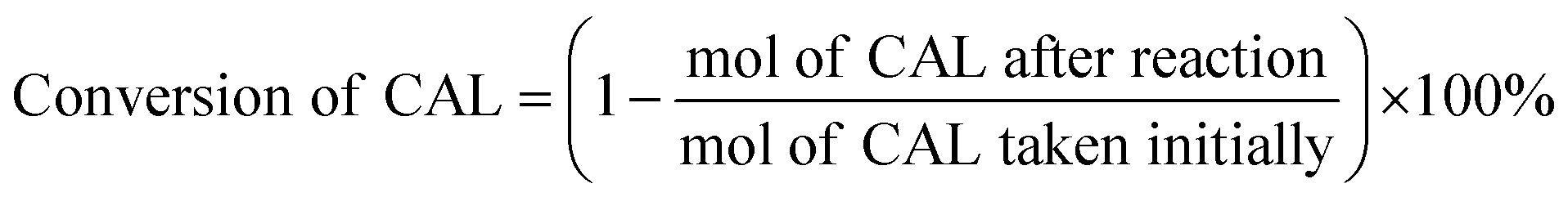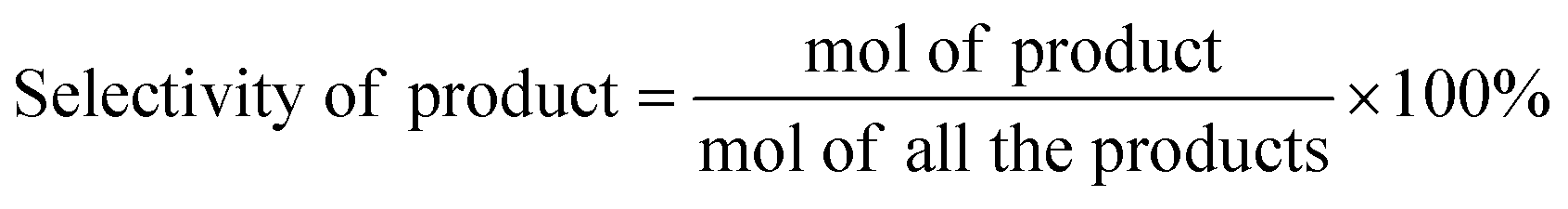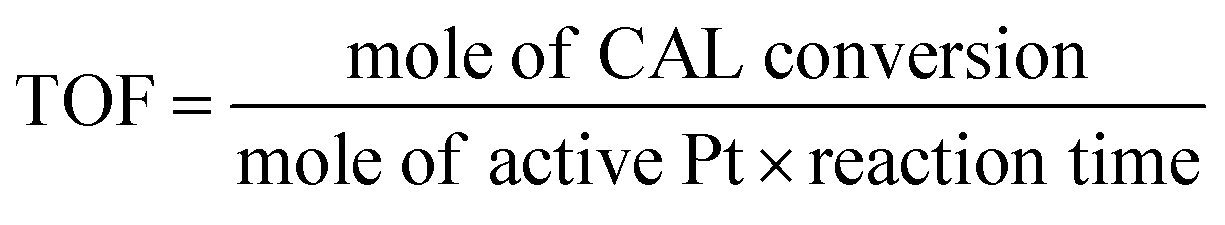DOI:
10.1039/C7RA02898A
(Paper)
RSC Adv., 2017,
7, 21107-21113
One-pot redox synthesis of Pt/Fe3O4 catalyst for efficiently chemoselective hydrogenation of cinnamaldehyde†
Received
10th March 2017
, Accepted 6th April 2017
First published on 12th April 2017
Abstract
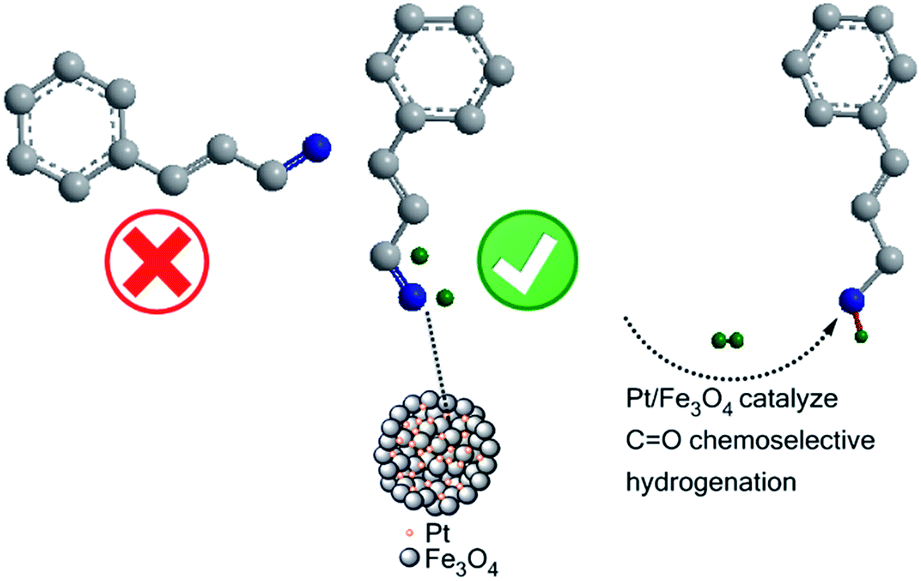
External additives (i.e., ionic electride donors) are usually necessary to obtain a high catalytic performance in an electron-density-dependent chemoselective hydrogenation reaction. Here, a sphere-shaped Pt/Fe3O4 catalyst is prepared through a one-pot redox reaction using Pt(IV) and Fe(II) precursors. The experimental results show that the prepared Pt/Fe3O4 catalyst with an Fe to Pt mole ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 can afford a high conversion of 94.2% towards cinnamaldehyde (CAL) and good selectivity of 92.2% towards cinnamyl alcohol (COL) under the mild conditions of 303 K, 5 bar H2 for 150 min in the chemoselective reduction of CAL. The excellent catalytic performance could be attributed to the dissociation of H2 and adsorption of CAL via a vertical configuration modulated by the electronic effect between Pt and Fe3O4, and the abundant available active sites induced by uniformly dispersed Pt nanoparticles. Additionally, the intrinsic magnetism of Fe3O4 nanospheres endows the catalyst with a rapid separation property, favourable for repeated use of a catalyst.
:1 can afford a high conversion of 94.2% towards cinnamaldehyde (CAL) and good selectivity of 92.2% towards cinnamyl alcohol (COL) under the mild conditions of 303 K, 5 bar H2 for 150 min in the chemoselective reduction of CAL. The excellent catalytic performance could be attributed to the dissociation of H2 and adsorption of CAL via a vertical configuration modulated by the electronic effect between Pt and Fe3O4, and the abundant available active sites induced by uniformly dispersed Pt nanoparticles. Additionally, the intrinsic magnetism of Fe3O4 nanospheres endows the catalyst with a rapid separation property, favourable for repeated use of a catalyst.
1. Introduction
Chemoselective hydrogenation of α,β-unsaturated aldehydes to relevant unsaturated alcohols is of great concern because of the importance on the synthesis of pharmaceutical and flavour intermediates in both chemical and energy industries.1–5 One of the typical reactions is the reduction of cinnamaldehyde (CAL) to corresponding cinnamyl alcohol (COL). However, chemoselective hydrogenation of the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in CAL is particularly challenging because of the higher activation energy of the CO bond than that of the CC bond. In addition, the competitive adsorption mechanism between CO and CC bonds can also influence the selectivity of the target product. Over the past decades, considerable endeavors have been devoted to selective hydrogenation of the CO bond over supported-metal catalysts.5–10 It has been reported the nature of supported-metal particles plays a vital role in improving selectivity to COL.11,12 Zhu and co-workers developed a bimetallic PtxCoy nanoparticles (NPs) with high-index curved faces, on which the contact of hydrogen atom with active sites was more favorable than on the low-index planes due to the smaller diffusion energy barrier.13 In addition to the natural property of active metal, the interaction of active metal and support also exercises significant influences in heterogeneous catalysis process, like influencing adsorption manner of reactants, promoting or restraining of catalyst activity, and/or varying product distribution etc. The interaction including geometric and electronic effects has been widely reported to improve the catalytic performance of catalyst with high conversion and selectivity.11,14–21 For example, amino-functionalized Zr-based metal–organic framework (UiO-66-NH2) encapsulated Pt nanoclusters showed high selectivity of COL because the steric confinement of characteristic porous channel in UiO-66-NH2 restricted CC planar adsorption.15 Regardless of this steric confinement, the change of adsorption manner of CAL from horizontal mode to vertical mode due to the electronic repulsion interaction between electron-enriched meal particles and benzene ring of CAL can also enhance the selectivity towards COL.22,23 For instance, an approximate 91% selectivity to COL at nearly complete CAL conversion could be achieved owing to electron-enriched Pt surfaces in the presence of TiO2–SiO2 system.16 Although many efforts were focused on modulating the surface electron density of active metal phase by using external modifiers (e.g., alkali compounds, surface organic ligands, or ionic electride donor), it is cost-expensive and environment-harmful in term of practical industrial application.14,24–27 Besides, the intrinsic interaction between metal and support is also very complicated in multi-component heterogeneous catalytic system, still under debate. Simultaneously, the complex separation of catalysts from the reaction mixture is still an issue, possibly resulting in the deactivation of catalyst. On the basis of the above discussion, there is an urgent need to develop the rational catalysts consisting of active metal and support for highly efficient selective hydrogenation of CAL with simple and rapid separation property.
O) in CAL is particularly challenging because of the higher activation energy of the CO bond than that of the CC bond. In addition, the competitive adsorption mechanism between CO and CC bonds can also influence the selectivity of the target product. Over the past decades, considerable endeavors have been devoted to selective hydrogenation of the CO bond over supported-metal catalysts.5–10 It has been reported the nature of supported-metal particles plays a vital role in improving selectivity to COL.11,12 Zhu and co-workers developed a bimetallic PtxCoy nanoparticles (NPs) with high-index curved faces, on which the contact of hydrogen atom with active sites was more favorable than on the low-index planes due to the smaller diffusion energy barrier.13 In addition to the natural property of active metal, the interaction of active metal and support also exercises significant influences in heterogeneous catalysis process, like influencing adsorption manner of reactants, promoting or restraining of catalyst activity, and/or varying product distribution etc. The interaction including geometric and electronic effects has been widely reported to improve the catalytic performance of catalyst with high conversion and selectivity.11,14–21 For example, amino-functionalized Zr-based metal–organic framework (UiO-66-NH2) encapsulated Pt nanoclusters showed high selectivity of COL because the steric confinement of characteristic porous channel in UiO-66-NH2 restricted CC planar adsorption.15 Regardless of this steric confinement, the change of adsorption manner of CAL from horizontal mode to vertical mode due to the electronic repulsion interaction between electron-enriched meal particles and benzene ring of CAL can also enhance the selectivity towards COL.22,23 For instance, an approximate 91% selectivity to COL at nearly complete CAL conversion could be achieved owing to electron-enriched Pt surfaces in the presence of TiO2–SiO2 system.16 Although many efforts were focused on modulating the surface electron density of active metal phase by using external modifiers (e.g., alkali compounds, surface organic ligands, or ionic electride donor), it is cost-expensive and environment-harmful in term of practical industrial application.14,24–27 Besides, the intrinsic interaction between metal and support is also very complicated in multi-component heterogeneous catalytic system, still under debate. Simultaneously, the complex separation of catalysts from the reaction mixture is still an issue, possibly resulting in the deactivation of catalyst. On the basis of the above discussion, there is an urgent need to develop the rational catalysts consisting of active metal and support for highly efficient selective hydrogenation of CAL with simple and rapid separation property.
It has been verified that the high-valent reducible positive ionic species (Feδ+, Ceδ+, Snδ+, Zrδ+), as support or promoter additives, can effectively modulate the surface electron density of active metal phase and activate of terminal CO bond.1,11,28 For instance, the Sn4+ promoter on the surface of Pd/Co3O4 catalysts can combine and weaken the conjugated CO bond of CAL molecule, leading to the insertion of H atom dissociated on Pd surface.23 The high selectivity towards relevant unsaturated alcohol is closely related to reducibility of support and electron density of metal particles over Fe-based supported Au catalysts in the selective hydrogenation of benzalacetone.29 Tang and co-workers have theoretically and experimentally demonstrated that the reaction energy of −2.15 eV for CO groups is lower than that of −1.89 eV for CC groups for MIL-101(Fe)@Pt catalyst.21 They draw a conclusion that this sandwich Fe-based nanostructure with Pt nanoparticles immobilized between the MIL-101 core and shell is very favorable for the hydrogenation of CO groups over CC groups.21 Ai and co-workers have reported that Zn–Fe Layered double hydroxides or oxides supported Pt catalyst show the high selectivity towards COL with a good CAL conversion in the presence of divalent and trivalent species.30 Although these types of catalysts can seemly treat as a potential candidate for chemoselective hydrogenation of CAL, the relevant heterogeneous catalysts generally suffer from the predicaments of complicated synthesis processes as well as discontented catalyst stability. Therefore, further effort is still required to develop highly active, selective and steady catalysts that should also be facilely prepared for large-scale practical use. To achieve this, a synthesis strategy is recently developed to fabricate hybrid nanomaterials (e.g., Ag or Pt/CeO2 system) by in situ interaction of support material and metal ions without the addition of surfactants or organic compounds, exhibiting superior performance of CO oxidation31 or catalytic reduction activity of nitrophenol.32 However, such synthesis strategy has not been applied to fabricate Pt–Fe3O4 composites with rapid separation property by magnetic field for highly efficient chemoselective hydrogenation of CAL to produce COL.
Upon considering that the high-valent reducible positive Fe ionic species can effectively reduce Pt ions, we report a facile one-pot route for preparation of Pt/Fe3O4 catalysts by an in situ redox method using Pt(IV) and Fe(II) salts as reaction precursors (Fig. S1, ESI†). Utilizing this spontaneous redox reaction between Pt(IV) and Fe(II) in aqueous solution, sphere-shape Fe3O4 structures with uniformly dispersed Pt nanoparticles can be obtained at 343 K to form Pt/Fe3O4 hybrid. Profiting from the excellent electron modulation property of the iron oxide between divalent and trivalent, the electron distribution and density of metal Pt surface could be also effectively regulated, thus resulting in significantly improved catalytic activity of Pt/Fe3O4 catalyst towards selective hydrogenation of CAL to COL. The experimental results show that the prepared Pt/Fe3O4 catalyst with Fe to Pt mole ratio of 100:1 can afford high conversion of 94.2% towards CAL and good selectivity of 92.2% towards COL under very mild reaction conditions of 303 K, 5 bar H2 for 150 min, simultaneously demonstrating high stability of the catalyst with rapid recycling property by magnetic field. Based on the experimental results, the process of chemoselective hydrogenation of CAL to COL on Pt/Fe3O4 catalyst has been proposed and discussed in detail.
2. Experimental section
2.1. Catalyst preparation
A series of Pt/Fe3O4 catalysts are prepared by a flexible one-pot method using the redox reaction between Pt(IV) and Fe(II) precursor. The diagram of preparation process has been depicted in Fig. S1 (ESI†). Generally, a 75 mM aqueous solution of FeSO4·7H2O is added into the calculated aqueous solution of NaOH under N2 protection. After vigorous stirring for 15 min, a certain aqueous solution of H2PtCl6·6H2O (1.5–15 mM, Aladdin, 37.5% Pt) is added quickly into the mixture. After being treated at 343 K for 4 h, the powder is separated and collected simply by a rectangle magnet after washing with deionization water and ethanol for several times, respectively. Then it is dried at 333 K overnight in a vacuum oven. The as-obtained samples are denoted as Pt/Fe3O4(x), where x is the mole ratio of Fe to Pt atom.
2.2. Catalyst characterization
TEM images and energy dispersive X-ray spectra (EDS) are performed on a JEOL-2010 EX instrument operating at 200 kV. The crystal phase structure of samples is analyzed by XRD equipped with Cu-Kα (λ = 0.154 nm) radiation operating at 40 kV and 40 mA for 2θ angles ranging from 10° to 75°. XPS measurement is recorded on an ESCALAB 250 photoelectron spectrometer (Thermo-VG Scientific Co., LTD) with Al Kα X-ray radiation as the X-ray source for excitation. The bonding energy of C 1s peak (284.8 eV) is referenced as a calibration. The actual Pt content of catalysts is determined by the inductively coupled plasma atomic emission spectrometry (ICP-AES). Before analysis, a certain amount of catalysts are digested in an inverse aqua regia solution (3:1 mixture of HNO3:HCl) under the assist of microwave radiation. The Pt dispersion (D) is determined by O2–H2 pulse titration with an AutoChem 2910 apparatus equipped with a thermal conductivity detector (TCD) for analysis, supported by the National Engineering Research Centre of Chemical Fertilizer Catalyst of Fuzhou University. The Pt dispersion is obtained from the amount of H2-chemisorption assuming a stoichiometric ratio H2:metal = 2:3 and the average particle size of Pt nanocrystallite is calculated by the equation: d (nm) = 1.1/D.33 The textural properties of catalysts are probed by the N2 adsorption–desorption measurements at 77 K using a Micrometrics Tristar 3020M. The pore volumes and pore size are obtained using the Barret–Joiner–Halenda (BJH) method.
2.3. Catalytic test
Typically, certain amount of substrate, 15 mg catalyst, and 5 mL solvent are added and sealed in 25 mL Teflon-lined steel autoclave vessel. After flushing with N2 for three times, the autoclave is pressurized to 5 bar H2 at room temperature and stirred under a continuous mechanical stirring equipped with a circle condensation device. The hydrogenation reaction is executed at 303 K for 150 min. After that, the liquid products are identified by gas chromatography mass spectrometry (GC-MS, Thermo Fisher Scientific-TXQ), and are quantitatively analyzed by GC (Shimadzu, GC-2010 Plus) using n-octanol as an internal standard. The used catalyst can be easily collected by an external magnet, washed by ethanol and acetone, and dried at 333 K for overnight in a vacuum oven. The recycling test is carried out under the condition of 303 K, 5 bar H2 for 10 min. In each run, the Pt/Fe3O4(x) catalyst is used directly without any pre-reduction treatment. The carbon balance has been checked in every run and it is found to be higher than 95%. CAL conversion and COL selectivity are calculated on the basis of equations as follows:| |
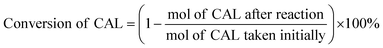 | (1) |
| |
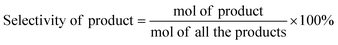 | (2) |
| |
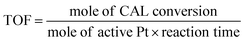 | (3) |
3. Results and discussion
3.1. Structural and textural properties of catalysts
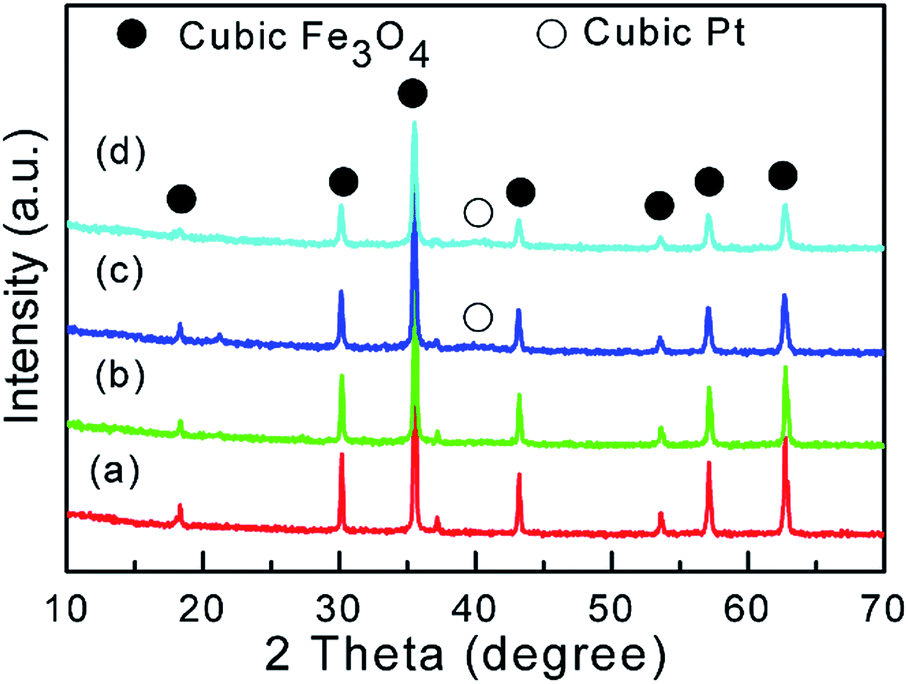
The hybrid Pt/Fe3O4(x) catalysts are fabricated by a facile redox reaction and denoted as Pt/Fe3O4(x), where x is the mole ratio of Fe to Pt atom. Briefly, the reducible FeO(OH)δ+ with partial positive charge can gradually attract the introduced negative ion of PtCl4− via their mutual electrostatic interaction under the strong alkaline medium. Deoxygenation process by the protection of N2 during this synthesis is very important in order to exclude the possibility that Fe2+ is oxidized by O2 instead by introduced Pt precursor. For the prepared sample, it almost consists of crystalline-like cubic Fe3O4 phase (JCPDS 88-0866), as confirmed by XRD patterns in Fig. 1. It is suggested that oxidation reaction of Fe(II) precursor takes place during the synthesis procedure. Contrast to obvious Fe3O4 phase, only a faint diffraction peak of (111) Pt plane located at 2θ of ∼40° can be observed over the Pt/Fe3O4(50) and Pt/Fe3O4(100) samples.
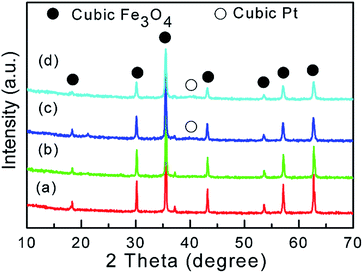 |
| | Fig. 1 XRD diffraction patterns of Pt/Fe3O4 catalysts: (a) Pt/Fe3O4(500); (b) Pt/Fe3O4(200); (c) Pt/Fe3O4(100); (d) Pt/Fe3O4(50). | |
It could be related to the low content of Pt or small amorphous Pt particles beyond the detection of XRD technique.
The actual Pt contents of catalysts are determined by the ICP-AES measurement and the relevant results are tabulated in Table 1. As can be seen, the real of Pt loading is 1.83%, 2.98%, and 6.52% for the Pt/Fe3O4(200), Pt/Fe3O4(100), and Pt/Fe3O4(50). Moreover, the real Pt loadings are slight higher than the nominal data due to the leaching of unreacted Fe(II) precursor in synthesis process.
Table 1 The textural properties of Pt/Fe3O4 catalysts
| Entry |
Catalysts |
Pt content wt% |
dFe3O4b (nm) |
DPtc (%) |
dPtd (nm) |
SBET (m2 g−1) |
Vp (cm3 g−1) |
| Nominal |
Actuala |
| Determined by ICP-AES analysis. Calculated by the Scherrer equation at 2θ of ∼35°. Determined by the H2–O2 pulse titration measurement. Calculated by the equation: dPt (nm) = 1.1/DPt. |
| 1 |
Pt/Fe3O4(200) |
1.24 |
1.83 |
55.7 |
16.2 |
6.8 |
20.2 |
0.3 |
| 2 |
Pt/Fe3O4(100) |
2.46 |
2.98 |
39.7 |
52.6 |
2.1 |
139.4 |
0.4 |
| 3 |
Pt/Fe3O4(50) |
4.80 |
6.52 |
34.7 |
37.5 |
2.9 |
72.6 |
0.3 |
The H2–O2 titration is considered as an effective method to determine dispersion and particle size of Pt NPs. As shown in Table 1, the catalyst with low content of Pt, for instance Pt/Fe3O4(200) sample, displays a poor dispersion of Pt NPs (DPt). It could be attributed to the inefficient adsorption of H2 on the surface of low Pt content, which is agreement with the results of Pd/CeO2 catalyst reported in our earlier work.34 With the increase of Pt content, the enhanced redox reaction between Pt(IV) and Fe(II) precursor can provide sufficient Pt active site, which is beneficial to H2 adsorption. Meanwhile, based on the dispersion of Pt, the mean particle size of Pt NPs (dPt) can also be provided.33 As displayed in Table 1, it is obvious found that the value of dPt gradually declines as the increase of Pt content. It can be deduced that the formed Fe3O4 derived from the oxidation of Fe(II) with Pt(IV) precursor can effectively encapsulate Pt NPs into its matrix simultaneously to prevent migration and agglomeration of Pt NPs into large particles.
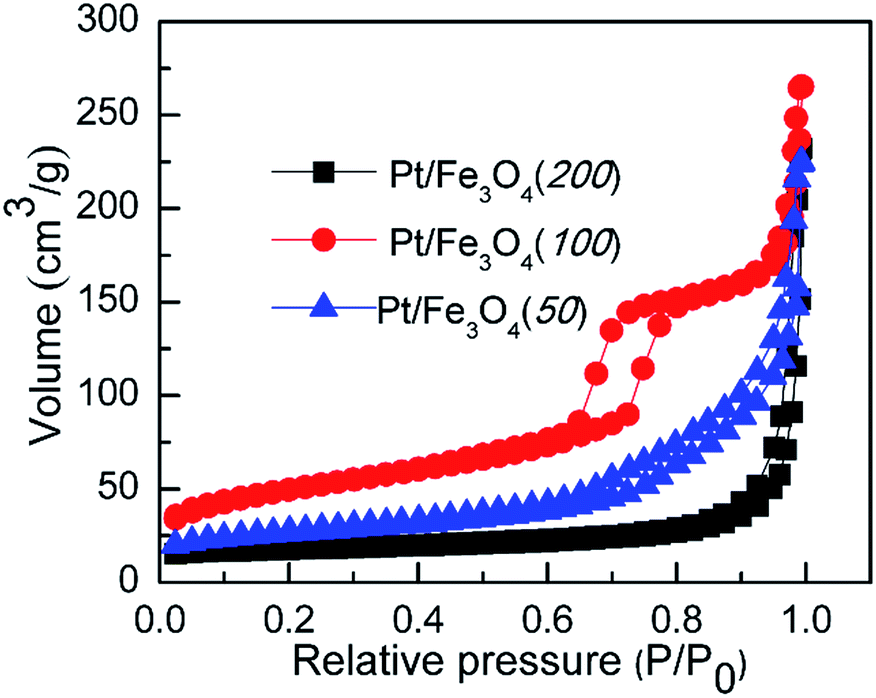
A typical IV adsorption isotherm and obvious hysteresis loop can be observed over Pt/Fe3O4 samples, as shown by the N2 adsorption–desorption isotherms (Fig. 2). It indicates that the presence of mesoporous in this nanostructure can provide available channels for effective diffusion of reactants trough the Pt/Fe3O4 sample, which is important for its catalytic applications. The Pt/Fe3O4(100) catalyst with the highest BET surface area of 139.4 m2 g−1 and pore volume of 0.4 cm3 g−1 (entry 2, Table 1), is most favourable for the dispersion of Pt NPs on the surface of Fe3O4 and exposure of catalytic active sites to improve its catalytic performance.
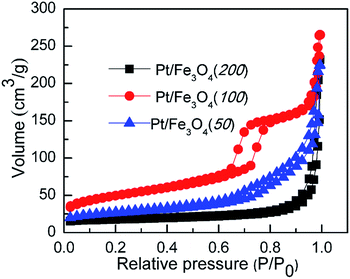 |
| | Fig. 2 N2-Adsorption and desorption isotherm curves of series of Pt/Fe3O4 catalysts at 77 K. | |
The morphology and geometric structure of self-assembled Pt/Fe3O4(100) composite are also investigated and corresponding TEM images are shown in Fig. 3. It presents a well-dispersed spherical-type composite with average diameter of 80–100 nm (Fig. 3a and b). The interplanar spacing of 0.22 nm, which is matched well with (111) crystal plane of Pt NPs, has also been observed in the inset HRTEM image (Fig. 3c). To more clearly distinguish the distribution of element component between Pt and Fe3O4 NPs, EDX mapping analysis has also been employed. The result indicates uniform distribution of Pt nano-crystallite in the matrix of Fe3O4. The similar geometric morphology can also be observed for Pt/Fe3O4 samples with different Pt contents (Fig. S2, ESI†). As can be seen, the morphology and particle distribution of this formed composite becomes more regular and uniform with increase of Pt content. It may be ascribed to the strong binding force between metallic Pt NPs and Fe3O4 NPs because the composite of Pd/Fe3O4 is fabricated by the interaction of Pt(IV) salt and Fe(II) salt without the addition of any other surfactants. Therefore, the morphology and particle distribution are highly correlated with the composition and content of Pt and Fe. At low Pt content, for instance Pt/Fe3O4(200) sample, the incomplete redox reaction between Pt(IV) and Fe(II) affords weak binding force which will influence the morphology and particle distribution of Pt/Fe3O4 (Fig. S2a and b, ESI†). As Pt content increase, the strong binding force between Pt NPs and Fe3O4 NPs facilitates the formation of Pt/Fe3O4 hybrids with uniform morphology and particle distribution (Fig. 3). However, the Pt NPs on the surface of composite will aggregate into chain-shape because of too strong binding force between Pt NPs and Fe3O4 NPs (Fig. S2c and d, ESI†). This is in accordance with the previous report in the Ag–CeO2 and Pd–CeO2 system.31,34
 |
| | Fig. 3 TEM and HRTEM image (up) and EDX mapping analysis (bottom) of Pt/Fe3O4(100) catalyst. | |
3.2. Surface chemical environment of catalysts
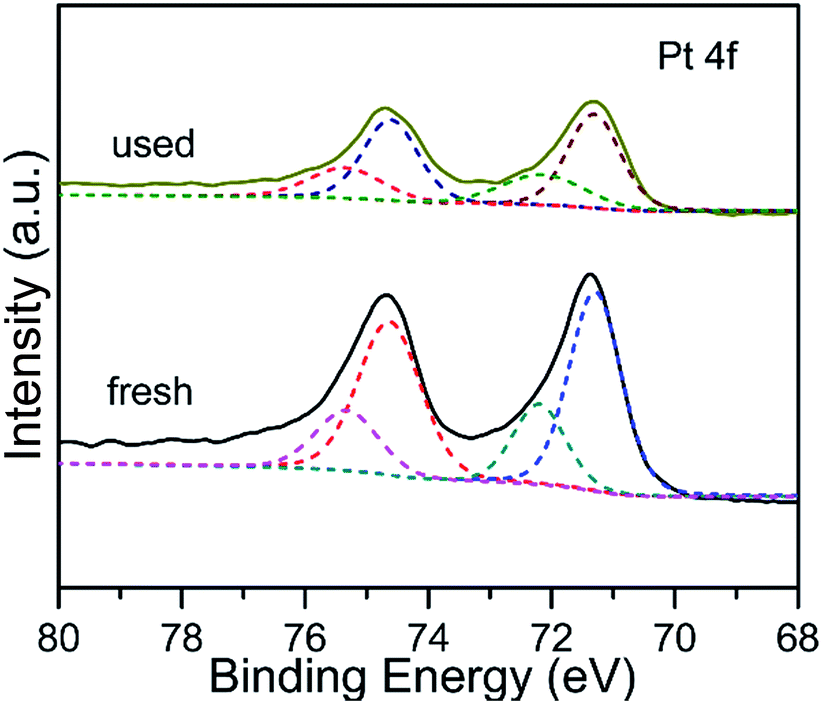
The electronic interaction between Pt and Fe3O4 NPs has been investigated by employing XPS technique, and the corresponding binding energy (BE) and fraction of various Pt species are displayed in Fig. 4 and Table S1 (ESI†). There are two apparent characteristic peaks located at 71.3 eV and 74.6 eV, owing to the 4f7/2 and 4f5/2 of metallic Pt, respectively.35 Interestingly, the BE value shifts to a lower orientation in comparison with previous reported Pt catalysts.36 It is most likely deduced by the fact that the electron density of Pt surface increases due to donating of Fe3O4 with variable valence, which is similar with the previous report.29 The Pt 4f characteristic XPS peak can be divided into two pairs of doublets corresponding to Pt(0) and Pt(II) species. By comparison, the content of Pt(II) for the used catalyst is a little higher than that of fresh one (Table S1, ESI†). It could be root in the electron transfer of metallic Pt for dissociation of H2 and activation of electrophilic CO group of CAL during hydrogenation reaction.16,25
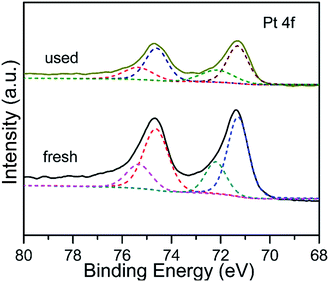 |
| | Fig. 4 Pt 4f XPS spectra of Pt/Fe3O4(100) catalyst. | |
3.3. Catalytic testing
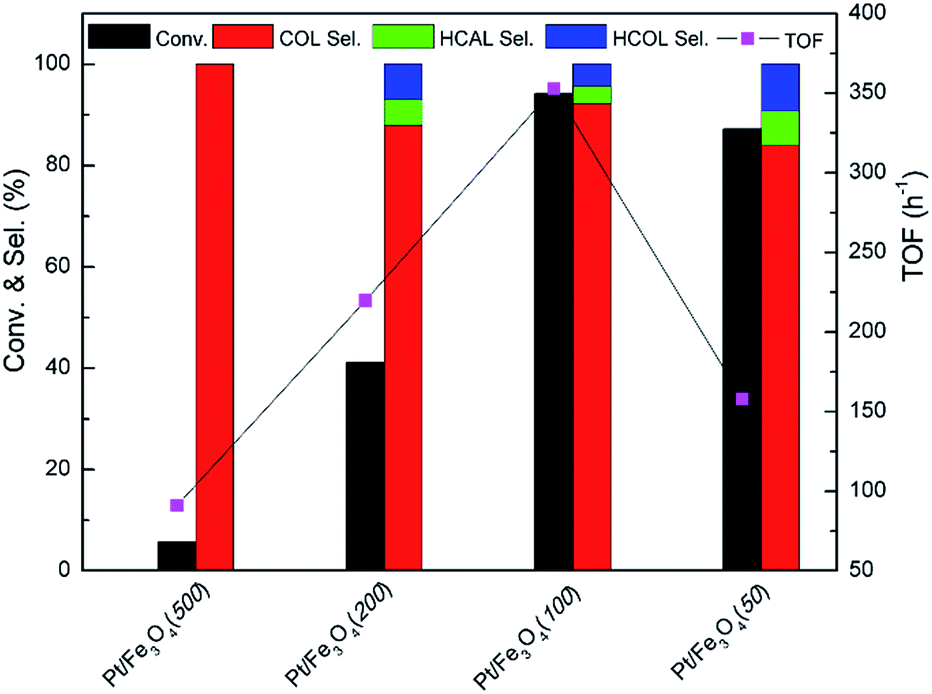
3.3.1. Screening of the catalysts. After characterization, the screening of Pt/Fe3O4 catalyst is carried out in the chemoselective hydrogenation of CAL to COL under the condition of 303 K, 5 bar H2 for 150 min. Generally, the by-products of CAL hydrogenation consist of hydrocinnamaldehyde (HCAL) and hydrocinnamyl alcohol (HCOL), which are obtained from the hydrogenation of CC and or/CO bond of CAL. The GC spectrum of various products for the hydrogenation of CAL over Pt/Fe3O4(100) sample is displayed in Fig. S3 (ESI†). As can be seen in Fig. 5, the superior CAL conversion can be obtained over Pt/Fe3O4(100) catalyst. The evident improvement of CAL conversion from 5.7% to 94.2% with the increase of mole ratio of Pt to Fe from 1:500 to 1:100 can be attributed to the improvements of dispersion and distribution of Pt NPs in the matrix of Fe3O4 (Fig. 3 and S2, ESI†). As observed in Fig. 1, only the Fe3O4 diffraction peak has be found for Pt/Fe3O4(500) sample. It cannot provide Pt active sites for activation of H2 and CAL substrate, thus results in a poor catalytic performance. With the mole ratio of Pt to Fe increasing to 1:100, it not only provides abundant exposure surface active sites of metallic Pt, but also gives more uniform distribution of Pt NPs on the surface of Fe3O4, as showed in Table 1. In addition, the highest catalytic activity of Pt/Fe3O4(100) catalyst also profits from the highest surface area (entry 2, Table 1). However, the catalytic performance will decline as the mole ratio of Pt to Fe surpasses 1:100. It may be attributed to agglomeration of Pt NPs, as marked with blue arrow in Fig. S2 (ESI†).
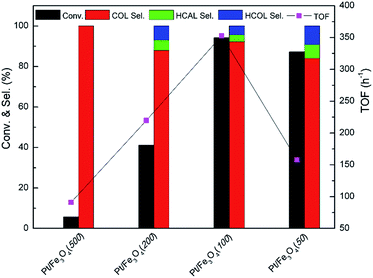 |
| | Fig. 5 Catalytic performance of Pt/Fe3O4 catalysts. Reaction condition: 303 K, 5 bar H2 and 150 min, 5 mL 2-proponal as solvent. | |
It is well known that turn of frequency (TOF) value of catalyst is one of important criterions to valuate catalytic performance in heterogeneous catalysis system. In Fig. 5, the Pt/Fe3O4(100) catalyst shows the highest value of 352.8 h−1 among all the Pt/Fe3O4 samples. It is suggested that moderate amount of Pt active sites can be available in Pd/Fe3O4(100) catalyst, leading to a rapid reaction rate in initial time. In comparison to the other supported Pt-based catalysts reported in literatures, the Pt/Fe3O4(100) catalyst can achieve comparative catalytic activity and selectivity (Table S2, ESI†).
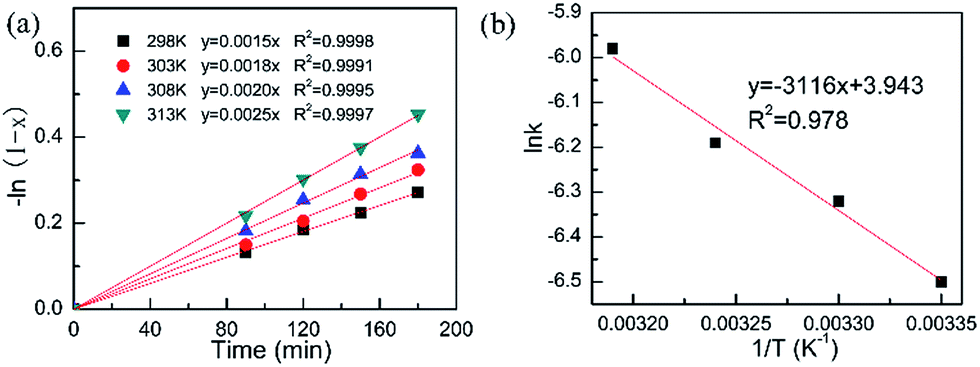
3.3.2. Experimental parameter and reaction intrinsic kinetics. The experimental parameters including substrate concentration and external diffusion could also influence the catalytic performance of heterogeneous catalysts. Interestingly, the COL selectivity is independent of the substrate concentration, inferring the excellent COL selectivity over the Pt/Fe3O4 catalysts. CAL conversion gradually decreases from 94.2% to 23.5% with increasing the concentration of substrate from 0.032 mol L−1 to 0.79 mol L−1 over the Pt/Fe3O4(100) catalyst (Table S3, ESI†). It can be inferred that the conversion of CAL strongly depends on the relative proportion of catalyst and substrate. It is found that the conversion of CAL decreases as the concentration of substrate increases because the available catalytic active sites is constant. The similar results have been reported in the previous literature.11 For the external diffusion effect, it can be ruled out by changing the stirring speed. As be presented in Table S4 (ESI†), the CAL conversion shows a remarkable increase as the stirring lifting from 200 rpm to 530 rpm. This can be explained by the improvement of mass transfer at the solid–liquid interface, resulting in a fast reaction rate. Once the stirring speed exceeding 530 rpm, the CAL conversion is almost invariable. It indicates that the effect of external diffusion has been nearly excluded. Therefore, the stirring speed has been fixed at 530 rpm for the subsequent investigation.Generally speaking, the selective hydrogenation of CAL can be simply depicted by the equation as below: CAL + H2→ COL, thus the rate equation can be described as r = k[CAL]m[H2]n. In this work, the pressure of H2 is fixed as a constant. So the above-mentioned equation can be abbreviated as r = k[CAL]m. As shown in Fig. 6a, the −ln(1 − x) is proportional to the reaction time t, where x represents CAL conversion. This plot is in accordance with the definition of a first order reaction. On the basis of this curve, the rate constants k at different temperatures can be calculated and the results are listed in Table 2. As we all known, the Arrhenius equation is expressed as lnk = lnA − Ea/RT, where A and Ea represent pre-exponential factor and reaction apparent activation energy, respectively. The value of Ea is 24.7 kJ mol−1, which is calculated based on the slope of curve (Fig. 6b). The much lower Ea value suggests that the Pt/Fe3O4 catalyst is quite suitable for selective hydrogenation of CAL comparing with iridium–carbon catalyst reported by Breen and co-workers.37
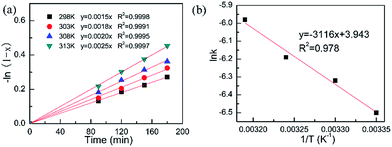 |
| | Fig. 6 Reaction intrinsic kinetics test: (a) plot of −ln(1 − x) vs. time (min) to calculate rate constant k; (b) Arrhenius plot to calculate Ea. | |
Table 2 Rate constant (k) for selective hydrogenation of cinnamaldehyde at different temperature
| Entry |
T (K) |
1/T (K−1) |
Rate constant k (min−1) |
−lnk |
| 1 |
298 |
0.00336 |
0.0015 |
−6.50 |
| 2 |
303 |
0.00331 |
0.0018 |
−6.32 |
| 3 |
308 |
0.00324 |
0.0020 |
−6.21 |
| 4 |
313 |
0.00319 |
0.0025 |
−5.99 |
In term of the above experimental and characterization results, we propose a plausible reaction mechanism for the chemoselective hydrogenation of CAL over Pt/Fe3O4(100) catalyst (Fig. 7). It has been proved theoretically and experimentally that the adsorption mode of substrate via vertical configuration of CO bond strongly depends on the electron density of metal surface.38,39 In the XPS results, the dense electron of Pt metal over the Pt/Fe3O4(100) sample is confirmed, which can improve the affinity of Pt and CO group of substrate. Besides, the Fe oxide species with facile variable valence can act as electrophilic sites to attenuate the CO bond.40 In this work, H2 is dissociated as nonpolar H atom on the Pt surface. Simultaneously, the terminal CO group of CAL can contact intimately with the activated H atom to form the desired product of COL. Although the specific mechanism needs to be illuminated, the electron interaction between active Pt and Fe3O4 NPs plays a crucial role in improving the catalytic performance in chemoselective reduction of cinnamaldehyde.
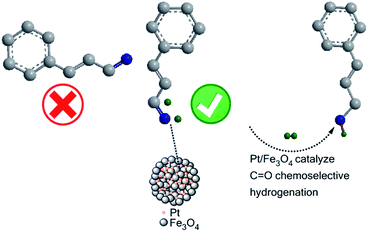 |
| | Fig. 7 Catalytic mechanism of selective hydrogenation of CAL over Pt/Fe3O4 catalysts. | |
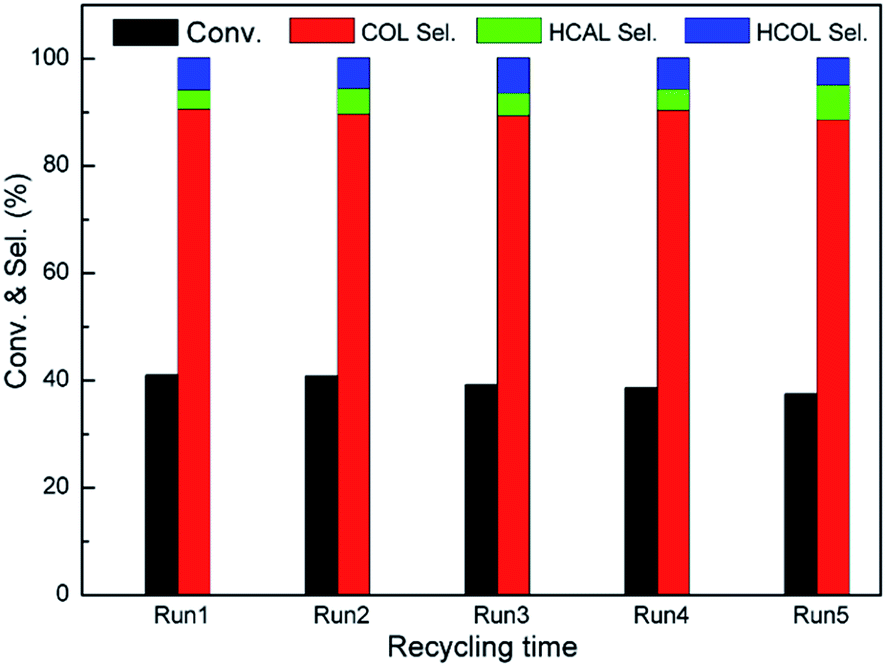
3.3.3. Recycling test. We further explore the reusability of Pt/Fe3O4(100) catalyst. The used catalyst can be readily collected from mixture by an external magnetic field. Prior to each run, the used catalyst is washed with deionized water and ethanol, and then dried overnight at 333 K in a vacuum oven. To our delight, both of CAL conversion and COL selectivity have only a slight loss after five cycles, as shown in Fig. 8. The leaching of Pt during reaction process is investigated by ICP-AES analysis. As can be seen, the Pt content in the used Pt/Fe3O4(100) catalyst is 2.92%, which is very close to one of the fresh catalyst (2.98%). It suggests the leaching of active Pt in the Pt/Fe3O4(100) catalyst can be effectively refrained during reaction process. Moreover, the morphology and element distribution can keep intact (Fig. S4, ESI†), further suggesting good stability of geometrical structure for this composite.
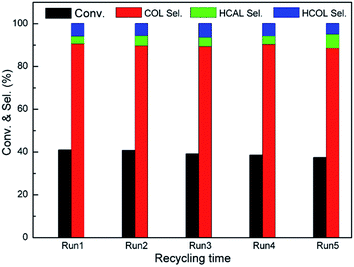 |
| | Fig. 8 Recycling evaluation of catalytic selective hydrogenation of CAL over Pt/Fe3O4(100) catalyst. Reaction conditions: 303 K, 5 bar H2 and 10 min, 5 mL 2-proponal used as solvent. | |
4. Conclusions
In summary, the hybrid composite Pt/Fe3O4 catalysts have been successfully fabricated by a facile one-pot redox reaction between Pt(IV) and Fe(II) precursor and evaluated in the chemoselective hydrogenation of CAL. The inherent variable electron transfer of Fe3O4 drives electron to metallic Pt nanocrystallites, which promote the adsorption of CAL on the surface of Pt metal via terminal CO group, showing prominent catalytic activity and high selectivity to COL under a mild condition of 303 K, 5 bar H2 for 150 min. The catalyst can be used for five cycles with a slight activity loss and separated rapidly via an external magnetic field. In view of practical application and cost-efficiency, it may provide a feasible avenue for designing and synthesis of novel heterogeneous catalysts to apply in selective reduction of unsaturated compounds.
Acknowledgements
We appreciate the Natural Science Foundation of China (Grant No. 51372248, 51432009 and 51502297), Instrument Developing Project of the Chinese Academy of Sciences (Grant No. yz201421), the CAS/SAFEA International Partnership Program for Creative Research Teams of Chinese Academy of Sciences, China.
References
- S. Bhogeswararao and D. Srinivas, J. Catal., 2012, 285, 31–40 CrossRef CAS.
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 21–33 CrossRef.
- C. H. Hao, X. N. Guo, Y. T. Pan, S. Chen, Z. F. Jiao, H. Yang and X. Y. Guo, J. Am. Chem. Soc., 2016, 138, 9361–9364 CrossRef PubMed.
- P. H. Z. Ribeiro, E. Y. Matsubara, J. M. Rosolen, P. M. Donate and R. Gunnella, J. Mol. Catal. A: Chem., 2015, 410, 34–40 CrossRef CAS.
- G. R. Bertolinia, C. I. Cabello, M. Munoz, M. Casella, D. Gazzoli, I. Pettiti and G. Ferraris, J. Mol. Catal. A: Chem., 2013, 366, 109–115 CrossRef.
- A. B. Dongil, B. Bachiller-Baeza, A. Guerrero-Ruiz and I. Rodríguez-Ramos, J. Catal., 2011, 282, 299–309 CrossRef CAS.
- Z. Guo, Y. Chen, L. Li, X. Wang, G. L. Haller and Y. Yang, J. Catal., 2010, 276, 314–326 CrossRef CAS.
- A. B. Merlo, B. F. Machado, V. Vetere, J. L. Faria and M. L. Casella, Appl. Catal., A, 2010, 383, 43–49 CrossRef CAS.
- J. Lenz, B. C. Campo, M. Alvarez and M. A. Volpe, J. Catal., 2009, 267, 50–56 CrossRef CAS.
- A. Nagendiran, V. Pascanu, A. Bermejo Gomez, G. Gonzalez Miera, C. W. Tai, O. Verho, B. Martin-Matute and J. E. Backvall, Chemistry, 2016, 22, 7184–7189 CrossRef CAS PubMed.
- W. O. Oduro, N. Cailuo, K. M. Yu, H. Yang and S. C. Tsang, Phys. Chem. Chem. Phys., 2011, 13, 2590–2602 RSC.
- A. J. Plomp, H. Vuori, A. O. I. Krause, K. P. d. Jong and J. H. Bitter, Appl. Catal., A, 2008, 351, 9–15 CrossRef CAS.
- Y. Gu, Y. Zhao, P. Wu, B. Yang, N. Yang and Y. Zhu, Nanoscale, 2016, 8, 10896–10901 RSC.
- B. Wu, H. Huang, J. Yang, N. Zheng and G. Fu, Angew. Chem., Int. Ed., 2012, 51, 3440–3443 CrossRef CAS PubMed.
- Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS.
- Q. Wu, C. Zhang, B. Zhang, X. Li, Z. Ying, T. Liu, W. Lin, Y. Yu, H. Cheng and F. Zhao, J. Colloid Interface Sci., 2016, 463, 75–82 CrossRef CAS PubMed.
- I. Nongwe, V. Ravat, R. Meijboom and N. J. Coville, Appl. Catal., A, 2016, 517, 30–38 CrossRef CAS.
- E. V. Ramos-Fernández, A. F. P. Ferreira, A. Sepúlveda-Escribano, F. Kapteijn and F. Rodríguez-Reinoso, J. Catal., 2008, 258, 52–60 CrossRef.
- X. Yang, L. Wu, L. Ma, X. Li, T. Wang and S. Liao, Catal. Commun., 2015, 59, 184–188 CrossRef CAS.
- A. Chirieac, B. Dragoi, A. Ungureanu, C. Ciotonea, I. Mazilu, S. Royer, A. S. Mamede, E. Rombi, I. Ferino and E. Dumitriu, J. Catal., 2016, 339, 270–283 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- R. Hirschl, F. Delbecq, P. Sautet and J. Hafner, J. Catal., 2003, 217, 354–366 CrossRef CAS.
- C. Espro, A. Donato, S. Galvagno and G. Neri, React. Kinet., Mech. Catal., 2016, 118, 223–233 CrossRef CAS.
- W. Lin, H. Cheng, L. He, Y. Yu and F. Zhao, J. Catal., 2013, 303, 110–116 CrossRef CAS.
- T.-N. Ye, J. Li, M. Kitano, M. Sasase and H. Hosono, Chem. Sci., 2016, 7, 5969–5975 RSC.
- M. G. Prakash, R. Mahalakshmy, K. R. Krishnamurthy and B. Viswanathan, Catal. Today, 2016, 263, 105–111 CrossRef CAS.
- H. Liu, L. Chang, L. Chen and Y. Li, ChemCatChem, 2016, 8, 946–951 CrossRef CAS.
- P. Concepcion, Y. Perez, J. C. Hernandez-Garrido, M. Fajardo, J. J. Calvino and A. Corma, Phys. Chem. Chem. Phys., 2013, 15, 12048–12055 RSC.
- C. Milone, R. Ingoglia, L. Schipilliti, C. Crisafulli, G. Neri and S. Galvagno, J. Catal., 2005, 236, 80–90 CrossRef CAS.
- Z. Tian, Q. Li, J. Hou, Y. Li and S. Ai, Catal. Sci. Technol., 2016, 6, 703–707 CAS.
- T. Kayama, K. Yamazaki and H. Shinjoh, J. Am. Chem. Soc., 2010, 132, 13154 CrossRef CAS PubMed.
- X. Wang, D. Liu, S. Song and H. Zhang, J. Am. Chem. Soc., 2013, 135, 15864–15872 CrossRef CAS PubMed.
- J. E. Benson and M. Boudart, J. Catal., 1965, 4, 704–710 CrossRef CAS.
- Y. Zhang, C. Chen, W. Gong, J. Song, H. Zhang, Y. Zhang, G. Wang and H. Zhao, Catal. Commun., 2017, 93, 10–14 CrossRef CAS.
- Z. Tian, C. Liu, Q. Li, J. Hou, Y. Li and S. Ai, Appl. Catal., A, 2015, 506, 134–142 CrossRef CAS.
- Z. Tian, Q. Li, Y. Li and S. Ai, Catal. Commun., 2015, 61, 97–101 CrossRef CAS.
- J. Breen, R. Burcha, J. Gomez-Lopeza, K. Griffinb and M. Hayes, Appl. Catal., A, 2004, 268, 267–274 CrossRef CAS.
- F. Delbecq, J. Catal., 2003, 220, 115–126 CrossRef CAS.
- F. Delbecq and P. Sautet, J. Catal., 1995, 152, 217–236 CrossRef CAS.
- W. Koo-Amornpattana and J. W. Winterbottom, Catal. Today, 2001, 66, 277–287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information of binding energy of Pt species of Pt/Fe3O4 catalyst; effect of kinetics parameter on catalytic performance; compared with the previous Pt-based supported catalysts; TEM and HRTEM image. See DOI: 10.1039/c7ra02898a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Wanbing Gongab,
Jieyao Songab,
Yanping Suab,
Haimin Zhang
*a,
Wanbing Gongab,
Jieyao Songab,
Yanping Suab,
Haimin Zhang