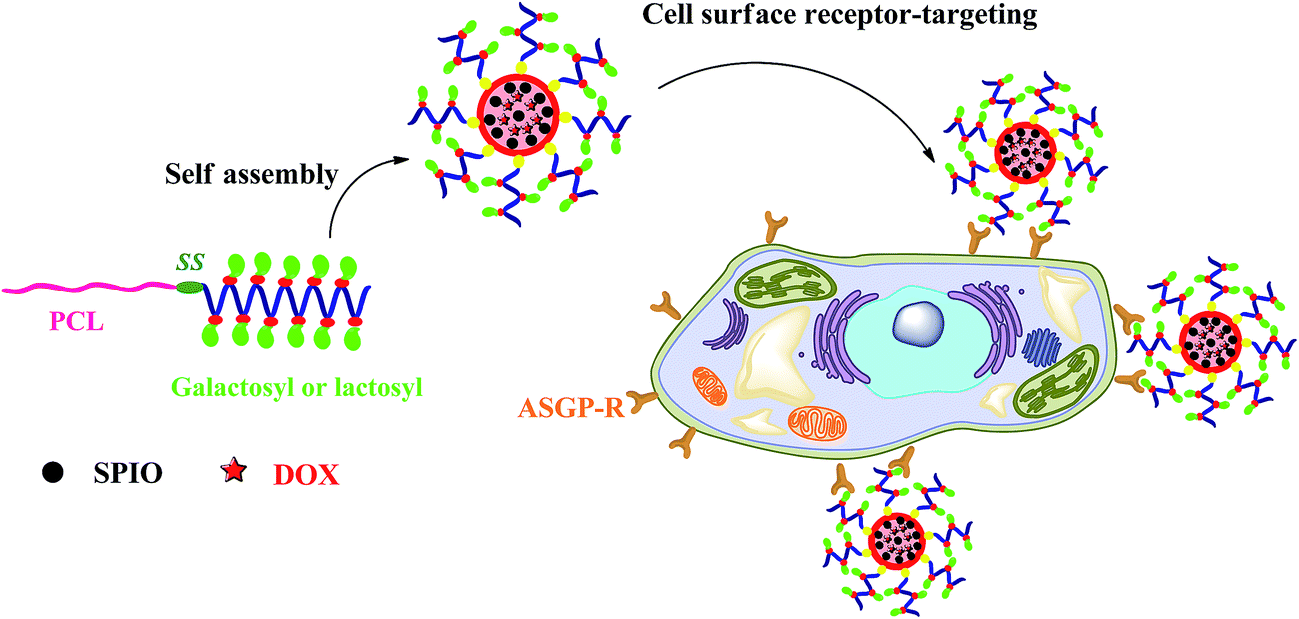
Herein, we report the synthesis of reduction-sensitive amphiphilic PCL–SS–GPPs diblock copolymers as SPIO and DOX nanocarriers for controlled drug delivery and efficient MRI contrast enhancement. To the best of our knowledge, reduction-responsive micelles combining amphiphilic glycopolypeptide and poly(ε-caprolactone) constructed via click chemistry have not yet been extensively explored for theranostic applications. The PCL–SS–GPPs composed of a hydrophobic PCL block and hydrophilic glycopolypeptides were prepared via click chemistry, where poly(ε-caprolactone)–SS–poly(2-azidoethyl-L-glutamate) was covalently conjugated with alkyne galactosyl and lactosyl sugar units. The self-assembly behaviors of amphiphilic PCL–SS–GPPs in solution were investigated using fluorescence spectroscopy, dynamic light scattering (DLS), and transmission electron microscopy (TEM). As shown in Scheme 1, the introduction of disulfide bonds was expected to adjust the release of DOX, due to the reduction-responsive disassembling behaviors of the micelles. Their lectin-recognition properties, intracellular drug delivery, and growth inhibition to HepG2 cells were investigated via fluorescence microscopy, MTT, and flow cytometry assay. Furthermore, the magnetic properties of the micelles containing SPIO were characterized to understand their efficacy as MRI probes.
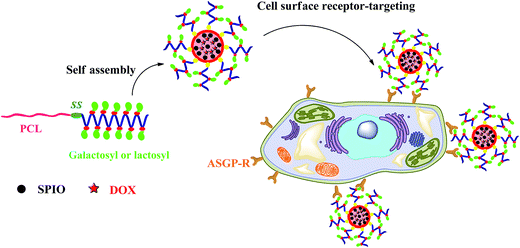 |
| | Scheme 1 Schematic of the reduction-responsive PCL–SS–GPPs micelles as nanocarriers for cancer chemotherapy and imaging. | |
2. Experimental
2.1 Materials
Copper sulfate (CuSO4·5H2O), tin(II) 2-ethylhexanoate (Sn(Oct)2, 95%), ε-caprolactone, sodium ascorbate (99%), and 1-dodecanol were purchased from Alfa Aesar. Doxorubicin hydrochloride, triphosgene, iron(III) acetylacetonate (99%), benzyl alcohol, N,N′-carbonyldiimidazole (CDI), L-glutamic acid (99%), and glutathione (GSH) were purchased from Aladdin Chemical Company. Sodium azide NaN3 (99%) and cystamine dihydrochloride were purchased from Aldrich. ε-Caprolactone and dimethylformamide (DMF, Shanghai Sinopharm Chemical Reagent Corp., 99.5%) were distilled from calcium hydride under reduced pressure and stored over molecular sieves. 2-Chloroethanol was purchased from Xiya Reagent. Tetrahydrofuran (THF, 99.5%) and toluene (99.5%) were refluxed and distilled over sodium benzophenone until a purple color was obtained. Lectin from Triticum vulgaris (wheat) FITC conjugate (LEC-FITC), and lyophilized powder were purchased from Sigma-Aldrich. Dulbecco's Modified Eagle Medium (DMEM), trypsin–ethylenediaminetetraaceticacid (Trypsin–EDTA), and fetal bovine serum (FBS) were purchased from Gibco-BRL (Canada). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Invitrogen Corporation (Washington, USA). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Beyotime Institute of Biotechnology (China). All the other reagents were of analytical grade and used without further purification. The human hepatocellular liver carcinoma (HepG2) cells line was purchased from the Animal Center of the Sun Yat-Sen University (Guangzhou, China). The detailed synthetic method for the SPIO was according to a previous publication.39,40
2.2 Synthesis of PCL–SS–NH2
2.2.1 Synthesis of PCL via ring-opening polymerization directly initiated from 1-dodecanol. A typical ring-opening polymerization procedure for ε-CL was as follows. A flamed glass ampule with a magnetic stirrer was charged with 1-dodecanol (0.028 g, 0.15 mmol), ε-CL (1.03 g, 9 mmol) and 5.0 mL of dry toluene. After 1.0 mL toluene was removed under vacuum to remove traces of water, Sn(Oct)2 (0.1 mL, 0.1 mol L−1 in toluene) was injected into the reaction mixture and the air was exchanged with nitrogen three times. The glass ampule was immersed in an oil bath at 110 °C with vigorous stirring to start the polymerization. After 24 h, the reaction mixture was rapidly cooled to room temperature and diluted with THF, and the crude product was precipitated in an excess of petroleum ether. The sediments were collected and dried in vacuum for 48 h until a constant weight (0.94 g, 87.2% overall yield, Mn = 5729, Mw/Mn = 1.18). 1H NMR analysis in CDCl3 revealed a degree of polymerization (DP) of 37 for the PCL–OH segments. Therefore, this polymer was denoted as PCL37–OH. 1H NMR (500.10 MHz, CDCl3, 298 K), δ, (ppm): 4.1–3.8 (–COOCH2CH2–), 3.6 (–CH2CH2OH), 2.4–2.2 (–CH2CH2COO–), and 1.17–1.12 (–COOCH2CH2CH2CH2CH2–) of PCL. FTIR (cm−1): 1727, 1601, 1489, 1453.
2.2.2 Synthesis of the PCL ester of carbonyl imidazole (PCL37–CI). Following the literature,41 200 mL anhydrous dichloromethane (DCM) and 1,1′-carbonyldiimidazole (CDI, 2.92 g, 18 mmol) were added into a 500 mL dry round-bottom flask. Then, PCL37–OH (0.90 g) dissolved in DCM was added dropwise over the course of 30 min. The obtained clear solution was stirred at room temperature for 2 h, washed with water (3 × 70 mL), dried over sodium sulfate, and finally evaporated to obtain a white solid (yield: 13.0 g, 87%). 8.16 (s, 1H, –NCHN–), 7.45 (s, 1H, –NCHCHN–) and 7.07 (s, 1H, –NCHCHN–) of the imidazole ring, 4.1–3.8 (s, 2H, –COOCH2CH2–), 3.6 (s, 2H, –CH2CH2OH), 2.4–2.2 (s, 2H, –CH2CH2COO–) and 1.17–1.12 (s, 2H, –COOCH2CH2CH2CH2CH2–) of PCL37.
2.2.3 End-group modification of PCL37–CI. Cystamine dihydrochloride was dissolved in deionized water then neutralized by slowly adding 4 M NaOH under stirring and extracted with dichloromethane to yield the cystamine. Then, PCL37–SS–NH2 was synthesized from PCL37–CI with the cystamine, as shown in Scheme 1. Briefly, cystamine (0.40 g, 2.63 mmol) and PCL37–CI (0.94 g, 0.164 mmol) were dissolved in 50 mL anhydrous DCM and subsequently stirred at room temperature for 48 h. After evaporation of the dichloromethane, 100 mL of 1.0 M NaH2PO4 (pH 4.0) was added. The aqueous solution was extracted with DCM (50 mL, 3 times), combined with the organic phase and then dried over sodium sulfate. After evaporating the DCM, a white solid was obtained (yield: 0.83 g, 88.3%). 1H NMR (500.10 MHz, CDCl3, 298 K), δ, (ppm): 8.19 (s, 1H, NCHN), 7.45 (s, 1H, NCHCHN), 7.10 (s, 1H, NCHCHN), 5.0 (s, 2H, OCH2C), and 2.63 (s, 1H, CCH).
2.3 Synthesis of γ-(2-azidoethyl)-L-glutamic acid-based N-carboxyanhydride (AELG-NCA)
To a solution of L-glutamic acid (15 g, 0.1 mol) suspended in anhydrous ethyl ether (100 mL) and 2-azide ethanol (20 mL) at 0 °C, 8 mL of sulfuric acid was added dropwise over the course of ca. 20 min. After removing the ethyl ether under reduced pressure, the reaction mixture was stirred at room temperature for 24 h. The solution was neutralized with pyridine. After storing in a refrigerator overnight, there was a white precipitate formed. The crude product was obtained after filtration. The crude product was washed with ethanol to afford γ-(2-azidoethyl)-L-glutamate as a white solid (5.0 g, 23.7%). 1H NMR (D2O, δ, (ppm)): 3.8 (t, 1H, NH2CH), 2.1 (m, 2H, –CHCH2CH2–), 2.6 (t, 2H, –COCH2CH2–), 4.5 (t, 2H, –OCH2CH2N3), 3.7 (t, 2H, ClCH2CH2–).
γ-(2-Azidoethyl)-L-glutamate (4.2 g, 20 mmol) and triphosgene (2.0 g, 6.74 mmol) were suspended in 60 mL of dry THF bubbling with a slow steady nitrogen flux in a flame-dry three-neck flask. The mixture was stirred in a 50 °C oil bath until the cloudy solution turned clear. The mixture was concentrated by reduced pressure distillation, and a yellowish oil was obtained, which was dissolved in 100 mL ethyl acetate, followed by washing with 50 mL 0.5% NaHCO3 ice-cold aqueous solution three times. The organic phase was then dried over anhydrous MgSO4 and evaporated to give AELG-NCA as a viscous oil (3.4 g, 70%).
2.4 Synthesis of PCL–SS–PAELG
Inside a glovebox, the initiator PCL37–SS–NH2 (0.40 g, 0.068 mmol) was dissolved in 15 mL DMF in a Schlenk tube. A solution of AELG-NCA (0.33 g, 1.36 mmol) in 5 mL dry DMF was added after PCL37–SS–NH2 was dissolved completely. The reaction mixture was stirred at room temperature for 72 h, and after that the reaction mixture was dialyzed against deionized water to remove the organic solution. A white precipitate appeared, which after filtration and drying under vacuum at 40 °C for 12 h afforded the final product as a white solid (0.62 g, 92%). 1H NMR analysis in CDCl3 revealed the degree of polymerization (DP) of 10 for the polypeptide block. Therefore, this polymer was denoted as PCL37–SS–PAELG10. 1H NMR ((500 MHz, CDCl3/CF3CO2D, v/v = 85/15, 298 K), δ, ppm): 0.88 (t, 3H, CH3CH2–), 1.26 (m, 2H, CH3CH2–), 1.34–1.43 (m, 2H, –CH2CH2CH2O–), 1.59–1.76 (m, 4H, –CH2CH2CH2CH2–), 1.91–2.25 (m, 2H, –CHCH2CH2–), 2.33–2.42 (m, 2H, –COOCH2CH2–), 2.47–2.60 (s, 2H, –CH2CH2COO–), 3.47–3.59 (s, 2H, –CH2CH2N3), 4.05–4.14 (t, 2H, –CH2CH2OCO–), 4.20–4.36 (s, 2H, –OCH2CH2N3), 4.50–4.67 (s, 2H, –COCHNH–).
2.5 Synthesis of polycaprolactone-block-poly(glutamategalactose) (PCL37–SS–PGluGal10) by click conjugation
The process for the click conjugation of propargylgalactose to the PCL37–SS–PAELG10 block copolymers started with 0.1 g of PCL37–SS–PAELG10 block copolymers containing about 0.20 mmol azide pendants and 0.126 g of propargylgalactose (0.54 mmol) being first dissolved in 10 mL of DMSO with argon bubbling. After the block copolymers and propargylgalactose dissolved completely, 15 mg of CuSO4·5H2O was introduced into the flask and the mixture was further bubbled with argon for about 10 min until 30 mg of sodium ascorbate (NaAsc) was added. Then the flask was sealed under argon and immersed in an oil bath at 40 °C for 3 days. The reaction medium was transferred to a 3.5 kDa molecular weight cut-off (MWCO) dialysis membrane and dialyzed first against ethylenediaminetetraacetic acid (EDTA) for 3 days, then against pure water for 3 days in order to remove the copper and ascorbate salts as well as excess propargylgalactose. The final product was obtained as a white solid by lyophilization. Yield: 0.126 g, 85%. 1H NMR (DMSO-d6): 0.88 (t, 3H, CH3CH2–), 1.26 (m, 20H, CH3CH2–), 1.34–1.43 (m, 2H, –CH2CH2CH2O–), 1.59–1.76 (m, 4H, –CH2CH2CH2CH2–), 1.91–2.25 (m, 2H, –CHCH2CH2–), 2.33–2.42 (m, 2H, –COOCH2CH2–), 2.47–2.60 (s, 2H, –CH2CH2COO–), 3.47–3.59 (s, 2H, –CH2CH2N3), 4.05–4.14 (t, 2H, –CH2CH2OCO–), 4.20–4.36 (s, 2H, –OCH2CH2N3), 4.50–4.67 (s, 2H, –COCHNH–), 5.1–3.4 t (Gla H-2, H-3, H-4, H-5, H-6), 8.07 ppm (s, 1H, –CONH–) and 8.15 ppm (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHN–).
CHN–).
2.6 Synthesis of polycaprolactone-block-poly(glutamatelactose) (PCL37–SS–PGluLac10) by click conjugation
Click conjugation between propargyllactose and the PCL37–SS–PAELG10 block copolymers started with 0.1 g of PCL–SS–PAELG block copolymers containing about 0.20 mmol azide pendants and 0.22 g of propargyllactose (0.54 mmol) being first dissolved in 10 mL of DMSO with argon bubbling. After the block copolymers and propargyllactose dissolved completely, 15 mg of CuSO4·5H2O was introduced into the flask and the mixture was further bubbled with argon for about 10 min until 30 mg of sodium ascorbate (NaAsc) was added. Then the flask was sealed under argon and immersed in an oil bath at 40 °C for 3 days. The reaction medium was transferred to a 3.5 kDa molecular weight cut-off (MWCO) dialysis membrane and dialyzed first against ethylenediaminetetraacetic acid (EDTA) for 3 days, then against pure water for 3 days in order to remove the copper and ascorbate salts as well as excess propargyllactose. The final product was obtained as a white solid by lyophilization. Yield: 0.146 g 82%. 1H NMR (DMSO-d6): 0.88 (t, 3H, CH3CH2–), 1.26 (m, 20H, CH3CH2–), 1.34–1.43 (m, 2H, –CH2CH2CH2O–), 1.59–1.76 (m, 4H, –CH2CH2CH2CH2–), 1.91–2.25 (m, 2H, –CHCH2CH2–), 2.33–2.42 (m, 2H, –COOCH2CH2–), 2.47–2.60 (s, 2H, –CH2CH2COO–), 3.47–3.59 (s, 2H, –CH2CH2N3), 4.05–4.14 (t, 2H, –CH2CH2OCO–), 4.20–4.36 (s, 2H, –OCH2CH2N3), 4.50–4.67 (s, 2H, –COCHNH–), 5.1–3.4 t (Lac H-2, H-3, H-4, H-5, H-6, H-7, H-8, H-9, H-10, H-11, H-12), 8.07 ppm (s, 1H, –CONH–) and 8.15 ppm (s, 1H, CCHN–).
2.7 Characterization
A Perkin-Elmer Paragon 1000 spectrometer was used to record the FTIR spectra of all the samples in transmission mode. Samples were ground with KBr and then compressed into pellets. The spectra were taken at the frequencies ranging from 500 to 4000 cm−1. 1H NMR spectra of the compounds were characterized on a Bruker 500 NMR spectrometer using deuterated chloroform (CDCl3-d) or deuterated dimethyl sulfoxide (DMSO-d6) as the solvent and tetramethylsilane (TMS) as the internal standard. The size and size distribution of the micellar aggregates were determined by dynamic light scattering (DLS) using the BI-200SM Goniometer particle size analyzer (Brookhaven Instruments Corporation, USA). Each analysis lasted for 300 s and the data were collected on an auto-correlator at 25 °C with a detection angle of scattered light at 90°. For each sample, the data from three measurements were averaged to obtain the mean ± standard deviation (SD). The morphology images of the micelles were obtained on a JEOL JEM-2100F transmission electron microscope (JEM-2100F, JEOL, Tokyo, Japan) at an accelerating voltage of 200 keV. In brief, 10 μL of 0.5 mg mL−1 micelle suspension on the copper grid was stained with a small drop of 1 wt% phosphotungstic acid solution (2 wt% in water). The grid was finally dried overnight at room temperature before TEM observation.
2.8 Self-assembly behavior of the amphiphilic PCL–SS–GPPs
To study the self-assembly behavior, the PCL–SS–GPPs in deionized water were prepared by the dialysis method as follows: 10 mg PCL–SS–GPPs, including PCL37–SS–PGluGal10 and PCL37–SS–PGluLac10, were first dissolved in 2 mL warm DMSO. Then, 4.0 mL of deionized water was added dropwise into the DMSO slowly via a syringe pump under stirring at room temperature. The organic solvent was removed by dialysis against PB for 24 h at 25 °C using a cellulose membrane bag (molecular weight cut-off, MWCO 3500–4000).
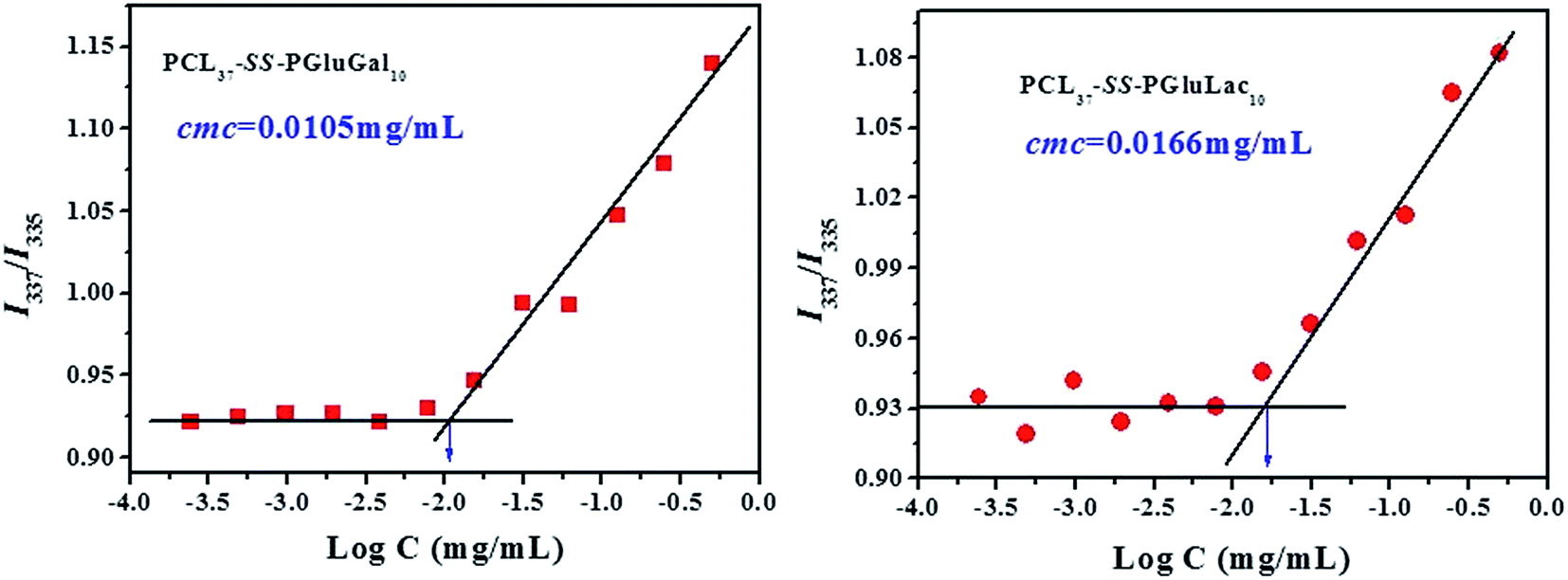
The critical micelle concentration (CMC) of the PCL–SS–GPPs was determined using pyrene as a fluorescence probe. The concentration of the PCL–SS–GPPs varied from 1.0 to 4.88 × 10−4 mg mL−1 and the final concentration of pyrene was fixed at 6.0 × 10−7 mol L−1. Before the measurement, the mixed solution of pyrene and PCL–SS–GPPs were kept on a shaker at 37 °C for 24 h to reach solubilization equilibrium in the dark. The fluorescence spectra were recorded using a fluorescence spectrometer (RF-5301 PC Shimadzu), where the excitation spectra of the PCL–SS–GPPs/pyrene solutions were scanned from 300 to 350 nm at room temperature, with an emission wavelength of 373 nm and a bandwidth of 5 nm. The intensity ratios of I337 to I335 were plotted as a function of logarithm of the PCL–SS–GPPs concentrations. The CMC value was estimated from the intersection of the tangent to the curve at the inflection with the horizontal tangent through the points at low concentrations.
2.9 Preparation of DOX and SPIO co-loading micelles
The hydrophobic DOX and SPIO could be loaded into the hydrophobic inner core of the micelles through a dialysis method. In brief, 10 mg PCL–SS–GPPs, 2 mg DOX·HCl, an equimolar amount of triethylamine (TEA), and 1.5 mg SPIO were dissolved in 2 mL DMSO. Next, 5 mL of deionized water was slowly added into the above mixed solution under moderate stirring. The mixed solution was dialyzed against deionized water with a molecular weight cut-off (MWCO) of 3500 Da at room temperature, with the dialysis water was changed every 8 h, to allow the formation of DOX/SPIO-loaded PCL–SS–GPPs micelles and to remove the DMSO and unencapsulated DOX and SPIO.
2.10 Determination of the DOX- and SPIO-loading contents
The DOX loading in the micelles was determined by fluorescence spectrophotometry. The DOX/SPIO-loaded PCL–SS–GPPs micelles were first broken in a mixed DMSO/PBS solvent (v/v, 70/30), then the sample was filtered to remove the insoluble SPIO and subjected to UV analysis (UV-3150, Shimadzu, Japan) at a wavelength of 480 nm. The loading amount of DOX was determined from the calibration curve of DOX. The drug-loading content (DLC) was calculated according to the following equation:| | |
DLC (%) = w1/w2 × 100
| (1) |
where w1 is the weight of loaded drug and w2 is the weight of the drug-loaded micelles.
The loading amount of SPIO NPs inside the PCL–SS–GPPs micelles was determined using an atomic absorption spectrophotometer (AAS). In brief, the DOX/SPIO-loaded PCL–SS–GPPs micelles were added to 1 M HCl solution to allow the disaggregation of micelles and the complete dissolution of the SPIO NPs. The Fe concentration was determined at the specific Fe absorption wavelength (248.3 nm) based on a previously established calibration curve. The SPIO-loading content was calculated as the ratio of SPIO to the total weight of the SPIO-loaded micelles.
| | |
SLC (%) = w3/w4 × 100
| (2) |
where
w3 is the weight of loaded SPIO and
w4 is the weight of SPIO-loaded micelles.
2.11 In vitro release of DOX
The in vitro release behavior of loaded DOX from the PCL–SS–GPPs micelles were studied at 37 °C in two different release media, where the DOX/SPIO-loaded PCL–SS–GPPs micelles were suspended in 3 mL of PBS with various GSH concentrations (0 and 10 mM), and then the micelle solution was immediately transferred into a dialysis bag (MWCO 3500 Da). The release experiment was initiated by placing the end-sealed dialysis bag in 27 mL of PBS containing 0 or 10 mM GSH with gentle shaking in a 37 °C water bath at 100 rpm. At predetermined time intervals, 3 mL release medium outside the dialysis bag was removed and subsequently replenished with 3 mL fresh buffered solution. The amount of released DOX was determined using a UV-vis spectrometer (UV-3150, Shimadzu, Japan) at 480 nm.| |
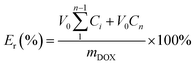 | (3) |
where V0 is the whole volume of the release media (V0 = 30 mL), Ve is the volume of the replace media (Ve = 3 mL), mDOX represents the amount of doxorubicin in the micelles, and Ci represents the concentration of DOX in the ith sample.
2.12 In vitro lectin recognition bioactivity
1 mL (1 mg mL−1) PCL37–SS–PGluGal10 and PCL37–SS–PGluLac10 micelles solutions were added into the 6-well plate, then after the water was evaporated off the PCL–SS–GPPs films were obtained. These films were treated with methanol for 1 h and then dried for 24 h. The insoluble PCL37–SS–PGluGal10 and PCL37–SS–PGluLac10 films were incubated with LEC-FITC solution (0.5 mg mL−1) at room temperature for 30 min. The films were washed with 100 mM phosphate buffer three times. The bioavailability of the galactose moieties on the PCL–SS–GPPs films with LEC-FITC were observed by a Carl Zeiss inverted fluorescence microscope.
2.13 Cell viability assays
Using a MTT assay, the in vitro relative cytotoxicities of the empty PCL–SS–GPPs micelles against human hepatocellular liver carcinoma (HepG2) cells were evaluated. Five multiple holes were set for every sample. The HepG2 cells were seeded in a 96-well plate at a density of 1 × 104 cells per well in 100 μL of DMEM and incubated under a 5% CO2 atmosphere at 37 °C for 24 h. After removing the culture medium, the PCL–SS–GPPs micelles solution was added to the wells at a specific concentration (0–500 μg mL−1). After co-incubation with the cells for 48 h, 20 μL of MTT solution in PBS (5 mg mL−1) was added to each well and the plate was incubated for another 4 h at 37 °C. After that, the medium containing MTT was removed, and 150 μL of DMSO was added to each well to dissolve the MTT formazan crystals. Finally, the plates were shaken for 10 min, and the absorbance of formazan product was measured at 490 nm by a microplate reader.| | |
Cell viability (%) = [A490 (sample)/A490 (control)] × 100
| (4) |
where A490 (sample) and A490 (control) represented the absorbances of the sample and control wells, respectively.
The in vitro antitumor activity of DOX/SPIO-loaded hepatoma-targeting PCL–SS–GPPs micelles against the asialoglycoprotein receptor (ASGP-R) overexpressing HepG2 cells was evaluated by MTT assay. Briefly, the HepG2 cells were respectively cultured onto 96-well plates at a density of 1 × 104 cells per well and incubated in a humidified atmosphere of 5% CO2 at 37 °C for 24 h. After that, the growth medium was replaced by 100 μL complete DMEM containing different DOX concentrations (0–10 μg mL−1 DOX) and further incubated for 24 h. After co-incubation with the cells for 24 h, 20 μL of MTT solution in PBS (5 mg mL−1) was added to each well and the plate was incubated for another 4 h at 37 °C. After that, the medium containing MTT was removed, and 150 μL of DMSO was added to each well to dissolve the MTT formazan crystals. Finally, the plates were shaken for 10 min, and the absorbance of the formazan product was measured at 490 nm by a microplate reader. The relative cell viability (%) was calculated based on the above method. Herein, the data are presented as the average ± SD (n = 3).
2.14 Cellular uptake studies
The cellular uptake and distribution of micelles were performed using flow cytometry (Guava easyCyte 5HT-2L, Merck Millipore, Germany) and inverted fluorescence microscopy (Axio Observer.Z1, Carl Zeiss, Germany). For flow cytometry, the HepG2 cells were seeded in 6-well plates at a density of 1 × 106 cells per well in 2 mL of complete DMEM and cultured at 37 °C in the 5% CO2 atmosphere overnight. Cells were then washed with PBS and incubated with the free DOX and DOX/SPIO-loaded PCL–SS–GPPs micelles (DOX concentration: 10 mg mL−1) at 37 °C for an additional 4 h. Cells without pretreatment were used for comparison. After the culture medium was removed, the cells were washed with PBS three times and treated with trypsin. Then, 2 mL of PBS was added to each culture well, and the cells were collected after centrifuging the solutions for 5 min at 3000 rpm. After removal of the supernatants, the cells were resuspended in 0.3 mL of PBS. A minimum of 2 × 104 cells were analyzed from each sample.
For the inverted fluorescence microscope studies, HepG2 cells were seeded in a 6-well plate (1 × 106 cell per well) in 2 mL of DMEM and cultured at 37 °C in the 5% CO2 atmosphere for 24 h. In order to observe the cells internalization, the culture medium was replaced with 2 mL of DMEM containing the free DOX and DOX/SPIO-loaded micelles (DOX concentration: 10 μg mL−1). After 4 h, the culture medium was removed and the cells were washed with PBS; the cells were then fixed with 4% formaldehyde for 30 min at room temperature, and the slides were rinsed with PBS three times. The cell nuclei were dyed with 4,6-diamidino-2-phenylindole (DAPI) for 30 min and washed with PBS three times. Finally, the samples were observed by the inverted fluorescence microscope, and the photos were obtained with a 20× objective. The samples were excited using a Mercury HBO Power instrument. The excitation wavelengths of DOX and DAPI were 546 ± 12 nm and 365 nm, respectively.
2.15 Relaxivity measurements
The T2 relaxivity of the DOX/SPIO-loaded micelles was measured by using a 3.0T MRI scanner (Verio, Siemens, Erlangen, Germany) at room temperature. The T2 mapping sequence (TR: 1000 ms, TE: 13.8/27.6/41.4/55.2/69.0 ms, flip angle: 180°, slice thickness: 3.0 mm, matrix 444 × 448) was used to measure the transverse relaxation time. The relaxation times were obtained by fitting the multi-echo data to a monoexponential decay curve using linearized least-squares optimization. The relaxivity values were calculated via linear least-squares fitting of 1/relaxation time (s−1) versus the iron concentration (mM Fe).
3. Results and discussion
3.1 Preparation and characterization of disulfide-bonds-linked PCL–SS–GPPs copolymers
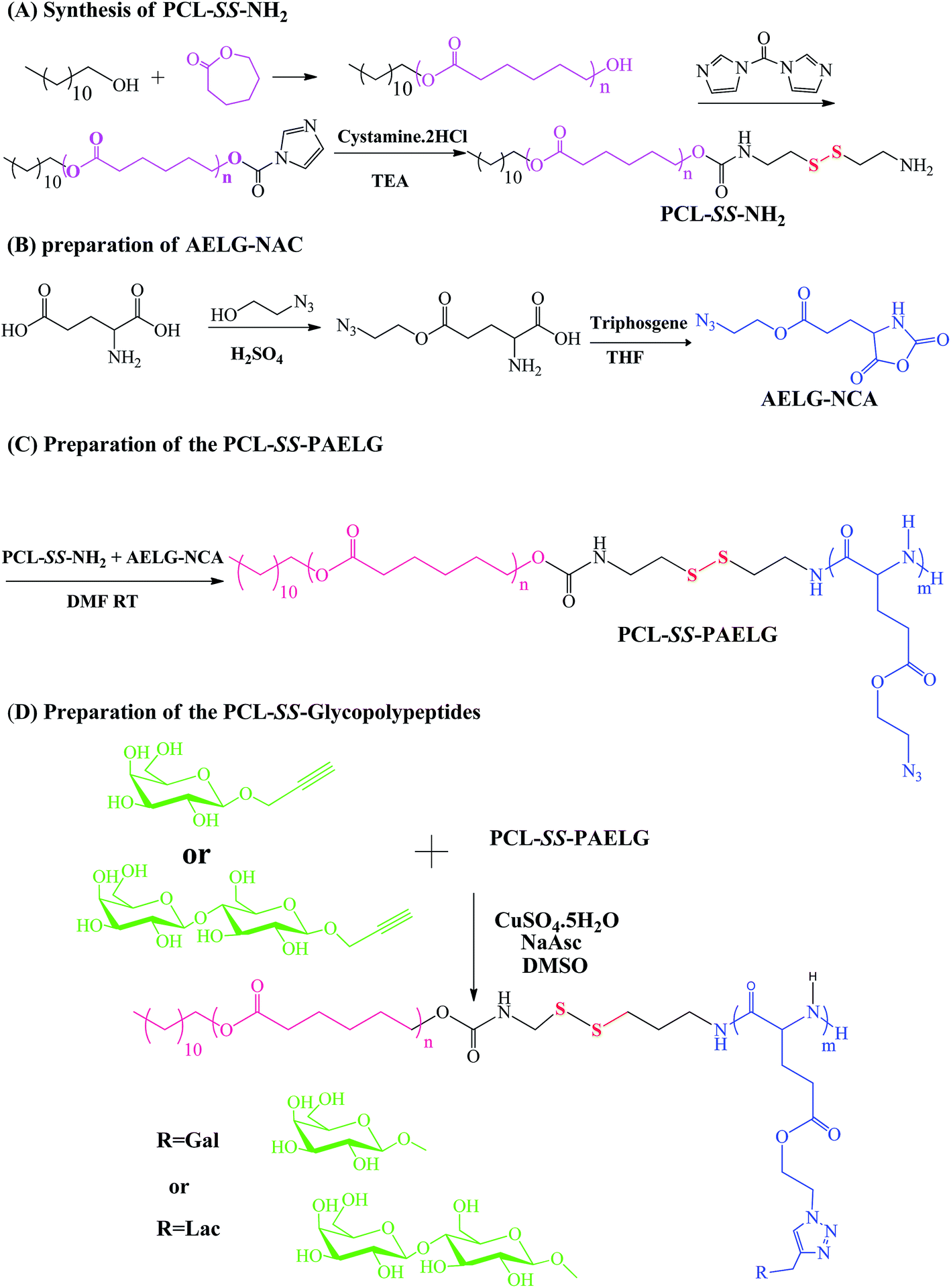
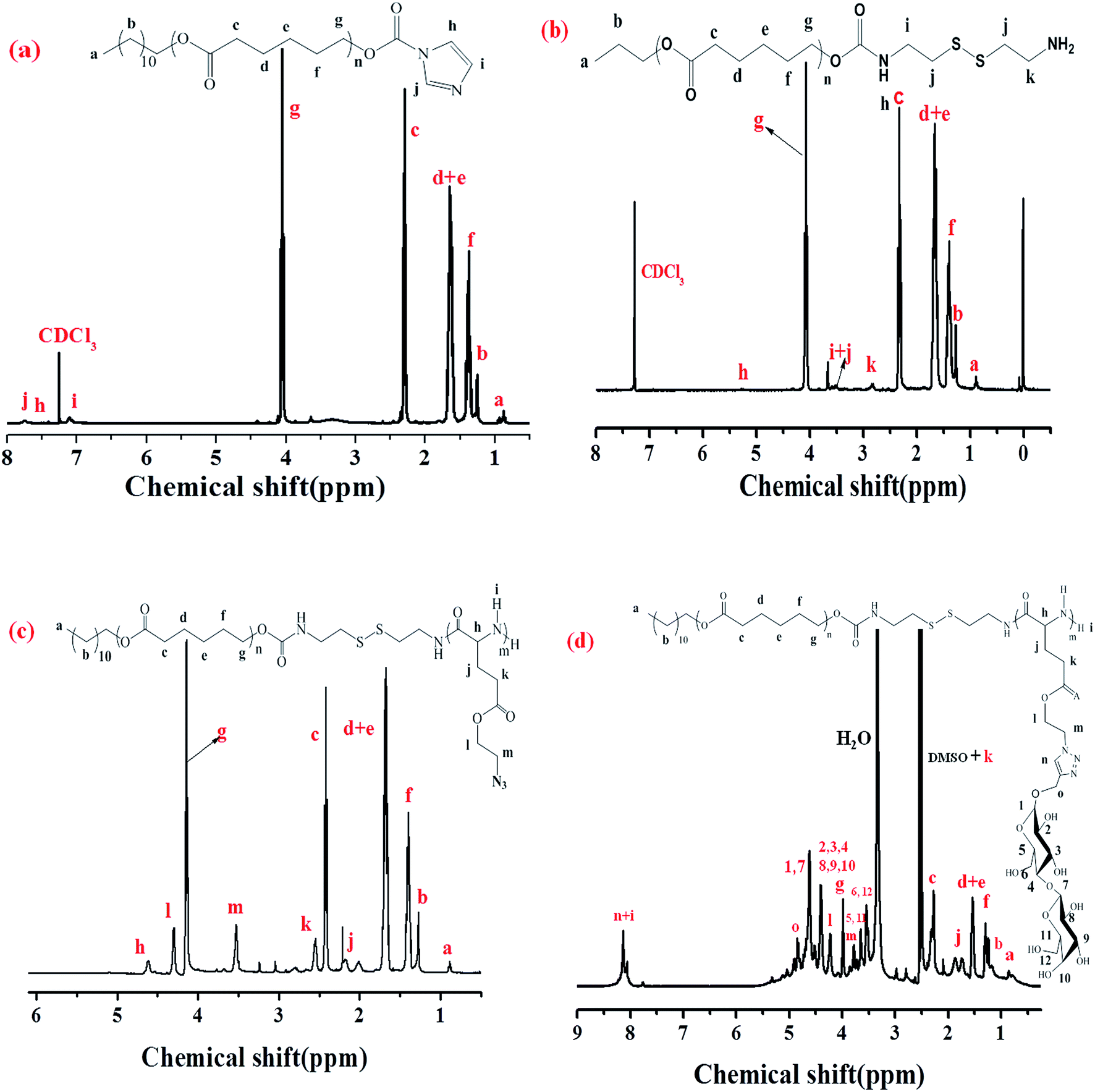
The diblock copolymers were synthesized by the combination of sequential ROP of the ε-CL and AELG-NCA monomers follow by the click conjugation of the saccharide units on the polypeptide blocks (see Scheme 2). Generally, PCL prepared by ε-CL ROP have a well-defined chain length, low polydispersity index, and a hydroxy end-group. The hydroxyl end-group can be modified into an amine group and thus acts as an initiator for NCA ROP. The precursors with the terminal amine group and disulfide bonds were prepared from the PCL–CI reacting with cystamine. The obtained PCL with the terminal amine were then reacted with AELG-NCA in DMF to obtain PCL–SS–PAELG. Fig. 1a and b display the 1H NMR spectra of PCL terminated with carbonylimidazole and amine groups, respectively. In this paper, the PCL–NH2 with a polymerization degree of 37 was used as a macroinitiator to polymerize AELG-NCA. As shown in Fig. 1c, the composition of the final block of PCL–SS–PAELG was estimated based on the integrals at 4.1 and 3.5 ppm from the corresponding spectrum for the PCL and PAELG residues, respectively. Alkyne and azide click conjugation was widely used as an effective synthetic method to form highly functionalized polypeptides. In our experiment, the PCL–SS–glycopolypeptides were obtained through a click conjugation between propargylgalactose/propargyllactose with PCL–SS–PAELG in DMSO at 45 °C using CuSO4/sodium ascorbate as a catalyst. 1H NMR spectra shown in Fig. 1d revealed new signals from the sugar. In addition, the characteristic signal at 8.0 ppm is attributed to the proton from the triazole ring. According to the 1H NMR spectrum of PCL37–SS–PGluLac10, the resonances for the triazole protons (h, Fig. 1d) are integrated in an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ratio relative to the polypeptide side chains (c–f of Fig. 1b), suggesting a nearly quantitative grafting efficiency. The high grafting efficiency observed from these polymers is consistent with the existing reports on the synthesis of glycolpolypeptides.
:6 ratio relative to the polypeptide side chains (c–f of Fig. 1b), suggesting a nearly quantitative grafting efficiency. The high grafting efficiency observed from these polymers is consistent with the existing reports on the synthesis of glycolpolypeptides.
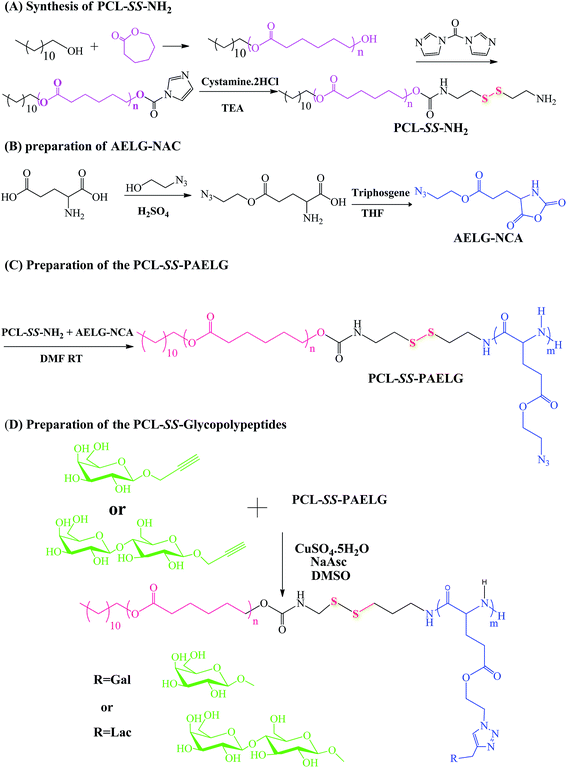 |
| | Scheme 2 Synthesis of amphiphilic PCL–SS–GPPs diblock copolymers. | |
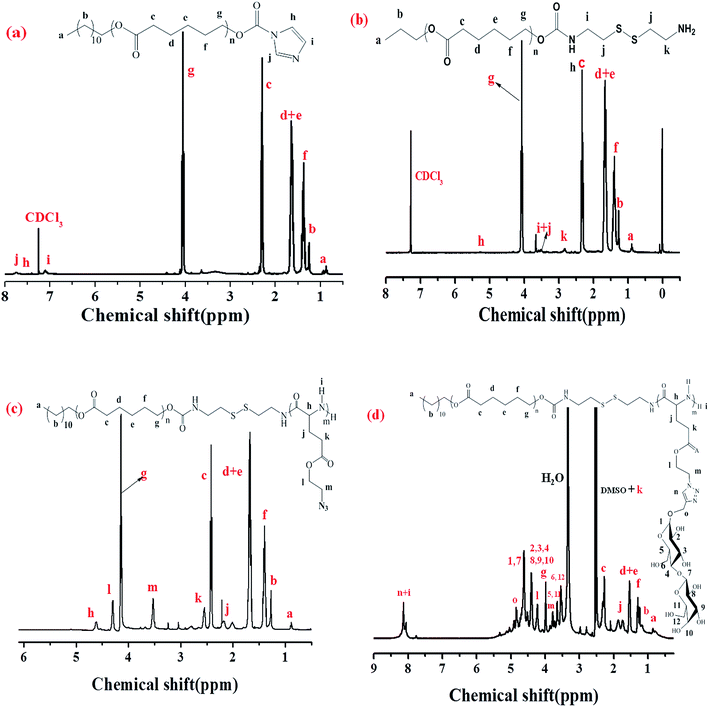 |
| | Fig. 1 1H NMR spectra of PCL–CI in CDCl3 (a), PCL–SS–NH2 in CDCl3 (b), PCL37–SS–PAELG10 in CDCl3 containing 15% TFA (c), and PCL37–SS–PGluLac10 in DMSO-d6 (d). | |
Fig. 2 shows the FTIR spectra of PCL–SS–GPPs and its precursors. As shown in the FTIR spectra, the characteristic absorption bands of PCL–SS–NH2 appear at 2934 cm−1 and 2841 cm−1 (ν C–H), and 1739 cm−1 (ν CO). The absorption bands attributed to PCL37–SS–PAELG10 appear at 3290 cm−1 (ν N–H), 2934 cm−1, and 2841 cm−1 (ν C–H), while the characteristic absorption peak of the azido group are located at 2115 cm−1 and 1739 cm−1 (ν CO), and the peaks at 1652 and 1547 cm−1 are ascribed to the amide I band (ν CO) and amide II band (ν N–H). The successful conjugation is evidenced by the fact that the characteristic azide absorbance peak at 2105 cm−1 disappeared in both spectra of PCL37–SS–PGluLac10 and PCL37–SS–PGluGal10, respectively, when compared with that of PCL37–SS–PAELG10. The characteristic absorption peak of the ester group at 1739 cm−1 from the PCL segment appears in the FTIR spectrum. Furthermore, the absorbance peak at 3400 cm−1 belongs to the hydroxy group of the galactosyl and lactosyl sugar units, which confirmed the click conjugation reaction. Taken together, both the FTIR and 1H NMR results confirmed the successful synthesis of the disulfide-bonds-linked PCL–SS–glycopolypeptides diblock copolymers.
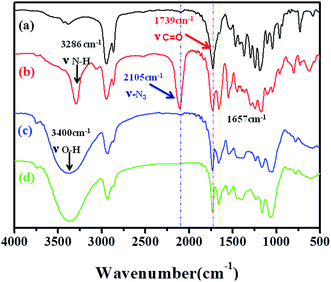 |
| | Fig. 2 FTIR spectra of PCL37–SS–NH2 (a), PCL37–SS–PAELG10 (b), PCL37–SS–PGluLac10 (c), and PCL37–SS–PGluGal10 (d). | |
3.2 Micelles formation and their properties
To evaluate the self-assembly of PCL-SS-GPPs diblock copolymers, the PCL–SS–GPPs micelles were prepared via the dialysis method. In this study, the self-assembly behavior in aqueous medium was investigated by using fluorometry in the presence of pyrene as a fluorescent probe, according to the method of Chen et al.42 It has been demonstrated that the variation in the intensity ratio (I337/I335) of the vibronic peak at 337 nm to the vibronic peak at 335 nm is quite sensitive to the polarity of the microenvironment where pyrene is located. The ratio of I337/I335 as a function of the concentration of the PCL–SS–GPPs micelles is shown in Fig. 3. It is shown that at lower micelles concentrations, the I337/I335 values remain nearly unchanged. As the micelles concentrations increase, the intensity ratio starts to increase, implying the formation of micellar self-aggregates with hydrophobic domains. The critical micelle concentration (CMC) could be determined by the crossover point of two straight lines, as indicated in Fig. 3. The CMC values for PCL37–SS–PGluLac10 was about 0.0166 mg mL−1, which was higher than the value of 0.0105 mg mL−1 for PCL37–SS–PGluGal10. In our experiment, the sugars were highly effectively grafted onto the same PCL37–SS–PAELG10. The lactose unit was more hydrophilic than the galactose unit, hence with a similar grafting efficiency, PCL37–SS–PGluLac10 was more hydrophilic than PCL37–SS–PGluGal10, resulting in a higher CMC value. The CMC results of our experiment are similar to the previous results reported by Lecommandoux et al.34
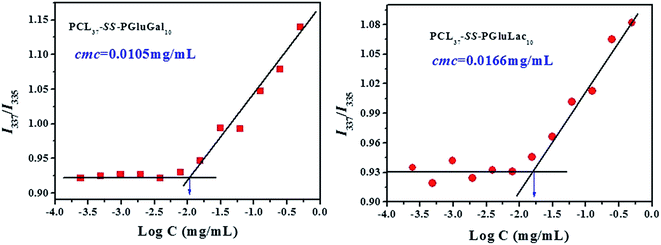 |
| | Fig. 3 Change in I337/I335 with the concentration of the PCL–SS–GPPs micelles. | |
Amphiphilic copolymers that can form reduction-sensitive micelles have attracted extensive attention for the active intracellular delivery of anticancer drugs and diagnostic agents. In this paper, the anticancer drug doxorubicin (DOX) and superparamagnetic iron oxide (SPIO) nanoparticles (NPs), as model therapeutic cargos and magnetic resonance imaging (MRI) contrast agents respectively, were simultaneously encapsulated in the hydrophobic core of the micelles by the dialysis method. The autocorrelation functions obtained from DLS measurements were analyzed using CONTIN analysis and were performed at a scattering angle varying from 40° to 130°. In Fig. 4, the dependence of the decay frequency, Γ, as a function of the square of the scattering vector q2 (q is defined as q = 4πnsin(θ/2)/λ, where n is the refractive index of the solution, q is the scattering angle, and λ = 632.8 nm is the wavelength of the incident laser light) was plotted. The linear variation of the relaxation frequency versus the squared scattering vector q2 passing through the origin is the hallmark of a translational diffusive process, confirming the presence of spherical objects with a pure diffusive mode.34,43 According to the multi-angle DLS analyses results, we could conclude that the PCL–SS–GPPs self-assembled in aqueous solution to form spherical micelles. Furthermore, for the SPIO and DOX-co-loaded PCL–SS–GPPs micelles, the linear variation of the relaxation frequency versus the squared scattering vector q2 passed through the origin too. Therefore, the PCL–SS–GPPs micelles still kept their the spherical form of the PCL-SS-GPPs micelles is well maintained in aqueous solution even after loading with SPIO and DOX.
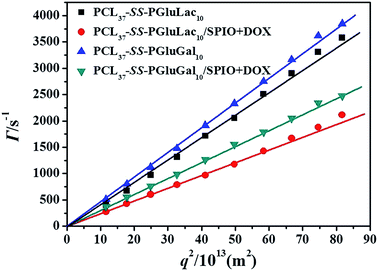 |
| | Fig. 4 Variation of decay rate vs. squared vector for aqueous systems of the blank and DOX/SPIO-co-loaded PCL–SS–GPPs micelles at the test angles ranging from 50° to 130°. | |
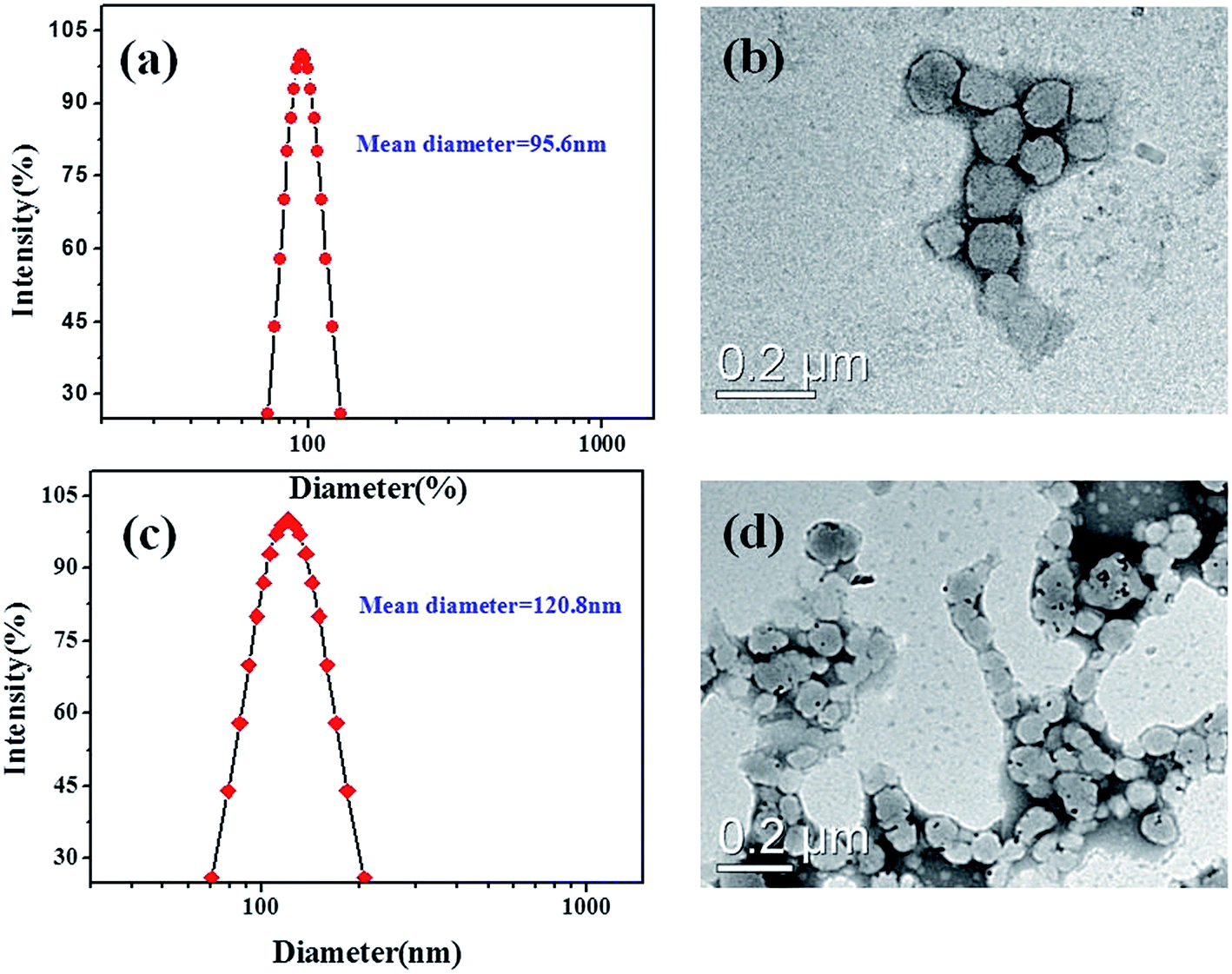
The hydrodynamic diameters of the resulting micelles were obtained by DLS analysis. As shown in Fig. 5a, the mean diameter (MD) for the empty PCL37–SS–PGluGal10 micellar aggregates was about 95.6 nm. TEM studies were performed to reveal the morphology of the micellar aggregates. Spherical-like aggregates were found in the TME observations (Fig. 5c), which is in good agreement with the multi-angle DLS analysis results. DLS and TEM measurements were also applied to determine the mean diameter and the morphology of the DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles. Dynamic light scattering (DLS) measurements (Fig. 5c) showed that the mean diameter was 120.8 nm for DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles. The mean diameter of the empty and the DOX/SPIO-loaded PCL37–SS–PGluLac10 was also determined by DLS (Table 1). The TEM image of the DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles (Fig. 5d) shows that they are uniform in shape and have uniform size distribution. From Table 1, we find that compared to the empty PCL–SS–GPPs micelles, the SPIO and DOX-co-loaded micelles shown a bigger size. The increase in micelle sizes might be due to the hydrophobic DOX and SPIO being encapsulated into the hydrophobic core of the micelles.
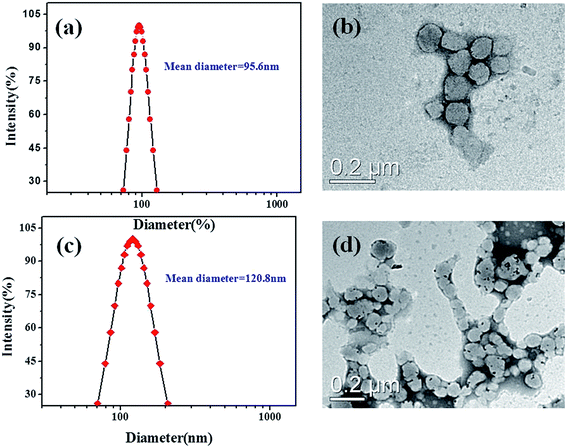 |
| | Fig. 5 Typical particle size distribution (a) and TEM image (b) of the blank PCL37–SS–PGluGal10 micelles; particle size distribution (c) and TEM image (d) of the DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles. | |
Table 1 Characteristics of the blank and DOX/SPIO-loaded micellesa
| Sample code |
Blank micelles |
DOX/SPIO-loaded micelles |
| Diameterb (nm) |
PDIb |
CMCc (mg mL−1) |
Diameterb (nm) |
PDIb |
DLC (%) |
SLC (%) |
| All the aggregate solutions had a final polymer concentration of 0.50 mg mL−1. Measured by dynamic light scattering (DLS). Determined by pyrene-based fluorescent spectrometry. |
| PCL37–SS–PGluGal10 |
95.6 ± 2.4 |
0.07 ± 0.01 |
0.0105 |
120.8 ± 1.7 |
0.12 ± 0.01 |
12.3 |
10.7 |
| PCL37–SS–PGluLac10 |
102.4 ± 0.7 |
0.19 ± 0.02 |
0.0116 |
150.1 ± 2.6 |
0.15 ± 0.03 |
10.2 |
11.3 |
3.3 Bioactivity assay
The bioactivity of the pendant galactosyl units in the PCL–SS–GPPs diblock copolymers were assessed by carbohydrate–lectin binding experiments. It has been reported that lectin would specifically and selectively bind to galactosyl groups.44 In order to observe the binding interaction between lectin and the galactosyl residues more directly, we chose lectin conjugated with FITC (LEC-FITC) for the binding study. The PCL–SS–GPPs films were prepared and treated with LEC-FITC solution and then the films were imaged with an inverted fluorescence microscope (Fig. 6). Green fluorescence due to the FITC-labeled LEC was observed from the PCL–SS–GPPs films, while no fluorescence was observed from the PCL–SS–GPPs films without treatment with LEC-FITC. Furthermore, the binding assay of the PCL–SS–GPPs films with FITC and PCL–SS–PAELG films with LEC-FITC were carried out. As displayed in Fig. S1,† these films did not exhibit green fluorescence as compared to the PCL–SS–GPPs/LEC-FITC. The binding results indicated that the glycopolypeptides can bind specifically to the lectin.45
 |
| | Fig. 6 Inverted fluorescence microscope images of cast films of PCL37–SS–PGluGal10 (a), PCL37–SS–PGluGal10/LEC-FITC (b), PCL37–SS–PGluLac10 (c), and PCL37–SS–PGluLac10/LEC-FITC (d). | |
3.4 In vitro stimuli-responsive drug release
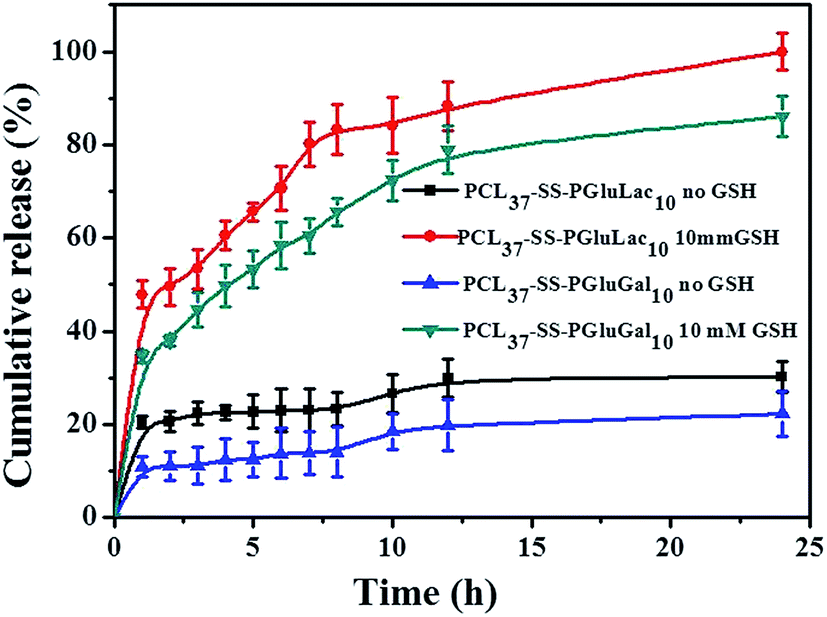
Recently, reduction-sensitive nanocarriers have attracted much interest for active intracellular cargo release due to a high GSH concentration in cancer cells.46 DOX was loaded into the nanocarriers, and the release behavior of the loaded DOX from the PCL–SS–GPPs micelles without and with the exposure to GSH was evaluated in PBS at pH 7.4, as shown in Fig. 7. In the case of without GSH, the DOX/SPIO-loaded micelles showed a sustained and slow release. For instance, only about 22% and 35% of the DOX were released in 24 h from the PCL37–SS–PGluLac10 and PCL37–SS–PGluGal10 micelles, respectively. In contrast, when 10 mM GSH was employed to trigger the release of DOX, a concentration that was similar to the intracellular GSH concentration, the DOX/SPIO-loaded micelles showed a burst release, in which 58.36% and 70.66% of the DOX was released in the initial 6 h. The amount of DOX release reached 81% and complete release was observed within 24 h. According to the above release results, the rapid release of DOX could be attributed to the cleavage of disulfide linkages by GSH consistent with results observed by Zhong and co-workers.46,47 This reduction control release behavior is of particular interest in achieving tumor-targeted DOX delivery. It is expected that the loaded drug will remain in the nanocarriers during the circulation time in the bloodstream, where the concentration of GSH is about 2 μM. After the vesicles are taken up by the cells into their cytoplasm where the GSH is about 1–10 mM, faster DOX release occurs once the disulfide bonds are cleaved by the GSH (Fig. 7).
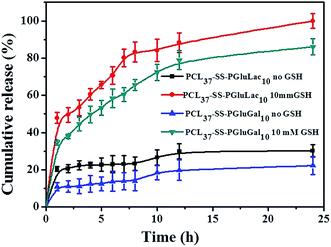 |
| | Fig. 7 The in vitro release behavior of loaded DOX from PCL–SS–glycopolypeptides micelles in the absence and presence of GSH (37 °C, pH 7.4). | |
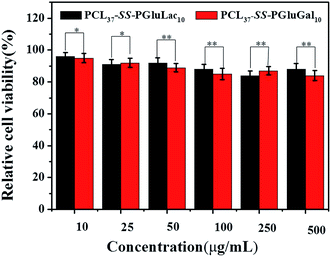 |
| | Fig. 8 In vitro cytotoxicity studies of blank PCL–SS–GPPs micelles against HepG2 cells lines at different concentrations after 48 h incubation period (*p < 0.05, **p < 0.01). | |
3.5 In vitro cell cytotoxicity and cellular uptake
MTT assay was performed to investigate the in vitro cytotoxicity of the blank PCL–SS–GPPs micelles toward HepG2 cell lines. The cells were incubated with PCL–SS–GPPs micelles at various concentrations for 48 h. The in vitro cell proliferation assay revealed that the viabilities of the HepG2 cells were over 80%, even with the PCL–SS–GPPs micelles concentrations up to 500 μg mL−1, confirming that these biodegradable PCL–SS–GPPs micelles have excellent biocompatibility (Fig. 8).
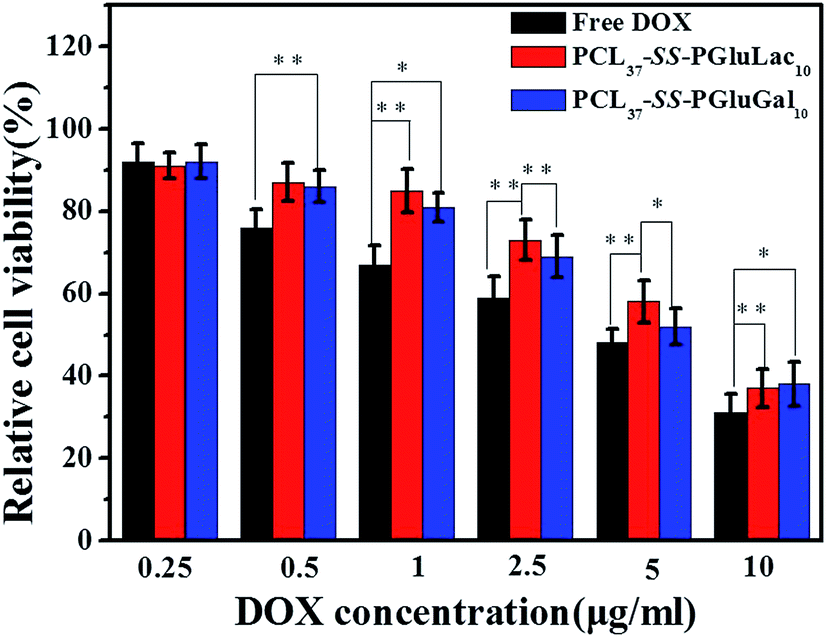
To demonstrate the potential antitumor utility of the PCL–SS–GPPs micelles, the use of HepG2 cell lines was employed to investigate the cytotoxicity of the DOX/SPIO-loaded PCL37–SS–PGluGal10 and PCL37–SS–PGluLac10 micelles, with free DOX tested as a positive control. HepG2 cells were treated with free DOX and DOX/SPIO-loaded micelles with various equivalent DOX concentrations from 0.01 to 10 μg mL−1. After incubation for 48 h, the cell viability was evaluated by MTT assay. From the MTT assay results, we could conclude that the viability of HepG2 cells was mainly dependent on the DOX concentrations. As the DOX concentrations increased, the viability of the HepG2 cells incubated with both free DOX and DOX/SPIO-loaded micelles decreased. Furthermore, compared to the DOX loaded into the micelles, the free DOX showed the highest cytotoxicity against the HepG2 cells, regardless of the DOX concentration. DOX is a widely used anticancer drug, but direct administration of DOX can induce many side-effects, such as cardiotoxicity.48 Consequently, DOX is loaded into micelles, which not only prolongs the release profiles, it gives higher therapeutic effects and reduced side-effects.49 Previous studies revealed that galactose ligand exhibits high specificity to asialoglycoprotein receptor (ASGP-R)-overexpressing hepatoma cells, including HepG2 cells.50 PCL–SS–GPPs micelles containing galactose or a lactose pendent unit can act as multifunction nanocarriers with the ability to target the ASGP-R-overexpressing hepatoma cells and to then release the antitumor drugs under the reduction microenvironment of the hepatoma cells (Fig. 9).
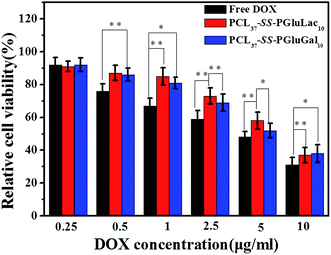 |
| | Fig. 9 Cytotoxicity of the free DOX, DOX/SPIO-loaded PCL37–SS–PGluGal10, and PCL37–SS–PGluLac10 micelles against HepG2 cells at different DOX concentrations (incubation time 48 h) (*p < 0.05, **p < 0.01). | |
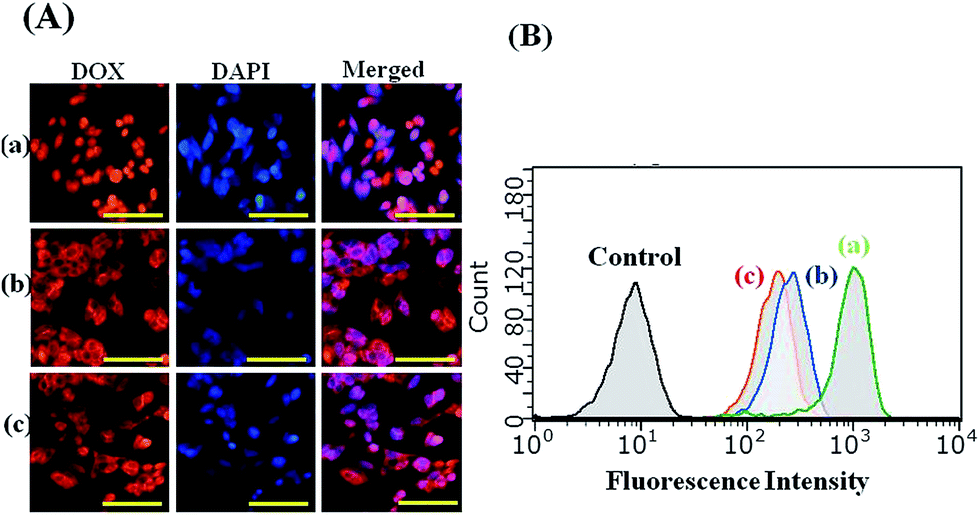
The fluorescence signal of DOX allows us to follow their internalization and intracellular distribution by using fluorescence microscopy and flow cytometry. For the fluorescence microscopy analysis, the HepG2 cells lines were incubated with free DOX and DOX/SPIO-loaded micelles for 4 h at a DOX concentration of 10 μg mL−1. As shown in Fig. 10, strong DOX fluorescence appeared in the perinuclear as well as in the nuclear regions of the cells, possibly due to the fast internalization of DOX inside the cells and intercalation of the chromosomal DNA. It should be noted that the HepG2 cells incubated with free DOX showed higher DOX fluorescence intensity than those incubated with DOX-loaded micelles. By merging the fluorescence microscopy pictures of DOX and DAPI, we found that free DOX mainly accumulated in the cell nucleus, whereas for the DOX delivered by PCL–SS–glycopolypeptides micelles, the DOX fluorescence mainly appeared in the cytoplasm and only partly in the cell nucleus. It has been reported that free DOX transported into cells occurs through a passive diffusion mechanism, while DOX-loaded micelles are internalized in the cells by an endocytosis mechanism.49,51 Hence, the PCL–SS–glycopolypeptides micelles could act as an efficient vehicle to transport DOX into the cytoplasm via an endocytic pathway.
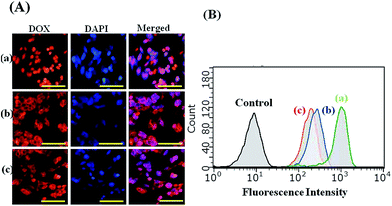 |
| | Fig. 10 (A) Fluorescence microscope images of HepG2 cells incubated with free DOX (a), DOX/SPIO-loaded PCL37–SS–PGluLac10 (b), and DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles (c) for 4 h at 37 °C. Scale bars represent 200 μm in all the images. (B) Flow cytometry plots for the control, free DOX (a), DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles (b), and DOX/SPIO-loaded PCL37–SS–PGluLac10 micelles (c) in HepG2 cell lines at 37 °C for 4 h, as measured by flow cytometry. | |
Flow cytometry was used for quantitative determination of the DOX levels in HepG2 cells. From the flow cytometry analysis results, we found that the highest fluorescence intensity was observed in the HepG2 cells incubated with free DOX, compared to the cells incubated with DOX-loaded micelles for 4 h, which was consistent with the cellular uptake behavior observed by fluorescence microscopy and this could be attributed to the different cellular uptake mechanisms. Moreover, HepG2 cells treated with DOX-loaded PCL37–SS–PGluGal10 micelles had a higher DOX level than those treated with PCL37–SS–PGluLac10 micelles.
In order to reveal the hepatocyte-targeting ability of the obtained PCL–SS–GPPs, a galactose competitive inhibition assay was performed. Invert fluorescence microscopy analysis (Fig. S2†) showed the red fluorescence of DOX in the HepG2 cells. The HepG2 cells without pretreatment with galactose showed the strongest red fluorescence, as compared to the pretreated galactose HepG2 cells. Furthermore, flow cytometry analysis (Fig. S3†) was carried to quantify the fluorescence intensity. The flow cytometry analysis results revealed that as the galactose concentration increased from 0, 10, 50, to 100 mM, the DOX fluorescence intensity decreased from 167.62, 101.16, 89.40, to 75.37, respectively.
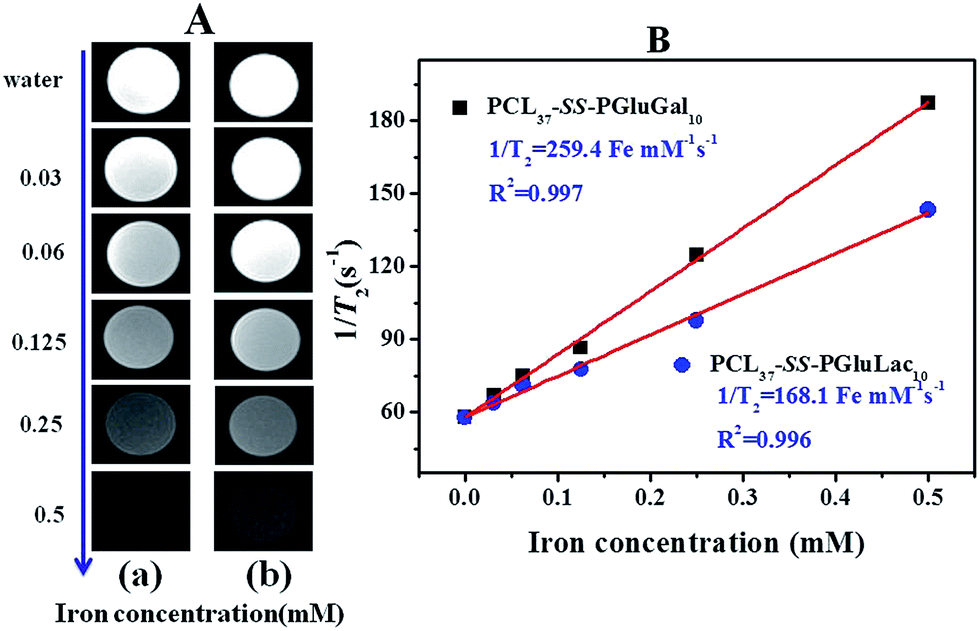
SPIO nanoparticles are capable of shortening the transverse relaxation time of water protons and their net effectiveness is expressed by T2 relaxivity (r2), which represents the reciprocal of the relaxation time per unit concentration of metal ions.52 In vitro MRI experiments were carried out in a 3.0T Siemens MRI scanner at room temperature. T2-Weighted images of the DOX/SPIO-loaded PCL–SS–GPPs micelles at various iron concentrations are shown in Fig. 11A, a remarkable darkening appeared with the increasing concentration of micelles. Fig. 11B displays the T2 rates of SPIO/DOX-loaded micelles over iron concentrations. The T2 relaxivity increased dramatically with iron concentration. Relaxivity can be calculated through the linear least-squares fitting of 1/relaxation time (s−1) versus the iron concentration (mM Fe). According to our measurements, we noted a steeper slope in Fig. 11B for PCL37–SS–PGluGal10 (r2 = 259.4 Fe m M−1 s−1) as compared to PCL37–SS–PGluLac10 (r2 = 168.1 Fe mM−1 s−1). The r2 value of the SPIO-loaded particles was affected by many factors, such as particle size, magnetic field, compositions of the SPIO nanocrystals, and the degree of SPIO clustering.53 Loading the SPIO into the hydrophobic inner core shortens the distant of each SPIO and leads to an increase of r2. SPIO/DOX-loaded PCL37–SS–PGluGal10 micelles thus showed a smaller size, hence they had higher r2 values as compared to SPIO/DOX-loaded PCL37–SS–PGluLac10 micelles. The r2 values for the DOX/SPIO-loaded PCL–SS–GPPs micelles were also larger than for Feridex® (111.5 Fe mM−1 s−1), a commercially available SPIO-based MRI contrast agent widely used in clinical MRI (Fig. 11).54
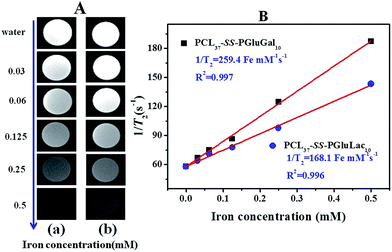 |
| | Fig. 11 (A) T2-Weighted MRI images of the DOX/SPIO-loaded PCL37–SS–PGluGal10 micelles (a) and PCL37–SS–PGluLac10 micelles (b). (B) Relaxation rates (s−1) of T2 as a function of iron concentration (mM) for the DOX/SPIO-loaded PCL37–SS–PGluGal10 and PCL37–SS–PGluLac10 micelles. | |
4. Conclusion
In this work, disulfide-bonds-linked PCL–SS–GPPs polymers were synthesized. The PCL–SS–GPPs, including PCL37–SS–PGluGal10 and PCL37–SS–PGluLac10, formed stable nano-sized micelles in an aqueous medium. The binding tests showed a specific recognition between the sugars on the surface of the PCL–SS–GPPs films with the FITC-labeled lectin. Furthermore, the T2 contrast agent hydrophobic superparamagnetic Fe3O4 and the anticancer drug doxorubicin were simultaneously loaded into the hydrophobic inner cores of the micelles. The DOX/SPIO-loaded micelles exhibited rapid drug release behavior in response to the intracellular level of reducing potential (10 mM GSH). MTT results of the DOX-loaded micelles showed an obvious proliferation inhibition effect on HepG2 tumor cells. Fluorescence images and flow cytometry tests revealed that DOX could be efficiently transported into HepG2 tumor cells by PCL–SS–GPPs micelles. The SPIO-loaded micelles showed increased MRI sensitivity and relaxivity. Consequently, these reduction-sensitive amphiphilic PCL–SS–GPPs showed significant potential as theranostic nanocarriers for MR imaging and chemotherapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81571665), the Natural Science Foundation of Guangdong Province (2014A030313647), the Science and Technology Plan Foundation of Guangzhou (201510010263, 1563000477, 201510010086, 201607010038), the Social Development Fund of Guangdong Province (2014A020212031). The authors thank Nian-Hua Wang for her assistance in data processing.
References
- D. T. Leong and K. W. Ng, Adv. Drug Delivery Rev., 2014, 79, 95–106 CrossRef PubMed.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., 2009, 48, 872–897 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- S. R. Croy and G. S. Kwon, Curr. Pharm. Des., 2006, 12, 4669–4684 CrossRef CAS PubMed.
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS PubMed.
- A. Zhang, Z. Zhang, F. Shi, J. Ding, C. Xiao, X. Zhuang, C. He, L. Chen and X. Chen, Soft Matter, 2013, 9, 2224–2233 RSC.
- X. Zhang, J. Chen, Z. Luo, D. Wu, X. Shen, S. Song, Y. Sun, P. Liu, J. Zhao and S. Huo, Adv. Healthcare Mater., 2013, 3, 133–141 CrossRef PubMed.
- X. Yang, Y. Chen, R. Yuan, G. Chen, E. Blanco, J. Gao and X. Shuai, Polymer, 2008, 49, 3477–3485 CrossRef CAS.
- X. Wang, Y. Yang, Y. Zhuang, P. Gao, F. Yang, H. Shen, H. Guo and D. Wu, Biomacromolecules, 2016, 17, 2920–2929 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 6065–6075 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, D. A. Steeber and S. Gong, Biomaterials, 2010, 31, 9065–9073 CrossRef CAS PubMed.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- N. Song, M. Ding, Z. Pan, J. Li, L. Zhou, H. Tan and Q. Fu, Biomacromolecules, 2013, 14, 4407–4419 CrossRef CAS PubMed.
- L. Y. Rizzo, B. Theek, G. Storm, F. Kiessling and T. Lammers, Curr. Opin. Biotechnol., 2013, 24, 1159–1166 CrossRef CAS PubMed.
- W. Wang, D. Cheng, F. Gong, X. Miao and X. Shuai, Adv. Mater., 2012, 24, 115–120 CrossRef CAS PubMed.
- U. Y. Lee, Y. T. Oh, D. Kim and E. S. Lee, Int. J. Pharm., 2014, 471, 166–172 CrossRef CAS PubMed.
- W. Chen, Y. Zou, J. Jia, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Macromolecules, 2013, 46, 699–707 CrossRef CAS.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS PubMed.
- P. Ma, S. Liu, Y. Huang, X. Chen, L. Zhang and X. Jing, Biomaterials, 2010, 31, 2646–2654 CrossRef CAS PubMed.
- W. Chen, Y. Zou, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2014, 15, 900–907 CrossRef CAS PubMed.
- D. Pati, A. Y. Shaikh, S. Das, P. K. Nareddy, M. J. Swamy, S. Hotha and S. S. Gupta, Biomacromolecules, 2012, 13, 1287–1295 CrossRef CAS PubMed.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, J. Am. Chem. Soc., 2014, 136, 2008–2016 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, Polym. Chem., 2013, 5, 671–682 RSC.
- K. Upadhyay, J. F. L. Meins, A. Misra, P. Voisin, V. Bouchaud, E. Ibarboure, C. Schatz and S. Lecommandoux, Biomacromolecules, 2009, 10, 2802–2808 CrossRef CAS PubMed.
- C. Schatz, S. Louguet, J. L. Meins and S. Lecommandoux, Angew. Chem., 2009, 48, 2572–2575 CrossRef CAS PubMed.
- J. R. Kramer, A. R. Rodriguez, U. Choe, D. T. Kamei and T. J. Deming, Soft Matter, 2013, 9, 3389–3395 RSC.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC.
- A. C. Engler, H. Lee and P. T. Hammond, Angew. Chem., 2009, 48, 9334–9338 CrossRef CAS PubMed.
- C. Bonduelle and S. Lecommandoux, Biomacromolecules, 2013, 14, 2973–2983 CrossRef CAS PubMed.
- T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed.
- C. Xiao, C. Zhao, P. He, Z. Tang, X. Chen and X. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef CAS PubMed.
- H. Tang and D. Zhang, Biomacromolecules, 2010, 11, 1585–1592 CrossRef CAS PubMed.
- K. Krannig, J. Sun and H. Schlaad, Biomacromolecules, 2014, 15, 978–984 CrossRef CAS PubMed.
- C. Gauche and S. Lecommandoux, Polymer, 2016, 107, 474–484 CrossRef CAS.
- J. Huang, C. Bonduelle, J. Thevenot, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2012, 134, 119–122 CrossRef CAS PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS PubMed.
- D. Pati, S. Das, N. G. Patil, N. Parekh, D. H. Anjum, V. Dhaware, A. V. Ambade and S. S. Gupta, Biomacromolecules, 2016, 17, 466–475 CrossRef CAS PubMed.
- Z. Wang, R. Sheng, T. Luo, J. Sun and A. Cao, Polym. Chem., 2017, 8, 472–484 RSC.
- H.-K. Yang, M. Qi, L. Mo, R.-M. Yang, X.-D. Xu, J.-F. Bao, W.-J. Tang, J.-T. Lin, L.-M. Zhang and X.-Q. Jiang, RSC Adv., 2016, 6, 114519–114531 RSC.
- T. Borase, T. Ninjbadgar, A. Kapetanakis, S. Roche, R. Oconnor, C. Kerskens, A. Heise and D. F. Brougham, Angew. Chem., 2013, 52, 3164–3167 CrossRef CAS PubMed.
- X. Jiang, J. Liu, L. Xu and R. Zhuo, Macromol. Chem. Phys., 2011, 212, 64–71 CrossRef CAS.
- S. Yu, C. He, J. Ding, Y. Cheng, W. Song, X. Zhuang and X. Chen, Soft Matter, 2013, 9, 2637–2645 RSC.
- J. Rodriguezhernandez, J. Babin, B. Zappone and S. Lecommandoux, Biomacromolecules, 2005, 6, 2213–2220 CrossRef CAS PubMed.
- R. Mildner and H. Menzel, Biomacromolecules, 2014, 15, 4528–4533 CrossRef CAS PubMed.
- S. Das, D. Pati, N. Tiwari, A. Nisal and S. S. Gupta, Biomacromolecules, 2012, 13, 3695–3702 CrossRef CAS PubMed.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS PubMed.
- Y. Zhong, W. Yang, H. Sun, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 3723–3730 CrossRef CAS PubMed.
- S. Turakhia, C. D. Venkatakrishnan, K. Dunsmore, H. R. Wong, P. Kuppusamy, J. L. Zweier and G. Ilangovan, Am. J. Physiol.: Heart Circ. Physiol., 2007, 293, H3111–H3121 CrossRef CAS PubMed.
- K. K. Upadhyay, A. N. Bhatt, A. K. Mishra, B. S. Dwarakanath, S. Jain, C. Schatz, J.-F. Le Meins, A. Farooque, G. Chandraiah, A. K. Jain, A. Misra and S. Lecommandoux, Biomaterials, 2010, 31, 2882–2892 CrossRef CAS PubMed.
- X. Wang, H. Sun, F. Meng, R. Cheng, C. Deng and Z. Zhong, Biomacromolecules, 2013, 14, 2873–2882 CrossRef CAS PubMed.
- X. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS PubMed.
- H. Su, Y. Liu, D. Wang, C. Wu, C. Xia, Q. Gong, B. Song and H. Ai, Biomaterials, 2013, 34, 1193–1203 CrossRef CAS PubMed.
- J. Lu, S. Ma, J. Sun, C. Xia, C. Liu, Z. Wang, X. Zhao, F. Gao, Q. Gong and B. Song, Biomaterials, 2009, 30, 2919–2928 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01440f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Guan-Hai Wangb,
Li-Ming Zhang*c and
Xin-Qing Jiang
b,
Guan-Hai Wangb,
Li-Ming Zhang*c and
Xin-Qing Jiang