
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PepBio: predicting the bioactivity of host defense peptides†
Saw Simeon‡
 ab,
Hao Li‡a,
Thet Su Win‡a,
Aijaz Ahmad Malika,
Abdul Hafeez Kandhroac,
Theeraphon Piachamd,
Watshara Shoombuatonga,
Pornlada Nuchnoic,
Jarl E. S. Wikberge,
M. Paul Gleesonf and
Chanin Nantasenamat*a
ab,
Hao Li‡a,
Thet Su Win‡a,
Aijaz Ahmad Malika,
Abdul Hafeez Kandhroac,
Theeraphon Piachamd,
Watshara Shoombuatonga,
Pornlada Nuchnoic,
Jarl E. S. Wikberge,
M. Paul Gleesonf and
Chanin Nantasenamat*a
aCenter of Data Mining and Biomedical Informatics, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand. E-mail: chanin.nan@mahidol.edu; Fax: +66 2 441 4371 ext. 2715; Tel: +66 2 441 4380
bInterdisciplinary Graduate Program in Bioscience, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand
cCenter for Research and Innovation, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
dDepartment of Clinical Microbiology and Applied Technology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand
eDepartment of Pharmaceutical Biosciences, Uppsala University, Uppsala SE751 24, Sweden
fDepartment of Biomedical Engineering, Faculty of Engineering, King Mongkut's Institute of Technology Ladkrabang, Bangkok 10520, Thailand
First published on 12th July 2017
Abstract
Host defense peptides (HDPs) represents a class of ubiquitous and rapid responding immune molecules capable of direct inactivation of a wide range of pathogens. Recent research has shown HDPs to be promising candidates for development as a novel class of broad-spectrum chemotherapeutic agent that is effective against both pathogenic microbes and malignant neoplasm. This study aims to quantitatively explore the relationship between easy-to-interpret amino acid composition descriptors of HDPs with their respective bioactivities. Classification models were constructed using the C4.5 decision tree and random forest classifiers. Good predictive performance was achieved as deduced from the accuracy, sensitivity and specificity in excess of 90% and Matthews correlation coefficient in excess of 0.5 for all three evaluated data subsets (e.g. training, 10-fold cross-validation and external validation sets). The source code and data set used for the construction of classification models are available on GitHub at https://github.com/chaninn/pepbio/.
1 Introduction
The emergence and increasing incidences of antibiotic resistance by pathogenic microbes poses a global threat.1 Due to their evolutionary conservation, components of the innate immune system are an interesting resource to look for novel antibiotics and thus remain effective for combating foreign pathogens. Host defense peptides (HDPs) are small cationic peptides (i.e. usually less than 100 amino acids in length), found ubiquitously in living organisms (i.e. fungi, plants, reptiles, mammals, etc.) and constitute an important component of the innate immune system.2 Most importantly, is the fact that the bactericidal activity of HDPs appears to be negligibly affected by the myriad of defensive mechanisms exerted by microbes against conventional antibiotics such as penicillin. Hence, HDPs can be considered to be an important and novel class of antibiotics that can address the threat of emerging and re-emerging diseases caused by drug-resistant pathogens.Another major contemporary health threat for which HDPs have shown great potential for, is the treatment of cancer.3 Despite advances in various therapeutic schemes, malignant neoplasm remains the leading cause of mortality. Chemotherapy is the mainstay of contemporary cancer treatments and are known to possess many shortcomings including low specific toxicity, the potential to induce secondary malignancies and the frequent emergence of multi-drug-resistant (MDR) cancer cell strains. The latter being the major cause for failure of chemotherapy and is a sign of poor prognosis for patients.4 In addition to their antimicrobial potential, HDPs have been demonstrated to be promising candidates as anticancer agents that possess high specificity, rapid and direct target neutralizing ability (i.e. especially those of MDR phenotype) and so far have no observed tendency of inducing resistance in all targeted cancer strains.5 In addition to the health threats posed by pathogenic microbes6 and malignant neoplasms,7 HDPs are not limited to the host defense system such as direct neutralization of pathogens8,9 but they have been demonstrated to be effective against an even wider spectrum of pathogens including viruses,10 parasites11 and fungi.12 In addition to direct pathogen neutralization, HDPs have been observed to be potent immunological modulators, regulating inflammatory responses13,14 as well as recruiting dendritic cells.15
HDPs are very diverse in nature and have been reported in almost all forms of life, from single celled microorganisms to more complex organisms such as humans. On the basis of their secondary structures, HDPs can be broadly classified into four classes as follows: (i) α-helical peptides (e.g. LL-37), (ii) β-sheet peptides stabilized by two to four disulfide bridges (e.g. lectofericin, androctonin, RTD-1 and hepcidin-20), (iii) αβ complex with one to three disulfide bridge (e.g. drosomycin) and (iv) non-αβ peptides with extended structures (e.g. indolicidin). Fig. 1 summarizes the structural diversity of HDPs. α-Helical peptides are most abundant and extensively well characterized owing to their small size and ease of chemical synthesis.16 In general, they are twelve to fifty amino acids in length with helical conformation and slightly bent at the center of the molecule. One of the characteristic property of α-helical peptides is that in aqueous solution they are usually unstructured but adopt the amphipathic helical structure upon interaction with the target cell membrane. This structural alignment of the polar and non-polar residues on the opposite side of the helical coat allows optimal interaction of the peptide with the host membrane. β-Sheet peptides are the second largest group of HDPs that are characterized by the presence of single, hairpin motif containing two to eight Cys residues in relatively defined positions involving one to four disulfide bonds for stabilization.17 The average length of residues is approximately twenty to thirty residues in length. αβ complex is also known as cysteine stabilized α-helical and β-sheets superfamily and it is characterized by the presence of an α-helix and generally three anti-parallel β-sheets that is stabilized by two to four disulfide bonds.18 Most peptides from this group have limited antimicrobial activity and are active against the filamentous fungi. Non-αβ peptides is comprised of very few peptides and they are characterized by the presence of higher proportion of certain amino acids such as Trp, Arg, Pro, Gly and His.19 However, these peptides have highly variable secondary structures that are mostly in the extended conformation.
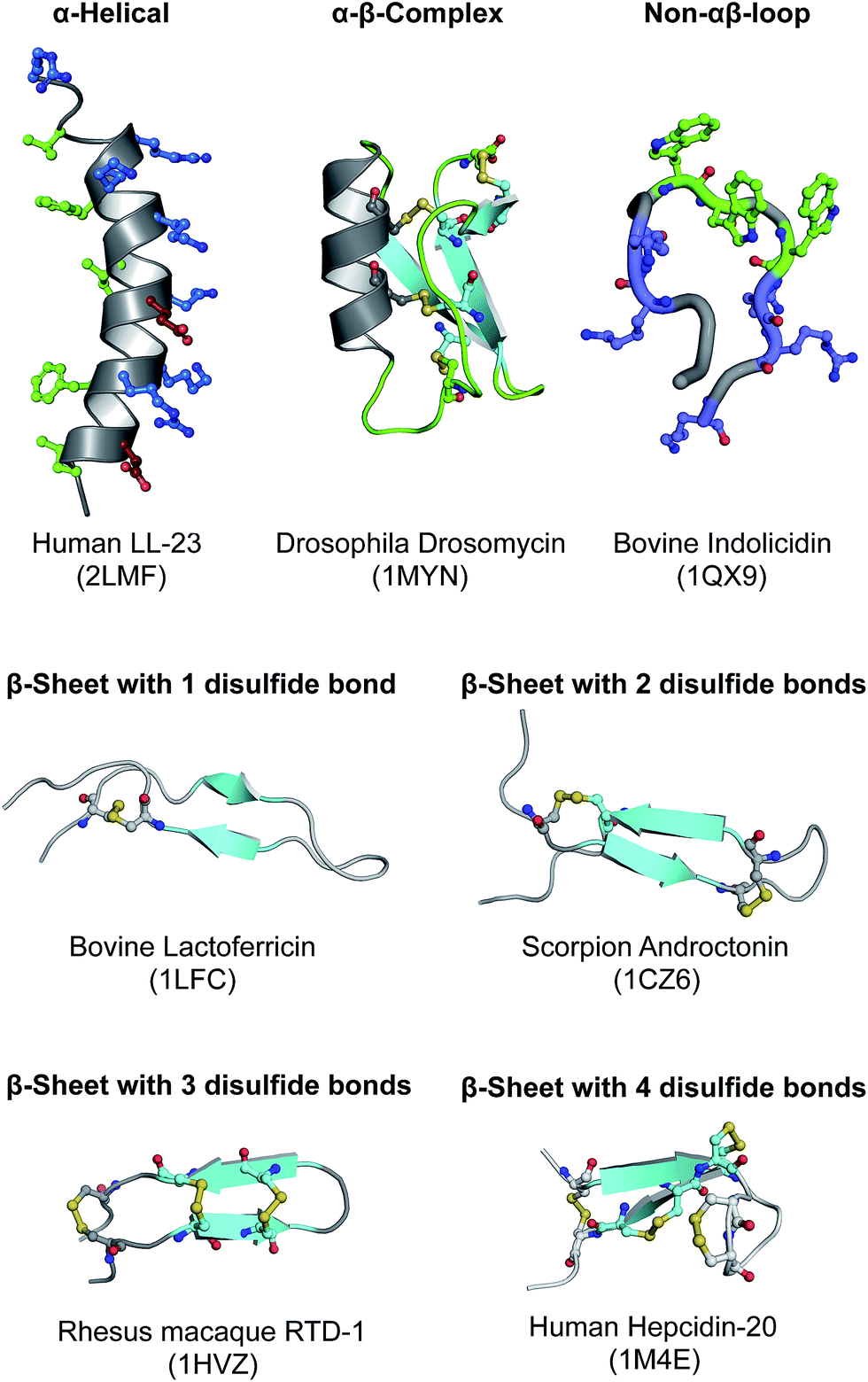 | ||
| Fig. 1 Overview of the structural diversity of HDPs. Residues are color coded green, blue, red and yellow to represent hydrophobic, positively-charged, negatively-charged and disulfide bridge, respectively. | ||
Quantitative structure activity relationship (QSAR) seeks to understand the correlation between the physicochemical properties of biomolecules with their observed bioactivities through the use of statistical or machine learning approaches.20,21 Although several QSAR studies have been reported for predicting a wide range of HDP bioactivities (e.g. antibacterial, anticancer, antifungal and antiviral), they may fall into the following situations: (i) models may be based on relatively small data sets,22–24 (ii) even if they are based on large data sets they are typically confined to modeling only one of the aforementioned bioactivities23,25–27 and lastly (iii) models may be predictive but are often not interpretable.28
In regards to the first point, the ability of QSAR models to predict unknown properties depends largely on the nature and size of the training set. Prediction accuracy and confidence for an unknown peptide sequence varies according to how well the training set represents the unknown peptides. Not only that, the stability and predictivity of the models are defined by the training set.29 Thus, one QSAR model will have a narrow applicability domain and low generalization capability if they are based on small and similar sequence. Secondly, predictive models based on large data sets may be of potential utility for any single bioactivity under investigation but may not be extrapolated to other bioactivities. As such, it is desirable to comparatively construct and analyze the predictive models for several HDP bioactivities at the same time so that comparisons and generalizations may be made.
In this study, QSAR models of the bioactivity of HDPs were constructed from large data sets constituting antibacterial, anticancer, antifungal and antiviral peptides. To the best of our knowledge, this study represents the first large-scale QSAR investigation spanning several classes of HDPs. Rather than exploring a single bioactivity type, this study explores multiple bioactivities of HDPs, so as to allow better contrast of key structural features governing the various bioactivities. Decision tree and random forest classifiers provided a robust performance as evaluated by statistical parameters derived from internal and external validations. The underlying features governing the origin of HDP bioactivities obtained from this study may be of potential use for the future design of novel HDPs with desired bioactivity.
2 Materials and methods
2.1 Data collection
The data set of HDPs along with their activity classes were taken from the Database of Antimicrobial Activity and Structure of Peptides (DBAASP),30 which is a manually curated database of HDPs with their therapeutic activity. The data set compiled from DBAASP constituted a total of 6195, 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 916, 102229 and 2503 peptides (as of September 3, 2015) against cancer, fungus, bacteria and virus, respectively. Redundant sequence strings were removed using the duplicated function from the which function to create an index in which those containing redundant sequence strings were removed with the matrix indexing operator. Consequently, this approach produced data sets of 597, 2582, 96052 and 540 HDPs with anticancer, antifungal, antibacterial and antiviral bioactivities, respectively. Non-canonical amino acids were removed using the protcheck function from the R package protr.31 This resulted in a total of 8413 HDPs with anticancer (466), antifungal (2179), antibacterial (5255) and antiviral (514) activities.
916, 102229 and 2503 peptides (as of September 3, 2015) against cancer, fungus, bacteria and virus, respectively. Redundant sequence strings were removed using the duplicated function from the which function to create an index in which those containing redundant sequence strings were removed with the matrix indexing operator. Consequently, this approach produced data sets of 597, 2582, 96052 and 540 HDPs with anticancer, antifungal, antibacterial and antiviral bioactivities, respectively. Non-canonical amino acids were removed using the protcheck function from the R package protr.31 This resulted in a total of 8413 HDPs with anticancer (466), antifungal (2179), antibacterial (5255) and antiviral (514) activities.
2.2 Negative HDP data set
As there are no source of experimentally proven non-HDPs, a more recent benchmark data set provided by Xiao et al.32 was used as a negative set. The translate function from the R package seqinr was used to convert the DNA strings to protein strings. Protein sequence containing stop codon of DNA strings during the translation were removed with protcheck function from R package protr. The final non-HDP data set contained a total of 1710 sequences.2.3 Data partitioning
One of the issues with random sampling is that each split may have a larger or smaller proportion of some classes. This is particular true for cases when a class (i.e. HDPs with anticancer properties) represents a very small proportion of a data set which may then lead the class to be omitted from the training data set. To address this, stratified random sampling was used to generate random partition that have approximately the same proportion of each class (i.e. HDPs with antibacterial properties, HDPs with anticancer properties, HDPs with antifungal properties and HDPs with antiviral properlies).The data set was divided into two groups, which are internal training set and external testing set. The createDataPartition function from caret R package was used to split the data in which 80% of the data set was used as a training set while the remaining 20% were used as the external testing set.
2.4 External sets
The aforementioned data subset constituting 20% from the full data set was taken from each of the 100 independent data splits and used as the external set. Furthermore, in order to truly assess the external predictability of models, an additional set of non-redundant peptides were obtained from the Dover Analyzer, which was developed by Aguilera-Mendoza et al.,33 and used as an additional external validation set whose peptides are not included in the training and test set derived from the aforementioned data partitioning. This additional external set consisted of 11028 unique HDP peptides.
2.5 Peptide descriptors
There are an abundance of available descriptor softwares that could potentially be used to represent protein sequences for performing QSAR studies.34–37 Of these, the composition of amino acids are simple, interpretable and yet robust descriptors. Thus, HDP sequences were encoded by amino acid composition (AAC) and dipeptide composition (DPC) descriptors. In addition, composition class (CC) descriptors were also employed for describing the sequence features of investigated HDPs.AAC is the proportion of each amino acid type (e.g. His, Thr, Tyr and so forth) within a protein sequence. The fractions of all 20 natural amino acids were calculated as:
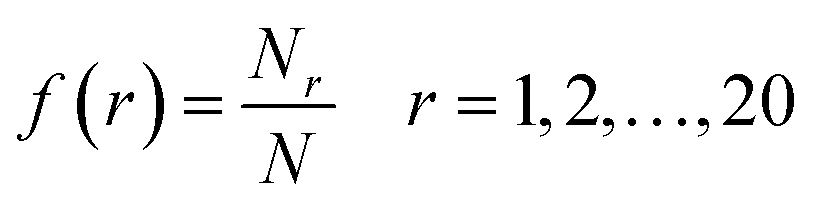 | (1) |
DPC is the fraction of dipeptides from a protein sequence which gives rise to 400 descriptors and can be defined as:
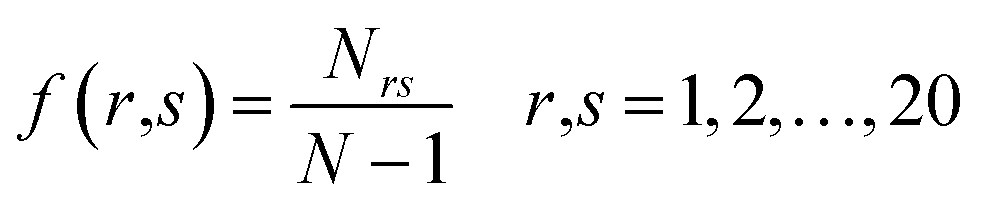 | (2) |
CC is defined as the global composition of the amino acid property in a protein as described by a set of 21 descriptors. CC descriptors were computed using the extractCTDC function from the R package protr.
2.6 Data set modelability
Practically, it is not always possible to build robust predictive models for all data sets. Thus, it is highly desirable to utilize a statistical criteria for a priori determination of the feasibility for building robust predictive models for any given data set. Recently, the modelability index (MODI) has been introduced by Golbraikh et al.38 for estimating the feasibility of a predictive model. The procedure of the calculation of MODI is briefly described below:![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) normalized) will be constructed as follows:
normalized) will be constructed as follows:
 | (3) |
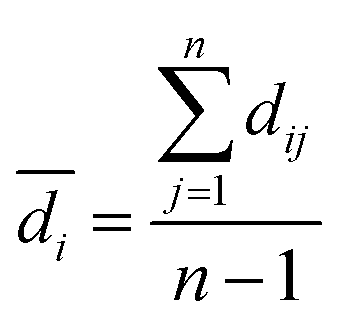 | (4) |
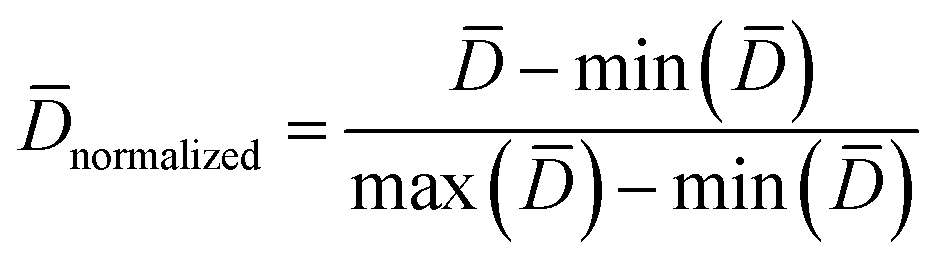 | (5) |
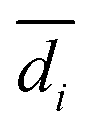 and n are the distance scores between two peptides, mean Euclidean distance and a number of peptides, respectively.
and n are the distance scores between two peptides, mean Euclidean distance and a number of peptides, respectively.
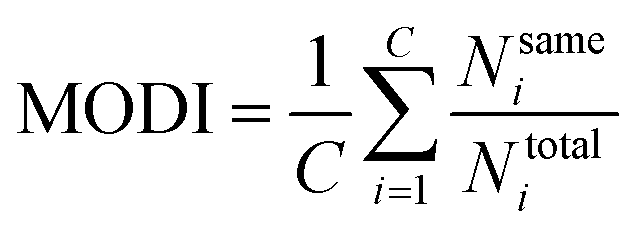 | (6) |
2.7 Data scaling
Often time, there is a great deal of variation in the range and distribution of each descriptor in the data set. This may create a problem for data mining. Thus, min–max normalization was applied where descriptors were rescaled to a standard range (e.g. 0–1). Furthermore, the family function named apply function with margin set at 2 was used to normalize the column set of descriptor block.2.8 Exploratory data analysis
So as to provide an overview on length of amino acid sequence, exploratory data analysis was performed using standard statistical methods. A total of HDPs having therapeutic effects against bacteria, cancer, fungus and virus were represented by histograms. All graphical figures and plots were made using the R statistical package ggplot2.392.9 Principal component analysis
Principal component analysis (PCA) provides a detail account on the structural information inside data structures. The two most useful features of PCA are loadings and scores. Loadings simultaneously reveals correlations between all descriptors whereas scores reveals the similarities and differences among samples. The fundamental assumption of PCA is that PC with a high explained variance is considered to possess systemic variances whereas PC with low explained variance is perceived as noise. Thus, it is important to decide on how many numbers of PC sufficiently represent the information presented in the data. By including the higher order PCs, it may over fit the model, which may in turn result in poor generalization of data structures. Thus, to obtain optimal PCs which were deemed enough to provide meaningful information, Horn's parallel analysis was applied.40 Descriptors with a variance close to 0 (i.e. less than one percent of variation in a column of a data frame) were removed with the function nearZeroVar and argument uniquecut set to 1 from the R package caret.41 The prcomp and kmeans functions from R package stats was used to perform PCA and K-means clustering, respectively.42 Prior to PCA analysis, all data were centered and scaled to have a unit variance using the argument of center and scale. The paran function with the argument of iterations set at 5000 from the R package paran was utilized to perform Horn's parallel analysis in order to determine the optimal number of PCs.432.10 Multivariate analysis
Decision tree (DT) is a transparent classifier that uses a tree-like structure to model the relationship between features and classes. The route toward modeling of activity classes of HDPs begins at the root node, whereby they are passed through decision nodes that require choices to be made based on features (i.e. a feature of amino acid composition). These outcomes split the data across branches that indicate potential class of a decision. At last, the final decision can be made where a predictive tree is terminated by leaf nodes, which provides a particular expected class, resulted from a series of decision. The J48 function from RWeka R package,44,45 an implementation of the Java-based machine learning package Weka,46 was utilized to build predictive models. To avoid the possibility of chance correlation arising from the random seed in the machine learning calculation, models were built for 100 times whereby the mean and their corresponding standard deviation of statistical parameters were reported.Random Forest (RF) is an ensemble classifier made up of several DTs. Similar to the DT classifier, classification starts at the root node where the data set is applied and splits according to the threshold values of each descriptor node (i.e. ACC and DPC) and subsequently flows outward until the decision leaf node (i.e. the class label) is reached. However, for each tree, bootstrap sampling is used to train the model thereby minimizing the variance. The RF classifier was generated using the R package ranger using a total of 500 trees.
It is worthy to note that two types of models were constructed in this study: (i) one multi-class model and (ii) several binary class models.
2.11 Validation of QSAR models
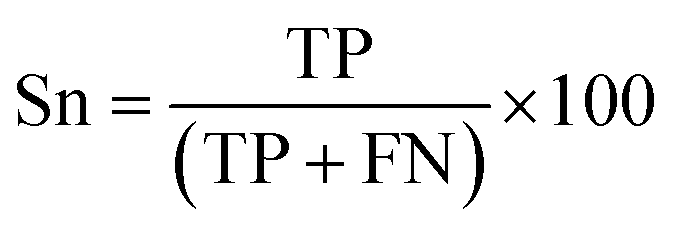
There are many statistical assessment tools that have been used to assess effectiveness and efficiency of predictive models. The following assessment parameters were used in this study: accuracy (Ac), sensitivity (Sn), specificity (Sp) and Matthews correlation coefficient (MCC), which corresponds to the percentage of correctly classified instances, the ratio of instances correctly classified as positive to all positive instances, the ratio of instances correctly classified as negative to all negative instances and the measure of the performance in terms of both positive and negative instances, respectively. These parameters can be calculated using the following equations:
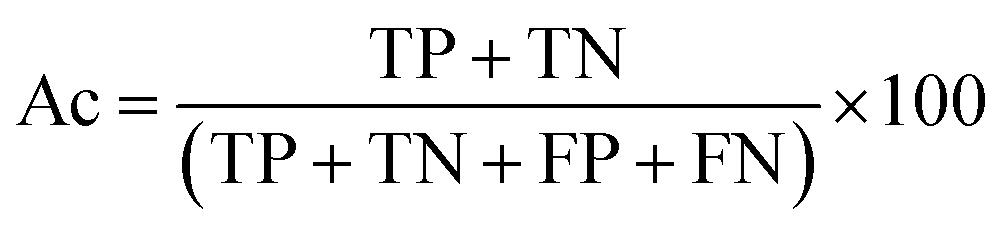 | (7) |
 | (8) |
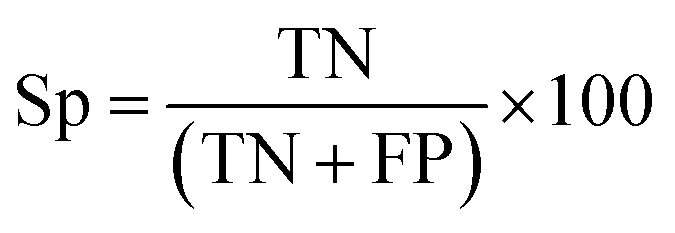 | (9) |
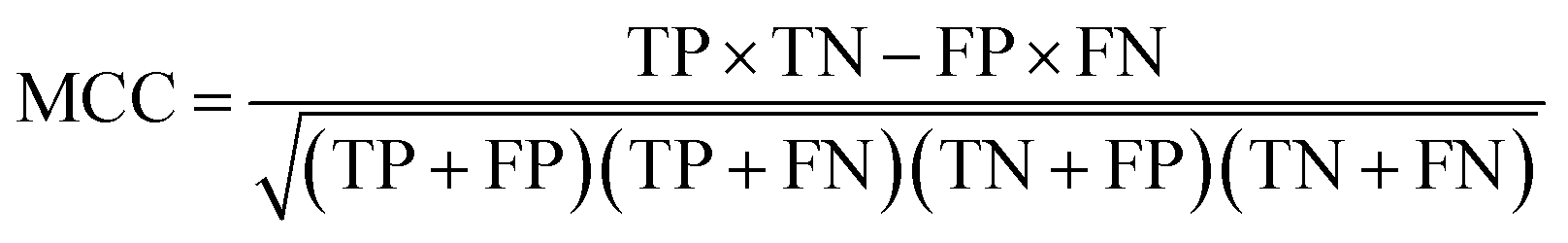 | (10) |
2.12 Applicability domain analysis
The applicability domain (AD) estimates the likelihood that the model reliably predict based on the extent of the feature space of the model. Although there are several methods for defining the applicability domain, k-nearest neighbors is a simple and robust approach for determining the AD of the model. Briefly, the Euclidean distance of each sample to its five nearest neighbors in the training set was calculated. Then, the normalized mean similarity of the sample to its neighbors were binned into four quantiles in which each quantile was plotted against the peptide's accuracy in the prediction. The aim is to look for a distance threshold in which the classification model can reliably predict the correct class of new peptides.2.13 Reproducible research
To facilitate the reproducibility of QSAR models described herein, the R source codes and associated data sets used to construct the models are made publicly available on GitHub at https://github.com/chaninn/pepbio/.3 Results and discussion
The notion that the biological activity of HDPs is governed by their physicochemical properties is the paradigm of QSAR. This study employs simple and interpretable descriptors for predicting the bioactivity (e.g. antibacterial, anticancer, antifungal and antiviral) of peptides. In the development of QSAR models, it is advisable to start from simple and interpretable descriptors along with machine learners and then gradually proceed up to complex descriptors. When the predictive performance between complex and simple machine learners are comparable, it is advisable to select simple models. For that reason, commonly used and highly interpretable protein descriptors (i.e. based on composition of amino acids) and interpretable DT and RF classifiers were selected for building the QSAR models. A schematic representation on the research framework performed herein is summarized in Fig. 2.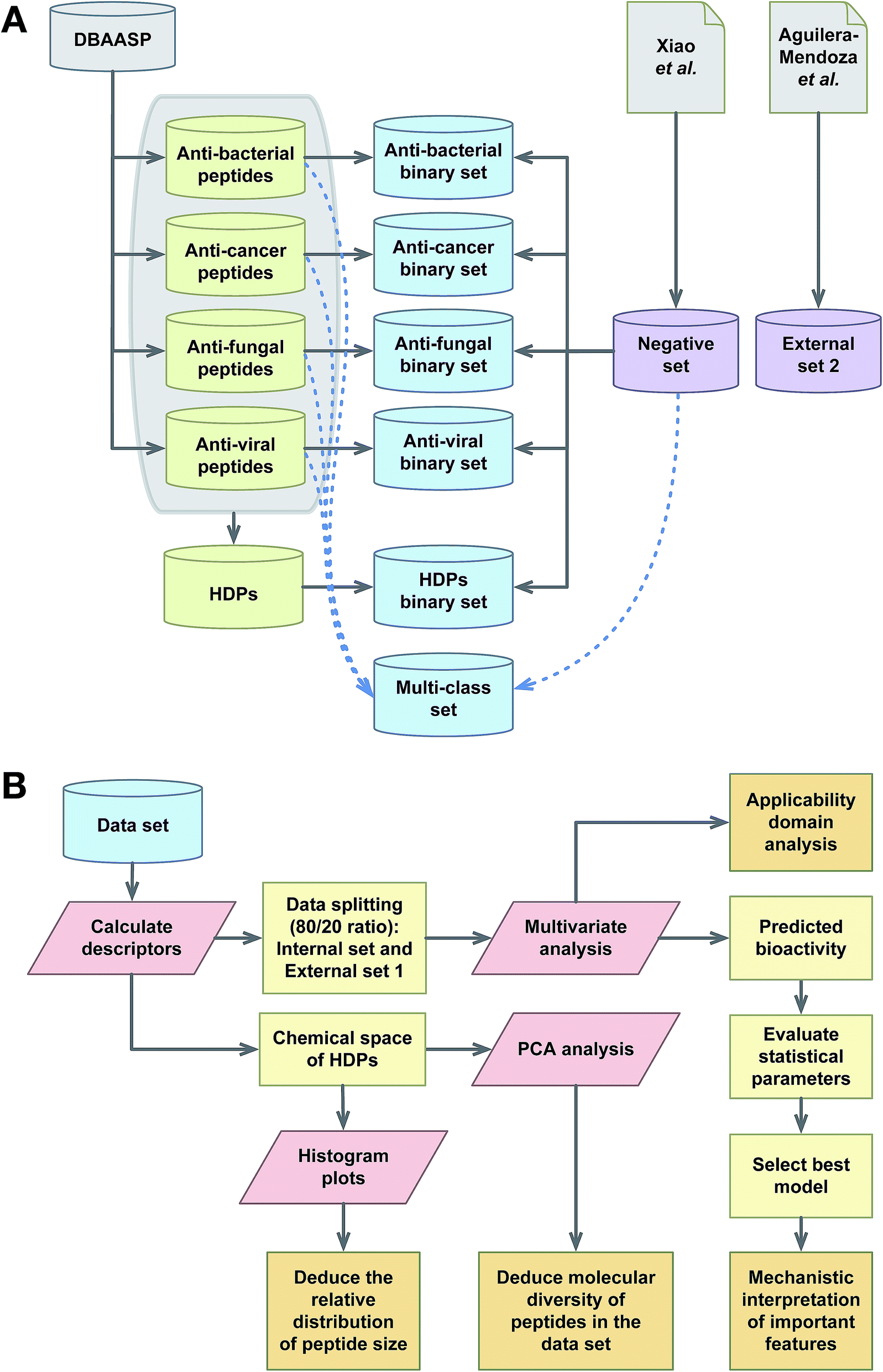 | ||
| Fig. 2 Schematic representation on the workflow of QSAR modeling of HDPs. | ||
3.1 Peptide space analysis
The peptide space of HDPs were explored so as to deduce the relative molecular diversity of the data set investigated herein. This was achieved by means of exploratory data analysis and PCA analysis.Firstly, exploratory data analysis was performed to discern the general characteristics of HDPs targeting bacteria, cancer, fungus and virus. A summary of the sequence length of these HDPs is provided in Fig. 3 as histogram plots. It can be observed that the region with the most count for all classes were within the range of 10 and 20. A close inspection revealed that the length of HDPs with antibacterial, anticancer, antifungal and antiviral activities were 21.63 ± 13.59, 19.23 ± 11.35, 23.91 ± 14.61 and 19.46 ± 12.14, respectively. Moreover, sequence length of the negative data set was also comparable with a value of 21.70 ± 8.82.
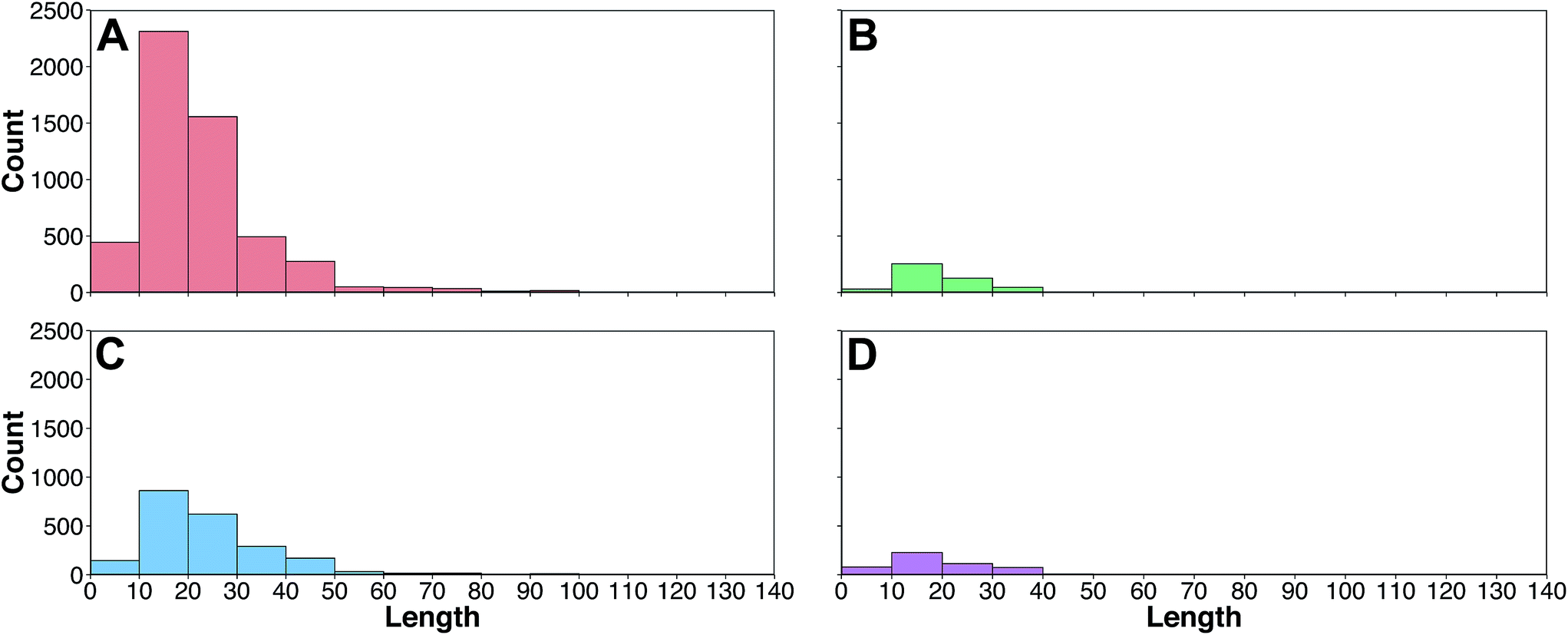 | ||
| Fig. 3 Histogram plots of the frequency distribution of the amino acid length of HDPs with antibacterial (A), anticancer (B), antifungal (C) and antiviral (D) bioactivities. | ||
Secondly, PCA analysis (Fig. 4) was performed to discern the relative molecular diversity of the constituent peptides in the investigated data set. The decision on how many principal component (PC) should be retained is an important issue in PCA analysis. The result from Horn's parallel analysis revealed that the adjusted eigenvalues of PC1, PC2 and PC3 were 1.23, 1.12 and 1.02, respectively thereby indicating that three PCs should be retained as it is over the threshold of 1. Particularly, the three PCs provided sufficient information for describing the data structure as the total explained variance for the first three PCs was 68.15%.
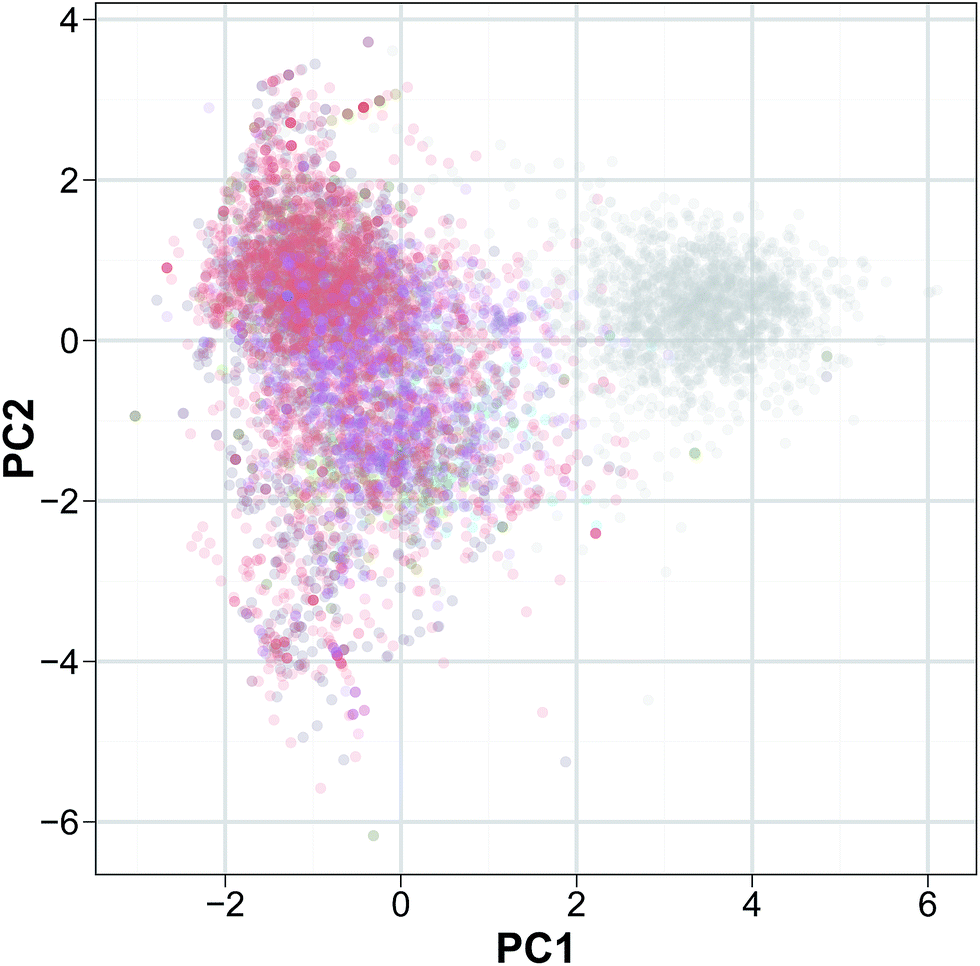 | ||
| Fig. 4 Peptide space of HDPs. Peptides are colored on the basis of their bioactivities: antibacterial (red), anticancer (green), antifungal (blue), antiviral (purple) and the negative set (gray). | ||
PC1 accounted for 25.28% of data variation, which is also the highest explained variance of all the PCs thus, it can be considered as the most informative PC. For the PC1, the loadings of the positive end is dominated by Lys and Leu while the negative end was dominated by Gly. PC2 accounts for the 22.54% of explained variance and the descriptors providing the highest loadings at the positive ends were Leu while the other end was dominated by Ile. PC3 accounted for 20.33% of the data variance in which the loading of PC3 stems Ala on the positive ends whereas Ile on the negative end.
3.2 Multivariate analysis
DT models for differentiating HDPs as having antibacterial, anticancer, antifungal or antiviral activity were built using either AAC or DPC as descriptors, which accounted for the composition of single amino acids and dipeptides, respectively. Two different approaches were taken in preparing the data sets for prediction. The first type entailed the generation of four separate binary class data sets in which each of the four bioactivities were combined with peptides obtained from Xiao et al.32 as the negative bioactivity set. The second type is a simple merger of all four bioactivities considered in this study in combination with the aforementioned negative set, termed herein as the multi-class model. The predictive performance of the resulting DT models were assessed via 10-fold cross-validation and external set using statistical parameters comprising of Ac, Sn, Sp and MCC.Prior to model construction, the modelability of the data set was evaluated using the MODI index. Particularly, antibacterial, anticancer, antifungal, antiviral and the combined HDP data sets built using AAC/DPC descriptors afforded MODI values of 0.942/0.941, 0.953/0.922, 0.942/0.941, 0.945/0.929 and 0.490/0.618, respectively. It can be clearly seen that nearly all data sets met the established cut-off of 0.65 for modelable data sets with the exception of the combined HDP data set. A closer look indicated that modelability of the HDP data set built using AAC descriptors provided poorer MODI value than DPC descriptors with respective values of 0.490 and 0.618.
Table 1 shows the overall performance of models built with AAC descriptors. It can be seen that all binary class models afforded good performance with Ac, Sn, Sp and MCC in excess of 96%, 91%, 92% and 0.89, respectively. In comparison to the binary class models, the multi-class models exhibited a decrease in the overall performance.
| Model type | Classes | Training set | 10-fold CV | External set | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ac (%) | Sn (%) | Sp (%) | MCC | Ac (%) | Sn (%) | Sp (%) | MCC | Ac (%) | Sn (%) | Sp (%) | MCC | ||
| Binary class | Antibacterial | 99.10 ± 0.12 | 97.26 ± 0.48 | 99.70 ± 0.08 | 0.98 ± 0.00 | 97.51 ± 0.18 | 93.69 ± 0.51 | 98.76 ± 0.16 | 0.93 ± 0.01 | 97.57 ± 0.40 | 93.81 ± 1.39 | 98.79 ± 0.40 | 0.93 ± 0.01 |
| Anticancer | 99.16 ± 0.21 | 99.55 ± 0.20 | 97.73 ± 0.66 | 0.98 ± 0.01 | 97.57 ± 0.28 | 98.46 ± 0.24 | 94.32 ± 0.95 | 0.93 ± 0.01 | 97.68 ± 0.75 | 98.61 ± 0.66 | 94.28 ± 2.90 | 0.93 ± 0.02 | |
| Antifungal | 98.91 ± 0.20 | 98.35 ± 0.40 | 99.35 ± 0.19 | 0.98 ± 0.00 | 96.86 ± 0.24 | 95.96 ± 0.38 | 97.56 ± 0.32 | 0.94 ± 0.01 | 97.02 ± 0.60 | 96.31 ± 1.10 | 97.57 ± 0.73 | 0.94 ± 0.01 | |
| Antiviral | 99.08 ± 0.27 | 97.91 ± 0.88 | 99.44 ± 0.26 | 0.97 ± 0.01 | 96.02 ± 0.44 | 91.23 ± 1.20 | 97.46 ± 0.38 | 0.89 ± 0.01 | 96.03 ± 0.88 | 91.49 ± 2.93 | 97.39 ± 0.87 | 0.89 ± 0.02 | |
| HDPs | 99.28 ± 0.09 | 99.82 ± 0.05 | 96.62 ± 0.55 | 0.97 ± 0.00 | 98.12 ± 0.14 | 99.23 ± 0.11 | 92.65 ± 0.56 | 0.93 ± 0.01 | 98.17 ± 0.29 | 99.28 ± 0.23 | 92.72 ± 1.48 | 0.93 ± 0.01 | |
| Multi-class | Overall | 95.52 ± 0.26 | 95.84 ± 0.25 | 89.89 ± 2.51 | 0.68 ± 0.02 | 93.16 ± 0.25 | 94.50 ± 0.17 | 69.70 ± 2.75 | 0.51 ± 0.02 | 93.40 ± 0.60 | 94.67 ± 0.45 | 71.51 ± 6.21 | 0.52 ± 0.05 |
| Antibacterial | 95.39 ± 0.29 | 95.75 ± 0.25 | 89.04 ± 2.99 | 0.67 ± 0.02 | 93.16 ± 0.27 | 94.51 ± 0.17 | 69.63 ± 2.91 | 0.51 ± 0.02 | 93.36 ± 0.60 | 94.70 ± 0.45 | 70.99 ± 5.85 | 0.53 ± 0.05 | |
| Anticancer | 95.32 ± 0.28 | 95.73 ± 0.27 | 88.27 ± 2.46 | 0.67 ± 0.02 | 93.18 ± 0.26 | 94.49 ± 0.16 | 70.04 ± 2.82 | 0.51 ± 0.02 | 93.42 ± 0.62 | 94.72 ± 0.46 | 70.99 ± 5.85 | 0.53 ± 0.05 | |
| Antifungal | 96.02 ± 0.18 | 96.18 ± 0.21 | 93.40 ± 1.64 | 0.72 ± 0.01 | 93.15 ± 0.26 | 94.51 ± 0.17 | 69.52 ± 2.69 | 0.51 ± 0.02 | 93.39 ± 0.66 | 94.62 ± 0.43 | 71.98 ± 5.55 | 0.52 ± 0.04 | |
| Antiviral | 95.33 ± 0.29 | 95.70 ± 0.29 | 88.85 ± 2.94 | 0.67 ± 0.02 | 93.15 ± 0.23 | 94.49 ± 0.17 | 69.61 ± 2.58 | 0.51 ± 0.02 | 93.39 ± 0.66 | 94.62 ± 0.44 | 71.79 ± 6.98 | 0.52 ± 0.05 | |
A closer look at results from both 10-fold CV and external sets revealed a mild decrease of Ac from 96–97% for binary class models to roughly 93% for the multi-class model. Similarly, the Sn of the multi-class model exhibited a slight decrease for some models (i.e. anticancer, antifungal and HDPs exhibited a drop in performance from 95–99% to roughly 94%) whereas a slight gain was seen in some (e.g. antibacterial and anticancer exhibited a gain in performance from 91–93% to 94%). Conversely, a steep decrease in Sp was observed where values dropped from 92–98% in binary class models to 69–71% in the multi-class model. Similarly, MCC also showed a sharp drop from 0.89–0.94 in binary class models to 0.51–0.53 in the multi-class model.
Table 2 summarizes the performance of models built with DPC descriptors. In comparison to models built with AAC descriptors, binary class models constructed as a function of DPC descriptors were found to afford a slight decrease in the prediction performance as can be seen from the 10-fold CV and external sets. Particularly, Ac decreased from 96–98% to 92–96%, Sn decreased from 91–99% to 78–98%, Sp decreased from 92–98% to 82–97% and MCC decreased from 0.89–0.94 to 0.78–0.88. As for the multi-class model, the performance did not differ significantly whether models were built with AAC or DPC descriptors. Particularly, Ac, Sn and MCC afforded no apparent difference while Sp was found to improve slightly from 69–71% to 73–75%.
| Model type | Classes | Training set | 10-fold CV | External set | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ac (%) | Sn (%) | Sp (%) | MCC | Ac (%) | Sn (%) | Sp (%) | MCC | Ac (%) | Sn (%) | Sp (%) | MCC | ||
| Binary class | Antibacterial | 97.37 ± 0.24 | 92.97 ± 0.79 | 98.81 ± 0.21 | 0.93 ± 0.01 | 94.80 ± 0.25 | 87.85 ± 0.74 | 97.07 ± 0.23 | 0.86 ± 0.01 | 94.97 ± 0.60 | 88.44 ± 1.63 | 97.09 ± 0.62 | 0.86 ± 0.02 |
| Anticancer | 97.01 ± 0.43 | 99.22 ± 0.32 | 88.89 ± 1.79 | 0.91 ± 0.01 | 94.49 ± 0.42 | 97.84 ± 0.39 | 82.20 ± 1.42 | 0.83 ± 0.01 | 94.60 ± 1.14 | 97.89 ± 1.02 | 82.51 ± 4.28 | 0.84 ± 0.04 | |
| Antifungal | 96.77 ± 0.52 | 98.02 ± 1.90 | 95.79 ± 0.95 | 0.94 ± 0.01 | 94.02 ± 0.40 | 95.68 ± 1.07 | 92.72 ± 0.73 | 0.88 ± 0.01 | 94.12 ± 1.00 | 95.51 ± 3.03 | 93.04 ± 1.71 | 0.88 ± 0.02 | |
| Antiviral | 96.49 ± 0.72 | 88.31 ± 2.92 | 98.95 ± 0.37 | 0.90 ± 0.02 | 92.36 ± 0.55 | 79.30 ± 1.91 | 96.30 ± 0.44 | 0.78 ± 0.02 | 92.27 ± 1.26 | 78.60 ± 5.05 | 96.35 ± 1.13 | 0.78 ± 0.04 | |
| HDPs | 98.30 ± 0.17 | 99.35 ± 0.12 | 93.15 ± 0.91 | 0.94 ± 0.01 | 96.35 ± 0.18 | 98.30 ± 0.14 | 86.77 ± 0.84 | 0.87 ± 0.01 | 96.44 ± 0.45 | 98.35 ± 0.40 | 87.05 ± 2.04 | 0.87 ± 0.02 | |
| Multi-class | Overall | 95.28 ± 0.27 | 95.62 ± 0.24 | 89.13 ± 3.33 | 0.66 ± 0.02 | 93.29 ± 0.24 | 94.28 ± 0.19 | 73.66 ± 2.87 | 0.51 ± 0.02 | 93.39 ± 0.54 | 94.39 ± 0.41 | 74.13 ± 6.70 | 0.51 ± 0.05 |
| Antibacterial | 95.30 ± 0.26 | 95.62 ± 0.22 | 89.54 ± 3.31 | 0.66 ± 0.02 | 93.27 ± 0.24 | 94.27 ± 0.21 | 73.39 ± 2.63 | 0.51 ± 0.02 | 93.48 ± 0.46 | 94.43 ± 0.42 | 74.86 ± 5.53 | 0.52 ± 0.04 | |
| Anticancer | 95.25 ± 0.27 | 95.63 ± 0.27 | 88.58 ± 3.42 | 0.66 ± 0.02 | 93.30 ± 0.25 | 94.28 ± 0.17 | 73.79 ± 2.85 | 0.51 ± 0.02 | 93.48 ± 0.50 | 94.47 ± 0.41 | 74.70 ± 6.51 | 0.52 ± 0.02 | |
| Antifungal | 95.28 ± 0.28 | 95.63 ± 0.23 | 88.90 ± 3.20 | 0.66 ± 0.02 | 93.31 ± 0.24 | 94.31 ± 0.18 | 73.75 ± 3.08 | 0.51 ± 0.02 | 93.48 ± 0.57 | 94.42 ± 0.42 | 75.39 ± 7.25 | 0.52 ± 0.05 | |
| Antiviral | 95.30 ± 0.26 | 95.62 ± 0.23 | 89.51 ± 3.38 | 0.66 ± 0.02 | 93.29 ± 0.24 | 94.26 ± 0.18 | 73.66 ± 2.93 | 0.51 ± 0.02 | 93.40 ± 0.58 | 94.37 ± 0.48 | 74.51 ± 7.24 | 0.51 ± 0.05 | |
The lower level of performance of the multi-class models when compared to that of binary class models could be attributed to the higher degree of complexity and the inherent heterogeneity of positive samples in the data set (i.e. the HDP class comprising of four bioactivities). Likewise, this contributed to the lower MODI value of the multi-class model (i.e. 0.490 and 0.618 for models built with AAC and DPC descriptors, respectively) when compared to those of the binary class models (i.e. 0.942–0.953 and 0.922–0.941 for models built with AAC and DPC descriptors, respectively).
Eriksson and Johansson47 established that when the R2 − Q2 margin is in excess of 0.2–0.3 then there is a possibility for chance correlation or the presence of outliers in the data set whereas if the R2 − Q2 margin is less than 0.2–0.3 then it is likely to be predictive and reliable. As the original concept was based on regression metrics (e.g. R2 − Q2), we will be extrapolating the concept to the classification problem by also considering the same magnitude of the margin where we will deem models to be reliable and predictive if the difference of statistical metrics (e.g. Ac, Sn and Sp) between the training and 10-fold CV sets as well as the training and external sets are less than 20–30%. On a similar note, the same margin magnitude of 0.2–0.3 was applied for the MCC metric.
In general, binary class models built with AAC and DPC descriptors afforded relatively low margins in the difference of statistical metrics (i.e. less than 10% for Ac, Sp and Sn while less than 0.1 for MCC) between the training set and the 10-fold CV set as well as the difference between the training set and the external set. A closer observation of the binary class models revealed that AAC models provided slightly lower margins than the DPC models.
As for the multi-class models of both AAC and DPC models produced lower margin than the binary class models for Ac and Sn whereas the Sp and MCC parameters of multi-class models afforded poorer results in which margins were about 2–6 folds higher than their binary class counterpart (i.e. Sp margin of 13–23% versus 1–6%, respectively, and MCC margin of 0.14–0.21 versus 0.04–0.12, respectively). Moreover, multi-class DPC models afforded lower margins than their AAC counterpart for all metrics evaluated. In summary, classification models based on AAC descriptors afforded the best performance as it could perform comparatively well on both binary and multi-class models.
In addition, classification models based on the combined use of AAC and DPC descriptors, termed herein as AAC + DPC, were also evaluated and their results are summarized in ESI Table S1.† This model performed on par with models built with AAC descriptors while affording slightly higher performance for multi-class models. Moreover, ESI Table S2† lists the classification performance of models built with CC descriptors and it was observed that binary class models yielded comparable performance with that of AAC models. However, the multi-class models were of poorer quality in which CV models produced a moderate drop in performance by 0.05–0.08 while the external set showed a significant loss in predictivity.
3.3 Benchmark against the RF classifier
In addition to the use of the DT classifier for constructing classification models, the RF classifier was also applied in the construction of classification models as to benchmark against the DT classifier using the same descriptor sets and the prediction results are summarized in ESI Tables S3–S6† for models built using AAC, DPC, AAC + DPC and CC descriptors. In general, it was found that classification models built using the DT classifier performed consistently well for both binary and multi-class models whereas the RF classifier performed extremely well on binary models while performing poorly on the multi-class model. Such substantial deterioration in its performance may be ascribed to the fact that RF models are built with bootstrapped samples for its constituent trees. Thus, there is a substantial chance that bootstrapped samples contain only a few to none of the minority class that would produce trees with poor performance in predicting the minority class.48,49 In light of the overall good performance and reliability of DT models, they were selected for further investigation on the underlying features that are important for the model's performance as well as their biological relevance in governing the observed bioactivity.3.4 Model extrapolation
Recently, Aguilera-Mendoza et al.33 compiled a set of non-redundant HDPs from 25 databases. Thus, to evaluate the model's extrapolation capability in a real-world setting, this large HDP set (i.e. peptides that are independent from the aforementioned training and test sets) was used as an additional external validation set. Particularly, all of these peptides were assigned the class label of HDP. Thus, a classification model was built in a similar fashion as mentioned earlier in which the original binary data set consisting of two class labels (i.e. HDP versus non-HDP) was used as the training set. Such trained model was then applied to predict the class label of this additional external data set as being HDP or non-HDP. Since all peptides in this external validation set was assigned the class label of HDP therefore a correct prediction for this external set would be to predict the class label as HDP. Results indicated that all classification models could well extrapolate on this additional external set as deduced from Ac values of 97.21, 94.89, 96.33 and 97.85% for models built with DT using AAC, DPC, AAC + DPC and CC descriptors, respectively. Similarly, Ac values of 98.39, 96.96, 98.11 and 98.45% were observed for models built with RF using AAC, DPC, AAC + DPC and CC descriptors, respectively.3.5 Applicability domain analysis
Table 3 reports the prediction accuracy as a function of the applicability domain in which sequences of the test set are binned into four quantiles as a function of the Euclidean distance (i.e. as averaged over five nearest neighbors). For the negative set, it can be seen that the accuracy deteriorated as the sequence neighbor-averaged Euclidean distance increases. In practice, a new peptide sequence showing a neighbor-average near 0.1 is likely to be predicted with good accuracy. On the other hand, the second quantile of inactives displayed an Ac of 97.6% thereby indicating that the model could not predict the three peptides that were negative. Nevertheless, these data show that estimating the accuracy for each peptide prediction constitutes a valuable source of information for understanding the classification model.| Quantile | Normalized Euclidean distance | N | Accuracy (%) |
|---|---|---|---|
| Q1 | 0.0–0.18 | 86 | 100% |
| Q2 | 0.18–0.27 | 85 | 97.6 |
| Q3 | 0.27–0.36 | 85 | 98.8 |
| Q4 | 0.36–1.00 | 86 | 80.2 |
3.6 Mechanistic interpretation of feature importance
Identifying and understanding the important features governing HDP bioactivities is an important first step towards designing HDPs with desirable properties. DT inherently possess a built-in function for revealing important features of data sets of interest. This study performed 100 independent data splits followed by constructing DTs for each split with important features based on information gain. Particularly, the most used feature is also deemed to be the most important feature. The feature importance of the four classes of HDPs are deduced from two sets of descriptors namely the (i) AAC and (ii) DPC descriptors. Interpretation of these descriptors are discussed and scrutinized in the forthcoming sections.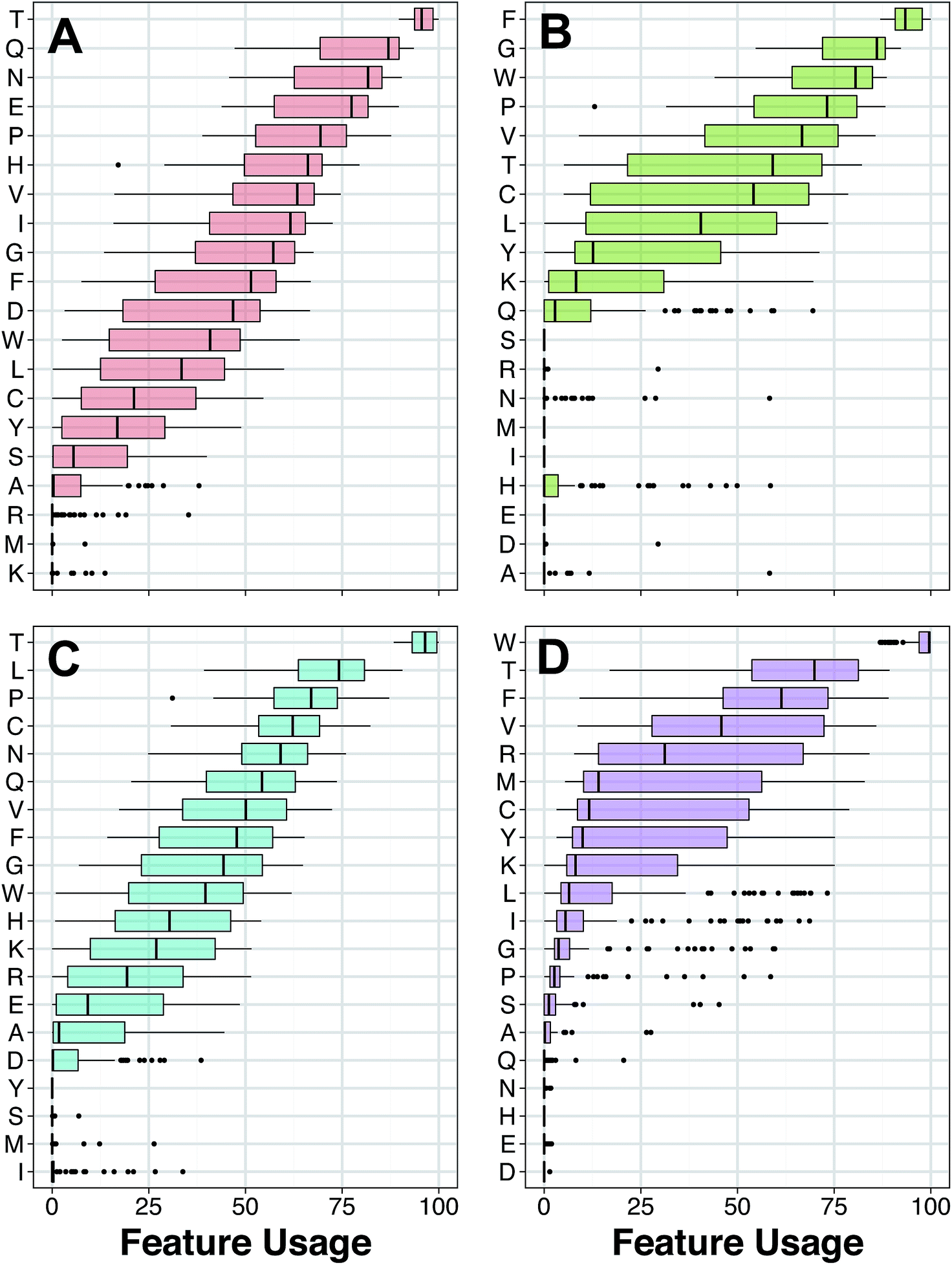 | ||
| Fig. 5 Box plot for AAC feature usage for HDPs with antibacterial (A), anticancer (B), antifungal (C) and antiviral (D) bioactivities. Features with the highest usage is deemed to be the most important. | ||
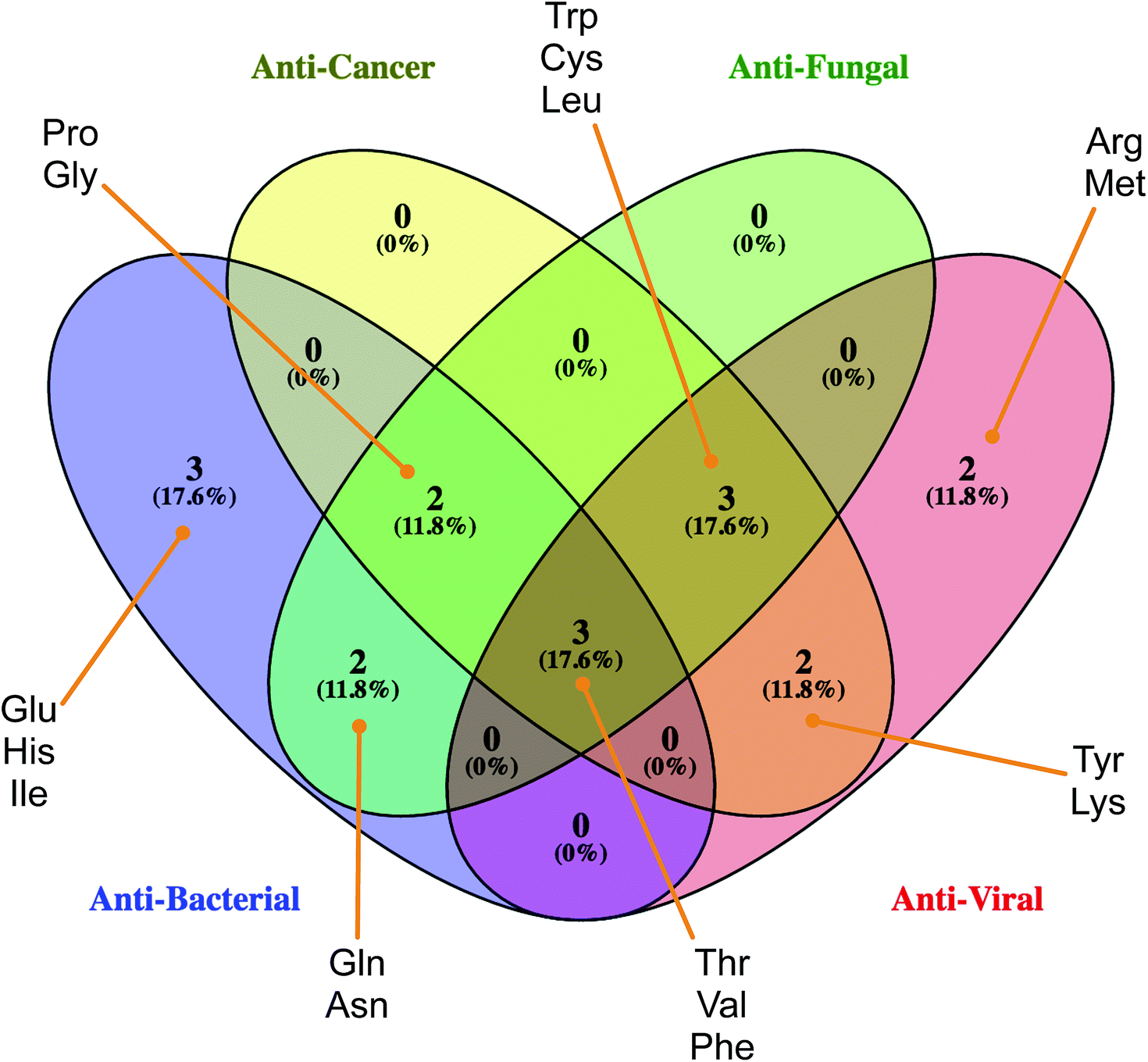 | ||
| Fig. 6 Venn's diagram of the common set of amino acids found amongst the four sets of bioactivity classes of HDPs. Image created using Venny 2.1.0 (http://bioinfogp.cnb.csic.es/tools/venny/). | ||
Amongst the important AACs shown in Fig. 5, Thr proclaims a significant role in all four bioactivity classes of HDPs. Thr is abundantly found in the intestinal mucin and plasma γ-globulin and are involved in many physiological and biochemical processes including promoting growth, enhancing immune mechanisms and stimulating lymphocyte proliferation.50–53 Thr takes part in the immune system by aiding the production of antibody as a major component of γ-globulin.50,54 The importance of Thr in the bioactivity of HDPs is related to its role in glycosylation, which is the most common form of post-translational modification involving the linkage between N-acetylactosamine (GalNAc) of membrane glycoproteins and the hydroxyl group of Thr residue. When a cell undergo tumorigenesis, it has the likelihood of being glycosylated. In this manner, anticancer peptides are rich in Thr residues and are thus more susceptible to induce cytotoxicity towards cancer cells.55,56 Moreover, Hara and Yamakawa57 reported that O-glycosylation of a Thr residue led to an increase in the antibacterial activity of lebocin. In addition, Thr substitution on the HIV protease inhibitory peptide resulted in a significant enhancement of its antiviral activity.58
Apart from Thr, Gln was also found to play an important role in affording the antibacterial activity of peptides with a high Gini index score. Gln is the most abundant free amino acid in human blood and was widely described for its contribution in the immune system. It was stated that Gln involves in improving the intestinal permeability to reduce the risk of systemic infections that originates in the gastrointestinal tract.59 Furthermore, Gln is required for stimulation of some immune cells such as lymphocytes and macrophages to defend against infections.60 The functional role of Gln in antibacterial peptides was proposed by Suarez et al.61 whereby Gln rich portions of Moringa oleifera seed-derived Flo peptide is crucial for antibacterial activity by mediating the aggregation and sedimentation of bacterial cells. Bactericidal process of this peptide is derived by aforementioned flocculation effect in conjunction with destabilization mechanism of hydrophobic loop structure. Their findings provide a notable importance of Gln residues in antibacterial peptides.
According to the Gini index, Phe is not only the top-ranking AAC for anticancer activity (Fig. 5A) but also a notable residue for other bioactivity of HDPs (Fig. 6). Phe is well recognized for its hydrophobic nature owing to the benzyl side chain. Because of its hydrophobic property, Phe-rich peptides exhibit potent antibacterial activity.62,63 Furthermore, the composition of Phe is relatively prominent in anticancer peptides (ACPs) rather than other antimicrobial peptides (AMPs)64,65 and have a noteworthy function on anticancer activity. In particular, the Phe residue has more favorable helix propensity than other aromatic residues.66 Thus, the findings of Shan et al.67 revealed that Phe substituted peptide analogs possess higher helical content which can be modulated to increase the anticancer activity of peptides.68,69
Aromatic amino acid, Trp, is found to be the most important AAC for antiviral activity (Fig. 5D). In general, Trp-rich peptides are well known for their powerful antimicrobial activity induced by their distinctive biochemical property to interact with and insert into biological membranes. Moreover, broad spectrum activities of Trp-rich peptides are in the range of antibacterial, antiviral, antifungal, antiprotozoal and anticancer activities.70,71 The mechanism behind the bioactivities of Trp-rich peptides is not clear yet, but the essential role of Trp residues was reported by Giannecchini and colleagues72 whereby the deletion of Trp-rich domain led to loss of antiviral activity of peptide 59. In addition, the work of Kliger et al.73 also explained that the Trp-rich region of DP178 peptide binds to the membrane of Human Immunodeficiency Virus type-1 (HIV-1) to inhibit cell fusion and viral entry. As stated in previous literatures, there is no doubt that the electrostatic interaction of Trp to phosphatidylcholine of biological membranes is dominated by its aromatic structure which serves as a membrane anchor.74,75 However, additional studies are needed to further understand the in-depth mechanism of Trp.
Another important AAC is Pro (i.e. a non-polar, aliphatic amino acid), which is one of the top-five AAC for both antibacterial and antifungal activity. Particularly, Pro-rich peptides represents a group of linear peptides and also a subgroup of antifungal peptides (AFPs) in the antimicrobial peptide database76 that is comprised of more than 30% Pro residues in their primary structure.77 Some of the Pro-rich peptides exhibit not only antifungal activity but also antibacterial activity.78,79 The prominent role of Pro residues in antifungal activity was discussed by Cabras et al.78 whereby Pro-rich peptides SP-B (i.e. APPGARPPPGPPPPGPPPPGP) are able to form an unusual secondary structure, polyproline helix type-II. Because of this unusual secondary structure, Pro-rich peptides fail to generate an amphipathic structure and this synergy is important to mention for its consequences on enhancing antifungal activity together with minimum hemolytic activity.80 In addition, Pro residues promote peptide entry into lipid membrane bi-layer without disrupting the cell membrane and allows subsequent interaction with specific target inside the cell which is essential for nontoxic antimicrobial activity.78,81
The results revealed that the aforementioned AAC descriptors consisting of Thr, Gln, Phe, Trp and Pro were the most significant features governing the antibacterial, anticancer, antifungal and antiviral activities of peptides as indicated in this study or in existing literature.
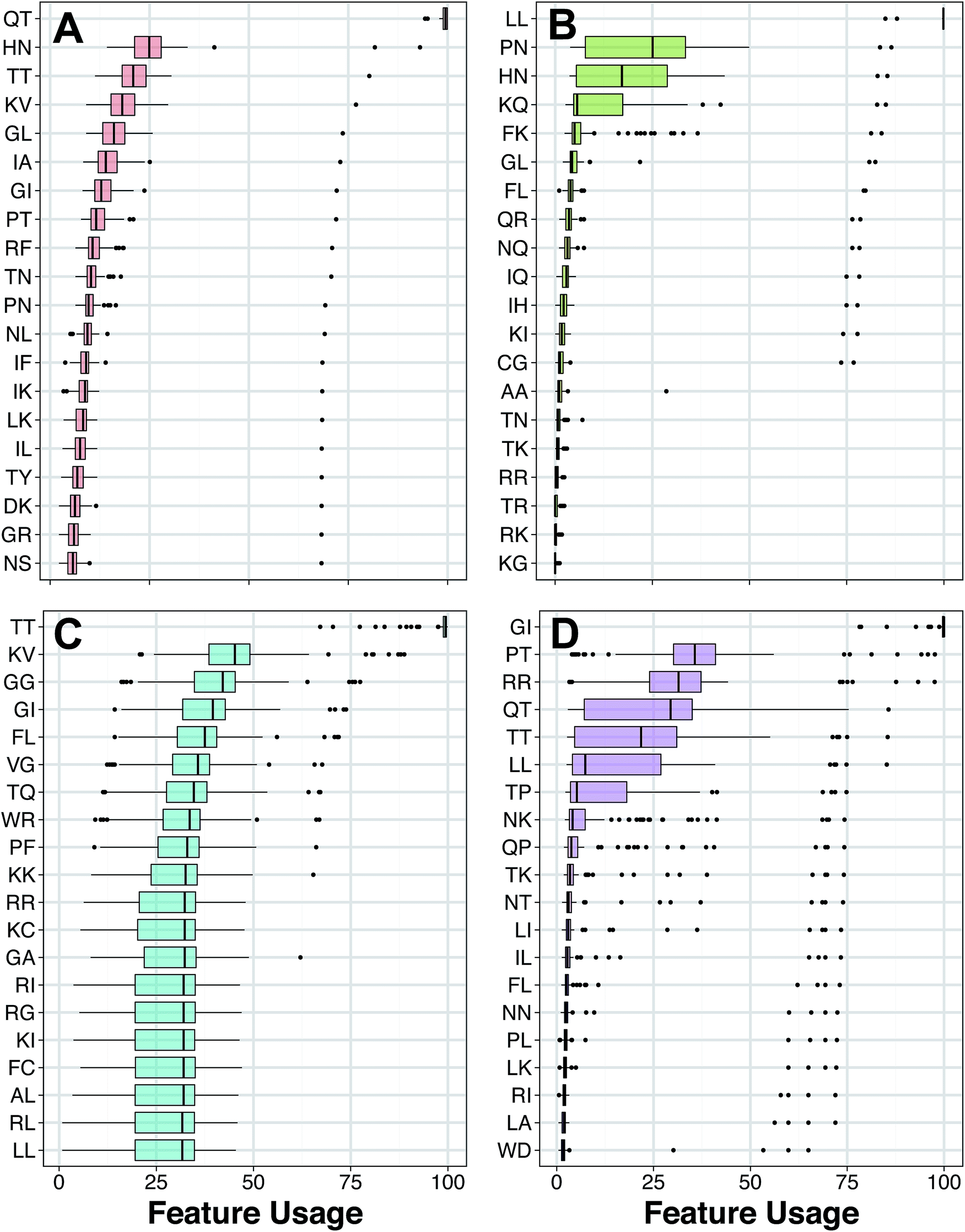 | ||
| Fig. 7 Box plots of DPC feature usage for HDPs with antibacterial (A), anticancer (B), antifungal (C) and antiviral (D) bioactivities. Features with the highest usage is deemed to be the most important. | ||
Each amino acid was assigned into one of three groups for each of the seven amino acid properties as proposed by Chothia and Finkelstein.82 For example, if a three residue peptide is composed of a hydrophobic, neutral and polar amino acid then its corresponding class composition would yield the string ‘123’. Furthermore, a three residue peptide composed solely of hydrophobic residues would afford the string ‘111’. Moreover, when dipeptides are converted into amino acid composition then there are nine possible combinations that exists for each property. The R statistical package protr provides a convenient way for calculating the amino acid class composition as well as providing a well compiled table that explains the class composition. However, it does not automatically provide the property statistics for calculated peptides. Thus, an in-house C++ was coded and used herein for the property analysis and the obtained results are provided in ESI Table S7.†
The top twenty most important dipeptides of HDPs with antimicrobial activity do not show significant bias towards a particular property composition. Dipeptides converted into attribute classes were found to be fairly and evenly distributed amongst the different possible combinations of the property composition. This observation is in line with the work of ref. 83, which states that the determining factors of AMPs at the/hlglobal level are hydrophobicity, charge and helicity. As such, effects of local sequence order are less important and are thus reflected by the absence of significant property composition at the dipeptide level.
ESI Table S8† describes the various characteristics of the amino acid properties considered in the dipeptide analysis. As four out of the twenty amino acids are non-neutral, therefore there is a high probability for dipeptides to consist of two consecutive neutral amino acids. On the other hand, HDPs with anticancer activity displayed some interesting property patterns in their twenty most distinguishing dipeptides. For the secondary structure property, six dipeptides consisted of two consecutive helical-forming amino acids while another six consisted of one helical-forming amino acid followed by one strand-forming amino acid whereas none of the other seven possible class combinations for the secondary structure property was exhibited by more than two dipeptides. Similarly, ten out of the twenty dipeptides were made of either two consecutive polar amino acids or one neutral amino acid followed by one polar amino acid. With the rest of the seven possible class combinations thinly spread. For the property of solvent accessibility, nearly all the dipeptides were concentrated in three possible class combinations. Five dipeptides were made of two consecutive buried amino acids, another five dipeptides were made of two consecutive exposed amino acids and five additional dipeptides were made of one intermediately exposed amino acid followed by one exposed amino acid. The remaining possible class compositions were thinly spread.
It has previously been discovered that unlike AMPs, the activity of oncolytic peptides were very sensitive to the effect of amino acid sequence.84 As such, the fact that many of the twenty most distinguishing dipeptides of the oncolytic peptides were concentrated in certain property class compositions may be a reflection of this activity dependency on the sequence order effect.
As for HDPs with antifungal activity, seven of their twenty distinguishing dipeptides were made of one high polarizable amino acid followed by one moderately polarizable amino acid. None of the other nine possible class combinations for the property of polarizability was exhibited by more than three dipeptides. Another noteworthy dipeptide property pattern for the antifungal peptides is that, there are eight dipeptides made of two buried amino acid and six dipeptides made of one exposed followed by one buried amino acid. Another dipeptide feature to be noted is that there were six dipeptides consisting of one positively-charged amino acid followed by a neutral one. This is in contrast to the other HDP classes, which had few of their twenty most distinguishing dipeptides consisting of anything but two neutral amino acids. Lastly, HDPs with antiviral activity did not seem to have significant preference for a particular class composition in any of the property attributes calculated, this is similar to the AMPs.
As can be seen in Fig. 5A and B, it was found that the top ranked features for HDPs with antibacterial and anticancer bioactivity, respectively, are distinctly different although, the amino acids (e.g. Pro) were similar for HDPs having antibacterial and anticancer properties. While there is no definitive consensus on whether the mechanism of AMP and OLPs are different,56 existing studies indicate that while AMPs and OLPs have an overall similar action pathways, they have numerous subtle yet important differences in both structure and activity mechanism.84,85 In addition to potent activity in combating two major contemporary health threats, namely pathogenic microbes and malignant neoplasm, HDPs have shown strong activity in combating other types of pathogens including, fungi and viruses. It would therefore be of great interest to compare whether different peptide structures are responsible for the activity against different pathogens or are the different activity types determined by a common structure. Thus, the results obtained will be beneficial for the identification of critical AMP and OLP structures and as a guide for the future development of HDPs as therapeutic for these classes of pathogens.
4 Conclusions
In spite of several decades of research into the structure–function relationship of HDP, we are far from fully understanding the implications of these large volumes of data that are often disparate and heterogeneous in nature. This is in concomitant with the inherent complexity of developing HDP-based therapeutic drugs. Therefore, to improve the chances of success, a map to guide the fine-tuning of structure–functional properties of HDP is needed. Thus, the results obtained from the learning classifiers provided general guidelines that may aid in the understanding of the HDP activity. We hope that the findings gained from this study would promote further research in the design and discovery of improved and efficient HDPs.Author's contributions
CN conceived the study and designed the experiments. SS compiled the data set and constructed the classification models. HL analyzed the contribution of DPC features on HDP bioactivities. TSW and AHK analyzed the contribution of AAC features on HDP bioactivities. AAM prepared figures of HDP structural diversity and reviewed the literature for notable HDPs with various bioactivity. AAM, TP, WS, PN, JESW, MPG and CN took part in discussion and analysis of results. MPG analyzed the applicability domain and contribution of AAC features on HDP bioactivity. SS, HL, TSW and CN drafted the manuscript. CN vetted and finalized the manuscript. All authors read and approved the final manuscript.Acknowledgements
This work is supported by the Goal-Oriented Research Grant (No. E09/2557) from Mahidol University. Partial support via the Swedish Research Links program (No. C0610701) from the Swedish Research Council is also acknowledged. SS and HL are graduate students grateful for research assistantship from Mahidol University. SS is also grateful for financial support from Department of Chemistry, Faculty of Science, Kasetsart University and the National Research University (NRU) for supporting his Ph.D. studies. Authors would also like to thank Dr Nathjanan Jongkon for critically reading the manuscript and providing useful comments.References
- H. C. Neu, Science, 1992, 257, 1064–1073 CAS.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- N. Papo and Y. Shai, Cell. Mol. Life Sci., 2005, 62, 784–790 CrossRef CAS PubMed.
- L. Amiri-Kordestani, A. Basseville, K. Kurdziel, A. T. Fojo and S. E. Bates, Drug Resist. Updates, 2012, 15, 50–61 CrossRef CAS PubMed.
- A. L. Hilchie, C. D. Doucette, D. M. Pinto, A. Patrzykat, S. Douglas and D. W. Hoskin, Breast Cancer Res., 2011, 13, R102 CrossRef CAS PubMed.
- G. Wang, M. L. Hanke, B. Mishra, T. Lushnikova, C. E. Heim, V. C. Thomas, K. W. Bayles and T. Kielian, ACS Chem. Biol., 2014, 9, 1997–2002 CrossRef CAS PubMed.
- S. Al-Benna, Y. Shai, F. Jacobsen and L. Steinstraesser, Int. J. Mol. Sci., 2011, 12, 8027–8051 CrossRef CAS PubMed.
- R. A. Cruciani, J. L. Barker, M. Zasloff, H.-C. Chen and O. Colamonici, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3792–3796 CrossRef CAS.
- Y. Park, D. G. Lee and K.-S. Hahm, Biotechnol. Lett., 2003, 25, 1305–1310 CrossRef CAS PubMed.
- W. Hong, T. Li, R. Song, R. Zhang, Z. Zeng, S. Han, Y. Zhang, W. Li and Z. Cao, Antiviral Res., 2014, 102, 1–10 CrossRef CAS PubMed.
- M. A. Lynn, J. Kindrachuk, A. K. Marr, N. Pante, M. R. Elliott, S. Napper, R. E. Hancock and W. R. McMaster, PLoS Neglected Trop. Dis., 2011, 5, 1–13 Search PubMed.
- B. Lemaitre, J.-M. Reichhart and J. A. Hoffmann, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 14614–14619 CrossRef CAS.
- J. Yu, N. Mookherjee, K. Wee, D. M. Bowdish, J. Pistolic, Y. Li, L. Rehaume and R. E. Hancock, J. Immunol., 2007, 179, 7684–7691 CrossRef CAS.
- K.-Y. G. Choi and N. Mookherjee, Front. Immunol., 2012, 3, 149 Search PubMed.
- P. Hubert, L. Herman, C. Maillard, J.-H. Caberg, A. Nikkels, G. Pierard, J.-M. Foidart, A. Noel, J. Boniver and P. Delvenne, FASEB J., 2007, 21, 2765–2775 CrossRef CAS PubMed.
- A. Giangaspero, L. Sandri and A. Tossi, Eur. J. Biochem., 2001, 268, 5589–5600 CrossRef CAS PubMed.
- D. Takahashi, S. K. Shukla, O. Prakash and G. Zhang, Biochimie, 2010, 92, 1236–1241 CrossRef CAS PubMed.
- Z. T. Zhang and S. Y. Zhu, Insect Mol. Biol., 2009, 18, 549–556 CrossRef CAS PubMed.
- A. Tossi and L. Sandri, Curr. Pharm. Des., 2002, 8, 743–761 CrossRef CAS PubMed.
- C. Nantasenamat, C. Isarankura-Na-Ayudhya, T. Naenna and V. Prachayasittikul, EXCLI J., 2009, 8, 74–88 Search PubMed.
- C. Nantasenamat, C. Isarankura-Na-Ayudhya and V. Prachayasittikul, Expert Opin. Drug Discovery, 2010, 5, 633–654 CrossRef CAS PubMed.
- M. A. Toropova, A. M. Veselinović, J. B. Veselinović, D. B. Stojanović and A. A. Toropov, Comput. Biol. Chem., 2015, 59, 126–130 CrossRef CAS PubMed.
- M. Torrent, D. Andreu, V. M. Nogués and E. Boix, PLoS One, 2011, 6, e16968 CAS.
- H. Jenssen, T. Lejon, K. Hilpert, C. D. Fjell, A. Cherkasov and R. E. W. Hancock, Chem. Biol. Drug Des., 2007, 70, 134–142 CAS.
- B. Vishnepolsky and M. Pirtskhalava, J. Chem. Inf. Model., 2014, 54, 1512–1523 CrossRef CAS PubMed.
- K. Y. Chang and J. R. Yang, PLoS One, 2013, 8, e70166 CAS.
- V. Frecer, B. Ho and J. Ding, Antimicrob. Agents Chemother., 2004, 48, 3349–3357 CrossRef CAS PubMed.
- A. Cherkasov and B. Jankovic, Molecules, 2004, 9, 1034–1052 CrossRef CAS PubMed.
- P. Gramatica, QSAR Comb. Sci., 2007, 26, 694–701 CAS.
- G. Gogoladze, M. Grigolava, B. Vishnepolsky, M. Chubinidze, P. Duroux, M.-P. Lefranc and M. Pirtskhalava, FEMS Microbiol. Lett., 2014, 357, 63–68 CrossRef CAS PubMed.
- N. Xiao, D. S. Cao, M. F. Zhu and Q. S. Xu, Bioinformatics, 2015, 31, 1857–1859 CrossRef CAS PubMed.
- X. Xiao, P. Wang, W.-Z. Lin, J.-H. Jia and K.-C. Chou, Anal. Biochem., 2013, 436, 168–177 CrossRef CAS PubMed.
- L. Aguilera-Mendoza, Y. Marrero-Ponce, R. Tellez-Ibarra, M. T. Llorente-Quesada, J. Salgado, S. J. Barigye and J. Liu, Bioinformatics, 2015, 2553–2559 CrossRef CAS PubMed.
- H. González-Díaz, R. Molina and E. Uriarte, Bioorg. Med. Chem. Lett., 2004, 14, 4691–4695 CrossRef PubMed.
- Y. Marrero-Ponce, E. Contreras-Torres, C. R. García-Jacas, S. J. Barigye, N. Cubillán and Y. J. Alvarado, J. Theor. Biol., 2015, 374, 125–137 CrossRef CAS PubMed.
- H. B. Rao, F. Zhu, G. B. Yang, Z. R. Li and Y. Z. Chen, Nucleic Acids Res., 2011, 39, W385–W390 CrossRef CAS PubMed.
- Y. B. Ruiz-Blanco, W. Paz, J. Green and Y. Marrero-Ponce, BMC Bioinf., 2015, 16, 162 CrossRef PubMed.
- A. Golbraikh, E. Muratov, D. Fourches and A. Tropsha, J. Chem. Inf. Model., 2014, 54, 1–4 CrossRef CAS PubMed.
- H. Wickham, Wiley Interdisciplinary Reviews: Computational Statistics, 2011, 3, 180–185 CrossRef.
- W. R. Zwick and W. F. Velicer, Psychol. Bull., 1986, 99, 432 CrossRef.
- M. Kuhn, J. Stat. Software, 2008, 28, 1–26 Search PubMed.
- R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2014 Search PubMed.
- A. Dinno, paran: Horn's Test of Principal Components/Factors, 2012 Search PubMed.
- K. Hornik, C. Buchta and A. Zeileis, Comput. Stat., 2009, 24, 225–232 CrossRef.
- I. H. Witten and E. Frank, Data mining: Practical machine learning tools and techniques, Morgan Kaufmann, 2005 Search PubMed.
- M. Hall, E. Frank, G. Holmes, B. Pfahringer, P. Reutemann and I. H. Witten, ACM SIGKDD explorations newsletter, 2009, 11, 10–18 CrossRef.
- L. Eriksson and E. Johansson, Chemom. Intell. Lab. Syst., 1996, 34, 1–19 CrossRef CAS.
- C. Chen, A. Liaw and L. Breiman, Using Random Forest to Learn Imbalanced Data, Department of Statistics, University of Berkeley technical report, 2004 Search PubMed.
- B. F. Huang and P. C. Boutros, BMC Bioinf., 2016, 17, 331 CrossRef PubMed.
- P. Li, Y.-L. Yin, D. Li, S. W. Kim and G. Wu, Br. J. Nutr., 2007, 98, 237–252 CrossRef CAS PubMed.
- H.-M. Habte-Tsion, M. Ren, B. Liu, X. Ge, J. Xie and R. Chen, Fish Shellfish Immunol., 2016, 51, 189–199 CrossRef CAS PubMed.
- H.-M. Habte-Tsion, X. Ge, B. Liu, J. Xie, M. Ren, Q. Zhou, L. Miao, L. Pan and R. Chen, Fish Shellfish Immunol., 2015, 42, 439–446 CrossRef CAS PubMed.
- H. Sepehri Moghaddam and M. Emadi, Int. J. Adv. Biol. Biomed. Res., 2014, 2, 756–763 Search PubMed.
- K. K. Bhargava, R. P. Hanson and M. L. Sunde, Poult. Sci., 1971, 50, 710–713 CrossRef CAS PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, in Biochemistry, ed. W. H. Freeman, New York, 5th edn, 2002 Search PubMed.
- D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 357–375 CrossRef CAS PubMed.
- S. Hara and M. Yamakawa, Biochem. J., 1995, 310, 651–656 CrossRef CAS PubMed.
- K. T. Chong, M. J. Ruwart, R. R. Hinshaw, K. F. Wilkinson, B. D. Rush, M. F. Yancey, J. W. Strohbach and S. Thaisrivongs, J. Med. Chem., 1993, 36, 2575–2577 CrossRef CAS PubMed.
- J. R. Rapin and N. Wiernsperger, Clinics, 2010, 65, 635–643 CrossRef PubMed.
- E. A. Newsholme and P. C. Calder, Nutrition, 1997, 13, 728–730 CrossRef CAS PubMed.
- M. Suarez, M. Haenni, S. Canarelli, F. Fisch, P. Chodanowski, C. Servis, O. Michielin, R. Freitag, P. Moreillon and N. Mermod, Antimicrob. Agents Chemother., 2005, 49, 3847–3857 CrossRef CAS PubMed.
- E. Lee, A. Shin, K.-W. Jeong, B. Jin, H. N. Jnawali, S. Shin, S. Y. Shin and Y. Kim, PLoS One, 2014, 9, e114453 Search PubMed.
- N. Ashwanikumar, N. A. Kumar, P. S. S. Babu, K. C. Sivakumar, M. V. Vadakkan, P. Nair, I. H. Saranya, S. A. Nair and G. S. V. Kumar, Int. J. Nanomed., 2016, 11, 5583–5594 CrossRef PubMed.
- B. Sah, T. Vasiljevic, S. McKechnie and O. Donkor, Compr. Rev. Food Sci. Food Saf., 2015, 14, 123–138 CrossRef CAS.
- Z. Wang and G. Wang, Nucleic Acids Res., 2004, 32, D590–D592 CrossRef CAS PubMed.
- C. N. Pace and J. M. Scholtz, Biophys. J., 1998, 75, 422–427 CrossRef CAS PubMed.
- Y. Shan, J. Huang, J. Tan, G. Gao, S. Liu, H. Wang and Y. Chen, Nanoscale, 2012, 4, 1283–1286 RSC.
- Y.-B. Huang, L.-Y. He, H.-Y. Jiang and Y.-X. Chen, Int. J. Mol. Sci., 2012, 13, 6849–6862 CrossRef CAS PubMed.
- S. R. Dennison, M. Whittaker, F. Harris and D. A. Phoenix, Curr. Protein Pept. Sci., 2006, 7, 487–499 CrossRef CAS PubMed.
- D. J. Schibli, R. F. Epand, H. J. Vogel and R. M. Epand, Biochem. Cell Biol., 2002, 80, 667–677 CrossRef CAS PubMed.
- N. Shagaghi, E. A. Palombo, A. H. Clayton and M. Bhave, World J. Microbiol. Biotechnol., 2016, 32, 31 CrossRef PubMed.
- S. Giannecchini, A. Di Fenza, A. M. D'Ursi, D. Matteucci, P. Rovero and M. Bendinelli, J. Virol., 2003, 77, 3724–3733 CrossRef CAS PubMed.
- Y. Kliger, S. A. Gallo, S. G. Peisajovich, I. Muñoz-Barroso, S. Avkin, R. Blumenthal and Y. Shai, J. Biol. Chem., 2001, 276, 1391–1397 CrossRef CAS PubMed.
- J. A. Killian, I. Salemink, M. R. de Planque, G. Lindblom, R. E. Koeppe and D. V. Greathouse, Biochemistry, 1996, 35, 1037–1045 CrossRef CAS PubMed.
- W.-M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2009, 37, D933–D937 CrossRef CAS PubMed.
- L. Otvos Jr, Cell. Mol. Life Sci., 2002, 59, 1138–1150 CrossRef CAS.
- T. Cabras, R. Longhi, F. Secundo, G. Nocca, S. Conti, L. Polonelli, C. Fanali, R. Inzitari, R. Petruzzelli, I. Messana, M. Castagnola and A. Vitali, J. Pept. Sci., 2008, 14, 251–260 CrossRef CAS PubMed.
- A. Matejuk, Q. Leng, M. Begum, M. Woodle, P. Scaria, S. Chou and A. Mixson, Drugs Future, 2010, 35, 197 CrossRef CAS PubMed.
- Y. Akkam, Jordan J. Pharm. Sci., 2016, 9, 51–75 CrossRef.
- K. Markossian, A. Zamyatnin and B. Kurganov, Biochemistry, 2004, 69, 1082–1091 CAS.
- C. Chothia and A. V. Finkelstein, Annu. Rev. Biochem., 1990, 59, 1007–1035 CrossRef CAS PubMed.
- Z. Oren and Y. Shai, Biochemistry, 1997, 36, 1826–1835 CrossRef CAS PubMed.
- N. Yang, M. B. Strøm, S. M. Mekonnen, J. S. Svendsen and Ø. Rekdal, J. Pept. Sci., 2004, 10, 37–46 CrossRef CAS PubMed.
- N. Papo and Y. Shai, Biochemistry, 2003, 42, 9346–9354 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures and tables on the analysis of dipeptide features. See DOI: 10.1039/c7ra01388d |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |