
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization and acid-catalysed depolymerization of condensed tannins derived from larch bark
Aibin Zhanga,
Jiongjiong Li a,
Shifeng Zhanga,
Youbing Mub,
Wei Zhang*a and
Jianzhang Li*a
a,
Shifeng Zhanga,
Youbing Mub,
Wei Zhang*a and
Jianzhang Li*a
aMOE Key Laboratory of Wooden Material Science and Application, Beijing Key Laboratory of Wood Science and Engineering, Beijing Forestry University, 35 Qinghua East Road, Haidian District, Beijing 100083, P. R. China. E-mail: zhangwei@bjfu.edu.cn; lijzh@bjfu.edu.cn; Tel: +86-10-62336912 Tel: +86-10-62338083
bCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
First published on 12th July 2017
Abstract
Condensed tannins from larch bark extracts are a natural renewable and eco-friendly material and are potential substitutes for phenolic petrochemicals. However, the wide application of tannin is restricted by its low reactivity. Therefore, the goal of this study was to enhance the reactivity of larch tannin by depolymerization and determine optimal reaction conditions. The structures of larch tannin and depolymerized larch tannin were characterized by Fourier transform infrared (FT-IR) spectroscopy, solid phase 13C-NMR and matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. The thermal stability of larch tannin before and after depolymerization was evaluated by thermogravimetric analysis (TGA). The results indicated that the monomeric units of larch tannin were mainly composed of catechin/epicatechin, gallocatechin/epigallocatechin, catechin–gallocatechin esters, and stilbene glucosides. The presence of a catechin gallate dimer that had lost both gallic acid residues and a hydroxy group and a small amount of fisetinidin units were also observed. Additionally, a series of peaks corresponding to oligomers of larch tannin of up to 11 repeating units were observed from the MALDI-TOF MS data. Depolymerization treatment, especially using 2-mercaptoethanol as a nucleophilic reagent, was found to be beneficial to the enhancement of thermal stability. The optimization of the depolymerization reaction allowed the reaction to be completed in two hours, at 60 °C, using 2-mercaptoethanol as a nucleophile and 0.1 mol L−1 HCl. Many compounds of molecular weight less than 600 Da, mainly dimers and monomers, were obtained under these reaction conditions.
1. Introduction
Non-renewable petroleum resources are gradually being depleted, which has sounded the alarm for humanity's main energy supply.1 Therefore, many countries have focused on the increased utilization of biomass to obtain energy, chemical resources, and materials, and as substitutes for non-renewable petroleum resources.Tannin is often more abundant than lignin in rapidly cycling soft tissues such as leaves, needles and barks.2,3 As a type of tannin, condensed tannins are mainly composed of oligomers or polymers formed by a variable number of flavan-3-ol subunits consisting of (+)-catechin and (−)-epicatechin and linked through interflavan C–C bonds,4,5 and have been widely studied as substitutes for phenol in adhesives and resins preparation.6,7 Many efforts have been made to substitute bisphenol A, phenol, or urea with condensed tannins for the synthesis of bio-based epoxy resins,8–10 tannin-phenol-formaldehyde resins (TPF),11–13 and tannin-urea-formaldehyde resins (TUF).14,15 However, the polymeric forms of tannins are not as good as the monomers for use in the production of tannin-based resins because they react differently or lack sufficient reactivity.4,5,16 This is because macromolecular condensed tannins exhibit greater steric hindrance and the available reactive sites are far apart.17,18 Moreover, the increased rate of tannin substitution leads to decreased bond strength and increased viscosity of tannin-based resins, resulting in a low utilization rate of the condensed tannins. Therefore, the depolymerization of polyhydroxyflavan-3-ol polymers to oligomers or monomers is a promising approach to decrease steric hindrance and enhance chemical reactivity.
The depolymerization reaction of condensed tannins is usually achieved by acid-catalysed cleavage of the interflavan bonds in the presence of a nucleophilic reagent.19 Generally, the A-rings of all the flavan-3-ol subunits are protonated and the interflavan bonds are cracked, releasing terminal subunits as neutral monomeric flavan-3-ols and extension subunits as carbocations. The carbocations are trapped by the nucleophilic reagent and transformed into the corresponding nucleophile adducts as shown in Fig. 1.
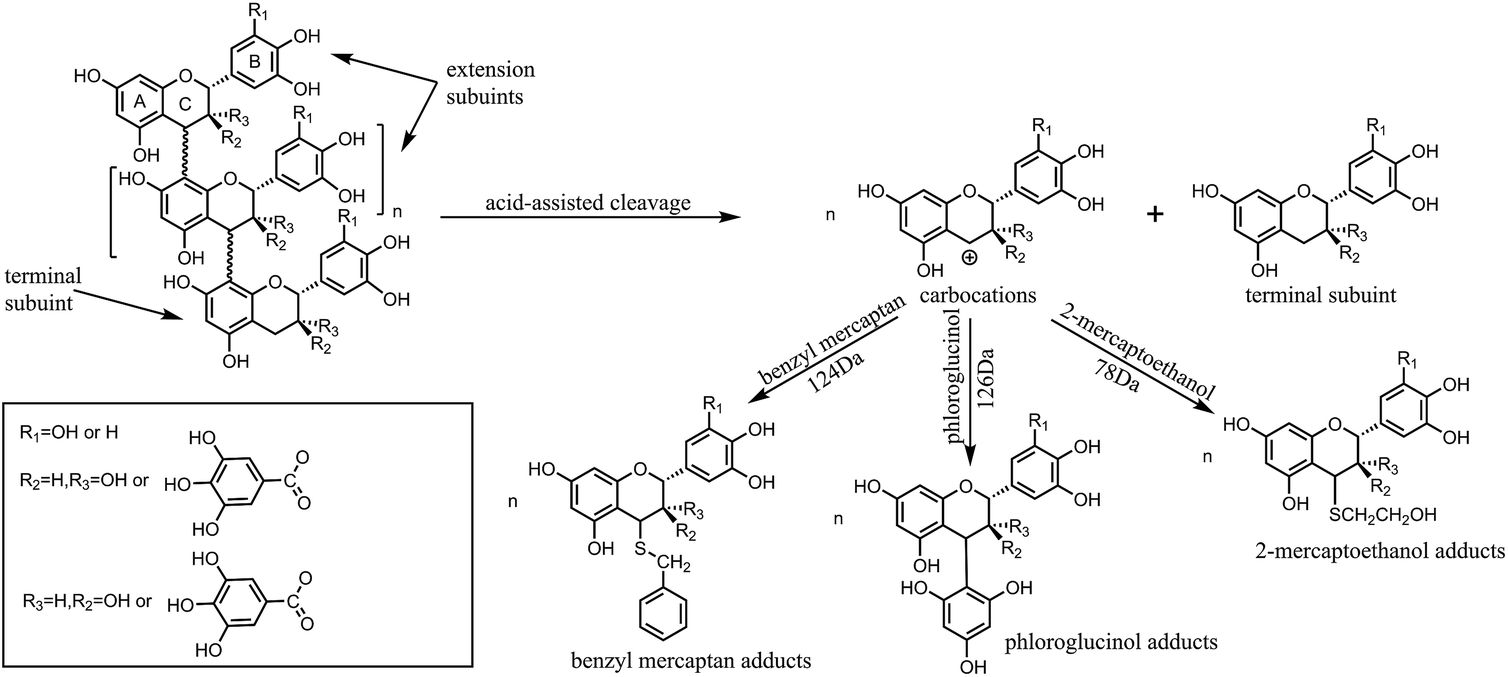 | ||
| Fig. 1 Acid depolymerization of condensed tannins in the presence of nucleophiles. | ||
Different strategies have been developed for the depolymerization of condensed tannins in the presence of nucleophilic reagents, and the use of phloroglucinol,20 thioglycolic acid,21 benzyl mercaptan,22 benzene-p-sulfinic acid, benzenethiol23 or 2-mercaptoethanol24 have already been reported. Among these nucleophiles, phloroglucinol and benzyl mercaptan are most commonly selected for the depolymerization of condensed tannins to determine the monomeric composition of tannins.20 Although good yields of monomeric phenolic synthons can be obtained with sulphur-containing compounds with unpleasant odor as nucleophilic reagents, the depolymerization process is also accompanied by the occurrence of some side reactions. Benzenethiol is easily oxidized in acidic medium and benzene-p-sulfinic acid has poor nucleophilicity for the depolymerization of polyphenols.23 Thioglycolic acid may polymerize into thioester and is unstable at room temperature.4 Therefore, compared to the other sulphur-containing compounds, it seems less ideal to use the three nucleophiles for the depolymerization of condensed tannins. Moreover, the 2-mercaptoethanol odour is much less pronounced compared to that of thioglycolic esters and esterification side reaction cannot occur in the case of 2-mercaptoethanol. Phloroglucinol is odorless and therefore has no special handling requirements. In previous studies, (+)-catechin, (−)-epicatechin, or (−)-epigallocatechin gallate were used as chain breakers to depolymerize condensed tannins.25,26 However, the goal of the present study is the production of phenolic synthons from renewable resources, so it seems inappropriate to use these monomers as nucleophiles. Hydrogen gas is another potential reducing agent that can be used for the depolymerization of tannins, but the hydrogenolysis reaction needs to be performed in a special reactor and the resulting yield of oligomers is low.5 Depolymerization under alkaline conditions is not ideal because of the competitive autoxidation of tannins and formation of catechinic acid, resulting in only a small percentage of condensed tannins being depolymerized.4,27 The effect of reaction conditions such as nucleophiles and temperature on the depolymerization of condensed tannins is significant.20,26,28 The depolymerization reaction is frequently applied to condensed tannins for structure elucidation, but studies of the optimal reaction conditions for the depolymerization of condensed tannins have been limited, and the yields for the conversion of condensed tannins into their constitutive subunits are low, with incomplete depolymerization. Therefore, to produce more monomers or oligomers, the conditions of the depolymerization reaction were investigated.
In this study, larch tannin (LT) (condensed tannins) extracted from larch bark was depolymerized to yield phenolic monomers. Mercaptoethanol (ML) was compared with benzyl mercaptan (BM) and phloroglucinol (PL) as depolymerization reagents for the depolymerization of larch tannin. The chemical structures of LT and depolymerized larch tannin (DLT) were detected using FT-IR, solid phase 13C-NMR and Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. Thermal properties of the LT and DLT were characterized by thermogravimetric analysis (TGA). The depolymerization conditions including temperature, time, and catalyst concentration were optimized to enhance larch tannin reactivity to facilitate the development and utilization of larch tannin as potential substitutes of phenolic petrochemicals for bio-based polymers and materials.
2. Experimental section
2.1. Materials
Larch tannin was purchased from Tian'guan Biotech Co., Henan province, China. Phloroglucinol, benzyl mercaptan, 2-mercaptoethanol and all other chemicals were obtained from Beijing Chemical Reagents Co., Ltd. (China). All chemicals were analytic grade and used without further purification.2.2. Optimization of the depolymerization reaction
Using a final concentration of larch tannin of 24 mg mL−1, all the depolymerization reactions were conducted in a nitrogen atmosphere to prepare DLT products. In each run, larch tannin was dissolved in absolute ethanol to a concentration of 60 mg mL−1, and 20 mL of the tannin stock solution was transferred to a 250 mL three-neck bottle loaded in a preheated water bath. To investigate the effect of nucleophiles on the depolymerization of tannins, different nucleophiles (5.2 g of phloroglucinol (PL), 4.9 mL of benzyl mercaptan (BM), or 2.9 mL of 2-mercaptoethanol (ML)) corresponding to 10 molar equivalents of catechin monomers were dissolved in 30 mL of absolute ethanol and injected into the 250 mL three-neck bottle. 414 μL of 37% HCl (i.e. 0.1 mol L−1 HCl) was applied and the reaction was performed at 60 °C under magnetic stirring at 500 rpm for 2 h. The obtained products were denoted as DLT-PL, DLT-BM, and DLT-ML, respectively.In order to study the effect of reaction temperature on the depolymerization of tannins, 414 μL of 37% HCl and 2.9 mL of 2-mercaptoethanol as nucleophile were added to 30 mL of absolute ethanol. The reaction was carried out at 40, 60, or 80 °C under magnetic stirring at 500 rpm for 2 h. The samples obtained under different reaction temperatures were labeled DLT-ML 40, DLT-ML 60, and DLT-ML 80, respectively.
To investigate the effect of reaction time, the temperature was set at 60 °C. The depolymerization reaction was performed 1, 2 or 3 h under magnetic stirring at 500 rpm. In the reaction, 414 μL of 37% HCl was used as the catalyst and 2.9 mL of 2-mercaptoethanol was placed into 30 mL of absolute ethanol. The obtained products were labeled DLT-ML 1, DLT-ML 2, and DLT-ML 3, respectively.
To study the effect of catalyst content on depolymerization, 207, 414, or 828 μL of 37% HCl (i.e. 0.05, 0.1, or 0.2 mol L−1 HCl, respectively) were added to 30 mL of absolute ethanol. The reaction was conducted under magnetic stirring at 500 rpm for 2 h at 60 °C, and with 2.9 mL of 2-mercaptoethanol in the reactor. The samples obtained under different catalyst contents were denoted as DLT-ML 0.05, DLT-ML 0.1, and DLT-ML 0.2, respectively. When each reaction was complete, the resulting solutions in bottle were collected. The solutions were stored in a refrigerator before further processing and analysis.
2.3. FTIR analysis
The reaction products were freeze-dried by placing them in a vacuum freeze-drier (Sihuan LGJ-25C, Sihuan Co., Ltd, Beijing, China) for 48 h. These freeze-dried samples were milled to 200 mesh meal and then recorded by FT-IR (Nicolet IS10, Thermo Fisher Scientific Corporation, Waltham, MA, USA) at a resolution of 4 cm−1. Each spectrum was obtained from 500 to 4000 cm−1 using 32 scans.2.4. Solid phase 13C-NMR analysis
The solid-state 13C CP-MAS NMR spectra of the freeze-dried samples were recorded on a Bruker 300 spectrometer at a frequency 75.47 MHz. Chemical shifts were calculated relative to TMS (tetramethylsilane). The rotor was spun at 12 kHz on a double-bearing 5 mm Bruker probe.2.5. MALDI-TOF-MS analysis
The MALDI-TOF mass spectra were performed on a MALDI-TOF instrument (AXIMA Performance, Shimadzu, Japan) equipped with a pulsed nitrogen laser (wavelength: 337 nm, laser pulse length 3 ns). The experimental conditions used were: polarity-positive, 100–150 pulses per spectrum, mass-high (20 kV acceleration voltage), and flight path-linear and 200–800 ns time of delayed extraction technique. Based on previous studies, the samples were first dissolved in acetone (4 mg mL−1). The sample solutions were mixed with an acetone solution consisting of 2,5-dihydroxy benzoic acid (DHB) as the matrix and acetone (10 mg mL−1 acetone). For the enhancement of ion formation, NaCl was added to the matrix. The samples of the solution and matrix were mixed in equal amounts, and 0.5–1 μL of the resulting mixture was placed on the MALDI target. The solvent was evaporated on the MALDI target before placing into the spectrometer.14,292.6. Thermogravimetric analysis (TGA)
Thermal decomposition was performed with a Q50 TGA analyzer (TA instruments, USA). To do this, 5–8 mg of freeze-dried sample was transferred to a platinum pan and the temperature ranged from 25 to 600 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere (40 mL min−1). The curves of the percentage of weight loss and the first time derivative were determined using the TGA software.3. Results and discussion
3.1. Characterization of larch tannin
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the galloyl unit on the epicatechin gallate.34,35 The bands between 1610 cm−1 and 1445 cm−1 were attributed to aromatic ring stretching vibration. These aromatic rings are no changes to the “core” of the tannin structure during the depolymerization procedures. The single peak at 1517 cm−1 indicated that the larch tannin predominantly consisted of procyanidin,36 and the high intensity of the C–C stretching peak at 1612 cm−1 indicated the presence of a high number of C4–C8 interflavonoid linkages.35 The bands observed from 1283 to 1060 cm−1 were the characteristic bands of C–O–C including aromatic C–O and aliphatic C–O, and bands in the range of 890–680 cm−1 were associated with C–H of benzene rings and O–H of alcohol vibration.37
O of the galloyl unit on the epicatechin gallate.34,35 The bands between 1610 cm−1 and 1445 cm−1 were attributed to aromatic ring stretching vibration. These aromatic rings are no changes to the “core” of the tannin structure during the depolymerization procedures. The single peak at 1517 cm−1 indicated that the larch tannin predominantly consisted of procyanidin,36 and the high intensity of the C–C stretching peak at 1612 cm−1 indicated the presence of a high number of C4–C8 interflavonoid linkages.35 The bands observed from 1283 to 1060 cm−1 were the characteristic bands of C–O–C including aromatic C–O and aliphatic C–O, and bands in the range of 890–680 cm−1 were associated with C–H of benzene rings and O–H of alcohol vibration.37
 | ||
| Fig. 2 FT-IR spectrum of larch tannin. | ||
O of the gallic residue linked in C3 to the heterocycle ring of a flavonoid structure. The band of high intensity at 154 ppm was assigned to C5, C7 and C9 of the aromatic A-ring. The band at 143 ppm was attributed to the C3′ and C4′. The split peak at 143 and 154 ppm (phenolic peak) showed the characteristic of tannins. The signals at 129 and 116 ppm can be respectively assigned to the C1′ and C5′ B ring sites. The peak at 116 ppm showed that larch tannin consists of both pyrogallol and catechol B-rings with the latter being more predominant. This was consistent with the finding from FT-IR analysis that larch tannin is predominantly consisted of procyanidin.
 | ||
| Fig. 3 Solid-state 13C CPMAS NMR spectrum of the larch tannin. | ||
The band at 104 ppm was assigned to the interflavonoid bonds C4–C8 and C4–C6. The absence of the C4–C6 interflavonoid band at 95 ppm could indicate that the predominant interflavonoid linkage was C4–C8, which was in accordance with the FTIR analysis that showed high number of C4–C8 linkages. The low band intensity at 98 ppm was corresponded to C6, C8 and C10. Peaks between 60 ppm and 85 ppm were assignable to carbohydrates present in the tannin. Resonance at 71 ppm could correspond to C3 in the chain interior and upper chain-ending positions. Its higher intensity could be due to the superposition of the oligomeric carbohydrates in the larch tannin. The peaks for C2 were covered by the C3 band but were visible as shoulder of the latter, especially the C2 (cis) at 80 ppm and C2 (trans) at 85 ppm. C4 of expansion units showed a peak at 37 ppm, while the terminal C4 showed a characteristic chemical shift at 28 ppm.
 | ||
| Fig. 4 The MALDI-TOF spectra of (a) larch tannin and (b) indication of the relevant 132 Da, 272 Da, 404 Da, and 440 Da repeat units. | ||
The spectra indicated that the identical larch tannin monomer constituents in accordance with the previous data were galloyl group, (epi)catechin, (epi)gallocatechin, and (epi)gallocatechin gallate with molecular weights (MW), respectively, of 152 Da, 290 Da, 306 Da, and 458 Da (not marked in Fig. 4b) (Fig. 5).14 A shift of approximately 440 Da among the peaks (e.g. 1610.5–1171.5 = 439 Da, 1874.7–1436.5 = 438.2 Da) was observed in the mass spectra. This mass change was associated with the loss of a (epi)catechin gallate unit (442.4 – 2H = 440.4 Da) (Fig. 6a).29,38 The peak at 905.0 Da was interpreted as a (epi)catechin gallate dimer (2 × (442.4 – 1H) + Na+ = 905.8 Da). The mass spectra indicated clearly that a shift of molecular mass of about 404 Da was observed, equal to the mass of stilbene glucoside astringin (Fig. 6b) when covalently bonded within a condensed tannin molecule (406.4 – 2H = 404.4 Da).41,42 The peak at 833.0 Da may reflect an astringin dimer (2 × (406.4 – 1H) + Na+ = 833.8 Da). There was therefore a reasonable possibility of astringin units as part of the building blocks of the larch tannin. The presence of stilbenes and their glucosides in the molecular structure of bark tannins from European larch was suggested previously.42 However, there was only a low amount of astringin units as indicated by the very low intensity of the peaks. More detailed analyses should be carried out to confirm the structure. Some low intensity peaks with mass increments of about 272 Da indicated that tetrahydroxy flavan units were possibly present in the structure of larch tannin (Fig. 6c). This unit could be formed by (epi)catechin gallate units in which the gallic acid residue has been removed.38 The mass spectra indicated that a main repetition unit of approximately 132 Da was observed, but no known flavonoid structure corresponds to that molecular weight. In previous studies, a similar mass spectrum for larch tannin was reported and suggested that the repeating unit is not 132 Da but appeared to be 528 Da (=4 × 132 Da) (Fig. 6d). The repeating unit is a dimer of structure C (Fig. 6c) with a –OH group missing. The unit has alcoholic hydroxyl groups that were lost at the separation of the gallic acid and a single phenolic hydroxyl group was also lost.38,42 These unusual structures were different from the commonly reported flavan-3-ols. Larch tannin with different polymerization degrees exhibited chemical structures that consisted of flavan-3-ol units and non-flavan-3-ol units, mainly including carbohydrates,42 which is in accordance with NMR analysis. Unfortunately, some peaks were not explained by structural units described above, suggesting that larch tannin had other monomer structures in addition to the observed structures. However, many different structural units in condensed tannins caused relevant MALDI peaks as reported by other researchers.29,39 Additionally, the MALDI-TOF spectra clearly showed different polymer chain length distributions of the larch tannin up to 11 repeating units.
 | ||
| Fig. 5 Molecular structures of the most common flavan-3-ols units in larch tannin. | ||
 | ||
| Fig. 6 The monomeric structures in larch tannin. | ||
 | ||
| Fig. 7 TG and DTG curves of larch tannin. | ||
3.2. Optimization of depolymerization process
 | ||
| Fig. 8 FT-IR spectra of DLT and PL. | ||
The solid state 13C NMR spectra of DLT-BM, DLT-PL and DLT-ML are shown in the Fig. 9. From Fig. 9a, the appearance of the new peaks, at 137 ppm, 134 ppm and 39 ppm in the case of DLT-BM compared to LT, was assignable to C of benzyl SH ring and C4–SCH2, respectively.47,48 The peaks at 128 ppm for DLT-BM and 129 ppm for LT were assigned to C1′. The increasing intensity of the C1′ peak of DLT-BM may be caused by the introduction of benzyl SH ring into the structure of tannin. These results illustrated the occurrence of the reaction between larch tannin and benzyl mercaptane. The depolymerization of tannins is usually achieved by acid-catalysed cleavage of the interflavan bonds C4–C8 and C4–C6 in the presence of a nucleophilic reagent. The C4 sites in the form of carbocations react with nucleophilic reagents. The intensity of the broad peak, around 104 ppm in the spectra of DLT-BM and DLT-ML, was largely weakened compared to that of LT. This peak was attributed to the interflavan bonds C4–C8 and C4–C6, and the weakening of its intensity indicated that great amounts of the interflavan bonds have been cleaved and turned into –C–S– groups. The band at 62 ppm was attributed to CH2OH of the mercaptoethanol.49,50 The peak at 37 ppm, which corresponds to interflavan C4, disappeared in the spectrum of the DLT-ML, and inversely, the higher intensity new peak at 32 ppm was obtained. These results indicated that larch tannin was successfully depolymerized by 2-mercaptoethanol and interflavan bonds were converted to C–S bonds.
 | ||
| Fig. 9 The solid-state 13C CPMAS NMR spectra of larch tannin depolymerized in the presence of different nucleophiles. (a) DLT-BM and DLT-ML, and (b) DLT-PL. | ||
From Fig. 9b, the two dominant peaks at 156 ppm and 95 ppm were assignable to the larch tannin and phloroglucinol carbons. The band at 105 ppm was due to the site of phloroglucinol connecting with the C4 of the tannin units, and the C4 was shown by the band at 33 ppm.20 These results indicated that larch tannin reacted with phloroglucinol under acidic condition. Based on the above analysis, the monomeric units, mainly catechin, epicatechin and flavanol-3-O-gallate, oligomers and corresponding derivatives co-existed in the larch tannin depolymerized by different nucleophiles, which are consistent with previous studies. The peak intensity at 30 ppm in NMR spectrum of DLT-ML was stronger than that of the DLT-BM and DLT-PL at 28 ppm, and the intensity of these peaks related to free C4 was weaker in the NMR spectra of the DLT-BM and DLT-PL than that of DLT-ML. These results indicated that 2-mercaptoethanol was the best nucleophile for the depolymerization of larch tannin.
The MALDI-TOF spectra were used to describe the molecular weight change of larch tannin depolymerized in the presence of different nucleophiles (Fig. 10). Most of the reaction product major peaks were differed those of the larch tannin. After depolymerization, the area of polymer peaks decreased and conversely, many low molecular weight peaks appeared in these spectra but were not present in the spectrum of larch tannin, suggesting that interflavan bonds were successfully broken down and the depolymerization of larch tannin was feasible under such reaction conditions. In Fig. 10a, several series with a repeating interval of 124 Da were noted in the DLT-BM spectra, the DLT-PL spectra displayed a main repetition unit of 125 Da, and DLT-ML samples showed a repetition unit of about 77 Da. These masses may be associated with a benzyl mercaptan unit (124 – 1H = 123 Da), a phloroglucinol unit (126 – 1H = 125 Da), and a mercaptoethanol unit (78 – 1H = 77 Da), respectively. Therefore, the three kinds of nucleophiles were successfully added to flavonoid structures, consistent with the FT-IR and NMR results. The mass spectra of 200 to 1000 Da clearly indicated dominant fragments at 391.7 Da (290.3 + 23 + 78 = 391.3 Da) for DLT-BM, 455.7 Da (378.4 + 78 = 456.4 Da) for DLT-PL, and 311.2 Da (290.3 + 23 = 313.3 Da) for DLT-ML (not marked in Fig. 10a), which may be due to a higher ratio of these peaks corresponding to the depolymerization products observed for these samples. As it can be seen from the spectra, the peaks sometimes did not include the sodium ion. The intense peaks of the mass spectra of the three samples were obviously different in the range from 500 to 3500 Da (Fig. 10b). The mass spectra of the DLT-BM and DLT-PL showed higher molecular mass peaks than those of DLT-ML. The mass spectra displayed the maximal molecular weight fragmentation peaks obtained at 1557.2 Da (pentamer) for DLT-BM, 1786.3 Da (hexamer) for DLT-PL and 1125.6 Da (trimer or tetramer) for DLT-ML. The DLT-ML exhibited the lowest polymerization degree followed by DLT-BM and DLT-PL, which was explained by the different nucleophilicity of different nucleophilic reagents in the acidified ethanol solution. In the depolymerization reaction, the difficulty is not the bond-breaking process of the interflavan C–C bonds, which can be readily cleaved under “mild” acidic conditions, but the trapping of the resulting carbocations. This indicates that the nucleophilicity of nucleophilic reagents is the key to the depolymerization reaction. Additionally, in the presence of flanking hydroxy groups at both the 3- and 5-positions, the bulky phloroglucinol and benzyl mercaptan cannot easily approach the carbocations.23 Therefore, we concluded that the 2-mercaptoethanol had better nucleophilicity than the two other reagents. These results were consistent with a previous report that benzyl mercaptan is a better reagent than phloroglucinol for the depolymerization reaction of tannins.20 Moreover, it can be concluded that the monomers, oligomers and corresponding nucleophile adducts co-existed in the reaction solutions from the MALDI-TOF spectra. These products with high reactivity are suitable as the substitutes of phenolic petrochemicals for bio-based polymers and materials, especially the resins and foams.51
 | ||
| Fig. 10 The MALDI-TOF spectra of larch tannin depolymerized in the presence of different nucleophiles. (a) 200–1000 Da, (b) 500–3500 Da, and (c) thermogravimetric degradation curves and the first derivatives of DLT-BM, DLT-PL, and DLT-ML. | ||
The TGA-DTG curves of DLT-BM, DLT-PL, and DLT-ML are shown in Fig. 10c. Under nitrogen atmosphere, the three DLT samples started to decompose at 160 °C for DLT-PL, 149 °C for DLT-BM and 140 °C for DLT-ML, which showed lower initial thermal stability than LT. Thermal degradation profiles of DLT-BM and DLT-ML revealed that the two samples had relatively lower decomposition onset than DLT-PL. This difference was due to the formation of carbon–sulfur bonds at the pyranyl ring (4-position) during the depolymerization reaction that promoted thermal instability. Similarly, the presence of a sulfonate group at the 4 position promoted thermal instability.52 In the second loss stage, the first decomposition peak of the DLT-PL and DLT-BM was slightly lower than that of the LT, while the first decomposition temperature of DLT-ML at 239 °C was higher than that of the LT and showed better thermal stability. Compared to the DLT-ML and DLT-BM, DLT-PL (207 °C) showed a low decomposition peak and exhibited the worst thermal stability in the second stage, which may be due to the decomposition of phloroglucinol as discussed in the FT-IR analysis section. For the DLT-BM, the shape of the curve and the position of the degradation steps were similar to those of the LT. The second decomposition peak of DLT-ML at the second weight loss stage at 304 °C was higher than that of LT, which indicated that the DLT-ML samples exhibited better thermal resistance than the LT. This indicates that the larch tannin was depolymerized into small molecules showing better thermal stability than the polymers, and no molecular bonds between the monomer structures of larch tannin were broken during degradation. The two distinct degradation peaks of DLT-ML were similar to those of catechin at 230 °C and 310 °C in the second stage,52 revealing that DLT-ML samples were composed of more monomers or oligomers. The TGA-DTG analysis clearly indicated that the thermal stability of depolymerized larch tannin was enhanced and the DLT-ML samples exhibited better thermal stability than LT and other depolymerization products. Based on the analysis data, we concluded that 2-mercaptoethanol was the best nucleophile for the depolymerization of larch tannin.
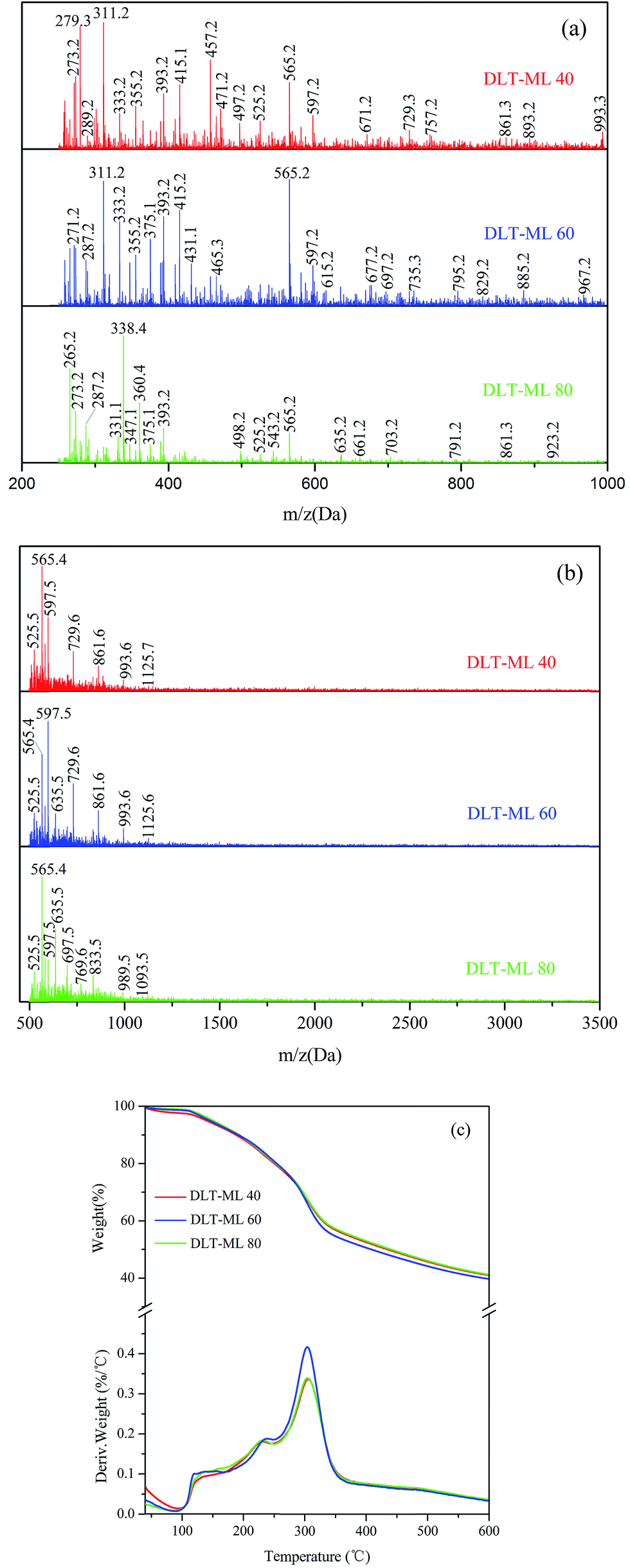 | ||
| Fig. 11 The MALDI-TOF spectra of larch tannin depolymerized with varying temperatures. (a) 200–1000 Da, (b) 500–3500 Da, and (c) thermogravimetric degradation curves and the first derivatives of DLT-ML prepared at different temperatures. | ||
The thermal curves of larch tannin depolymerized by 2-mercaptoethanol at different temperatures are shown in Fig. 11c. A similar degradation thermogram was observed for the three DLT-ML samples prepared at different temperatures. In the second degradation step, the first degradation peaks were observed at 231 °C, 239 °C, and 229 °C for DLT-ML 40, DLT-ML 60 and DLT-ML 80, respectively, and the second degradation peaks were fairly similarly positioned at around 304 °C. Additionally, the char yields obtained at 600 °C were about 41% for the three samples. The thermal decomposition behavior of DLT-ML 60 was dramatically decreased after 300 °C, reaching to the same level as that of the DLT-ML 40 and DLT-ML 80. The depolymerization temperature affected the thermal decomposition behavior of the DLT-ML samples prepared at different temperatures mainly in the low temperature range of 100–250 °C. The DLT-ML 60 showed a slightly higher thermal stability in comparison with DLT-ML 40 and DLT-ML 80 in the range of 100–250 °C. The curves of the depolymerized larch tannin at different temperatures indicated that the optimal depolymerization temperature of larch tannin was 60 °C.
 | ||
| Fig. 12 The MALDI-TOF spectra of larch tannin depolymerized with varying amounts of time. (a) 200–1000 Da, (b) 500–3500 Da, and (c) thermogravimetric degradation curves and the first derivatives of DLT-ML prepared under different amounts of time. | ||
Shown in Fig. 12c are the thermal degradation profiles of depolymerized larch tannin for 1, 2, or 3 h. During degradation, the two-step degradation thermograms of the DLT-ML samples prepared for different times were extremely similar, especially the second degradation peaks at ca. 304 °C. The thermal degradation of DLT-ML 2 was faster than that of the other two samples at temperatures higher than 250 °C. In the second degradation step, the first maximum degradation temperature for the samples subjected to depolymerization for 1 h, 2 h, or 3 h were 229 °C, 239 °C and 230 °C, respectively, indicating higher thermal stability for DLT-ML 2. However, higher residual mass percentage values at 600 °C were observed for DLT-ML 1 (42.3%) and DLT-ML 3 (43.8%), and the char yield for DLT-ML 2 was 39.7%. Based on the above analysis, we can easily conclude that the larch tannin was optimally depolymerized for 2 h.
 | ||
| Fig. 13 The MALDI-TOF spectra of larch tannin depolymerized with varying catalyst concentrations. (a) 200–1000 Da, (b) 500–3500 Da, and (c) thermogravimetric degradation curves and the first derivatives of DLT-ML prepared with different catalyst concentrations. | ||
The TGA-DTG curves of depolymerized larch tannin using 2-mercaptoethanol as nucleophile with different catalyst concentrations are shown in Fig. 13c. The first degradation peak (232 °C) of DLT-ML 0.2 in the second thermal event was higher than that of the DLT-ML 0.05 (227 °C). Compared to the DLT-ML 0.2, the DLT-ML 0.1 had a higher degradation temperature (239 °C) and showed better thermal stability. The second degradation peaks were fairly similarly positioned at around 304 °C for the three samples. The thermal degradation of the DLT-ML 0.1 and DLT-ML 0.2 was faster than that of DLT-ML 0.05 at temperatures above 250 °C. The residual mass percentage values obtained at 600 °C were about 39% for the DLT-ML 0.1 and DLT-ML 0.2, slightly lower than that of DLT-ML 0.05. From the MALDI-TOF-MS and TGA-DTG analysis, these results indicated that 0.1 mol L−1 HCl was the optimal catalyst concentration for the depolymerization of larch tannin.
4. Conclusions
The condensed tannins extracted from larch tannin with polymer chain lengths up to 11 units were mainly composed of procyanidins, prodelphinidins and flavan-3-ol gallates. The presence of the stilbene glucosides and small proportion of fisetinidin units was also suggested. Some oligomers appeared to contain a catechin gallate dimer that has lost both the gallic acid residues and a hydroxygroup at their terminal unit. These peculiar structures of larch tannin may affect behavior in resin production or as heavy metal scavengers. Thermogravimetric analysis revealed higher thermal stability of larch tannin compared to quebracho and mimosa tannins. Larch tannin was successfully depolymerized into monomers and/or oligomers under the acidic condition by different nucleophiles. The degradation products obtained using 2-mercaptoethanol as a nucleophilic reagent displayed better thermal stability than the un-depolymerized larch tannin. Furthermore, 2-mercaptoethanol was very effective in the depolymerization process of larch tannin. The optimized reaction conditions allowed the efficient depolymerization of larch tannin into monomers and/or oligomers at 60 °C, for 2 h, using 2-mercaptoethanol as the nucleophile, with 0.1 mol L−1 HCl. A high amount of the dimers and monomers were obtained under these optimized reaction conditions. Depolymerization of larch tannin was expected to increase their reactivity.These results provide a promising method to obtain bio-based phenolic monomers and the possibility for further high value-added utilization of larch tannin as the substitutes of phenolic petrochemicals for bio-based polymers and materials, especially the resins and foams.
Acknowledgements
This research was supported by the Fundamental Research Funds for the Central Universities (No. 2016ZCQ01), the CAS Key Laboratory of Bio-based Materials (No. KLBM2016002), and the Chinese National Science and Technology Support Program (2015BAD14B0302).References
- J. L. Yuan, Z. H. Gao and X. M. Wang, Pigm. Resin Technol., 2009, 38, 290–297 CrossRef CAS.
- P. J. Hernes and J. I. Hedges, Anal. Chem., 2000, 72, 5115–5124 CrossRef CAS PubMed.
- P. J. Hernes and J. I. Hedges, Geochim. Cosmochim. Acta, 2004, 68, 1293–1307 CrossRef CAS.
- L. Roumeas, C. Aouf, E. Dubreucq and H. Fulcrand, Green Chem., 2013, 15, 3268–3275 RSC.
- Z. Li, J. J. Zeng, Z. H. Tong, Y. J. Qi and L. W. Gu, Food Chem., 2015, 188, 337–342 CrossRef CAS PubMed.
- C. N. Aguilar and G. Gutiérrez-Sánchez, Food Sci. Technol. Int., 2001, 7, 373–382 CrossRef CAS.
- L. Wang, W. Y. Liang, J. Yu, Z. X. Liang, L. L. Ruan and Y. C. Zhang, Environ. Sci. Technol., 2013, 47, 5771–5777 CrossRef CAS PubMed.
- C. Aouf, C. Le Guernevé, S. Caillol and H. Fulcrand, Tetrahedron, 2013, 69, 1345–1353 CrossRef CAS.
- H. Nouailhas, C. Aouf, C. Le Guerneve, S. Caillol, B. Boutevin and H. Fulcrand, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2261–2270 CrossRef CAS.
- S. Benyahya, C. Aouf, S. Caillol, B. Boutevin, J. P. Pascault and H. Fulcrand, Ind. Crops Prod., 2014, 53, 296–307 CrossRef CAS.
- S. Jahanshaei, T. Tabarsa and J. Asghari, Pigm. Resin Technol., 2012, 41, 296–301 CrossRef CAS.
- A. Moubarik, A. Pizzi, A. Allal, F. Charrier and B. Charrier, Ind. Crops Prod., 2009, 30, 188–193 CrossRef CAS.
- E. C. Ramires and E. Frollini, Composites, Part B, 2012, 43, 2851–2860 CrossRef CAS.
- J. Z. Zhang, H. J. Kang, Q. Gao, J. Z. Li, A. Pizzi and L. Delmotte, J. Appl. Polym. Sci., 2014, 131, 547–557 Search PubMed.
- M. Zanetti, V. Causin, R. Saini, A. Cardin and R. Cavalli, Eur. J. Wood Wood Prod., 2014, 72, 385–392 CrossRef CAS.
- S. Deprez, I. Mila, J.-F. Huneau, D. Tome and A. Scalbert, Antioxid. Redox Signaling, 2001, 3, 957–967 CrossRef CAS PubMed.
- A. P. Barbosa, E. B. Mano and C. T. Andrade, For. Prod. J., 2000, 50, 89–92 CAS.
- A. Pizzi, J. Appl. Polym. Sci., 1979, 24, 1257–1268 CrossRef CAS.
- S. Matthews, I. Mila, A. Scalbert, B. Pollet, C. Lapierre, C. L. M. Hervé du Penhoat, C. Rolando and D. M. X. Donnelly, J. Agric. Food Chem., 1997, 45, 1195–1201 CrossRef CAS.
- J. A. Kennedy and G. P. Jones, J. Agric. Food Chem., 2001, 49, 1740–1746 CrossRef CAS PubMed.
- L. Mouls and H. Fulcrand, Tetrahedron, 2015, 71, 3012–3019 CrossRef CAS.
- J. Rigaud, J. Perez-Ilzarbe, J. M. R. Da Silva and V. Cheynier, J. Chromatogr. A, 1991, 540, 401–405 CrossRef CAS.
- B. R. Brown and M. R. Shaw, J. Chem. Soc., Perkin Trans. 1, 1974, 2036–2049 RSC.
- W. Chen, C. L. Fu, Y. Qin and D. J. Huang, Food Chem., 2009, 114, 874–880 CrossRef CAS.
- T. Esatbeyoglu, V. Wray and P. Winterhalter, J. Agric. Food Chem., 2010, 58, 7820–7830 CrossRef CAS PubMed.
- H. W. Liu, T. Zou, J. M. Gao and L. W. Gu, Food Chem., 2013, 141, 488–494 CrossRef CAS PubMed.
- B. L. White, L. R. Howard and R. L. Prior, J. Agric. Food Chem., 2010, 58, 7572–7579 CrossRef CAS PubMed.
- L. W. Gu, M. A. Kelm, J. F. Hammerstone, G. Beecher, J. Holden, D. Haytowitz and R. L. Prior, J. Agric. Food Chem., 2003, 51, 7513–7521 CrossRef CAS PubMed.
- C. W. Oo, A. Pizzi, H. Pasch and M. J. Kassim, J. Appl. Polym. Sci., 2008, 109, 963–967 CrossRef CAS.
- W. J. Lee and W. C. Lan, Bioresour. Technol., 2006, 97, 257–264 CrossRef CAS PubMed.
- S. Ben Mahmoud, H. Saad, B. Charrier, A. Pizzi, K. Rode, N. Ayed and F. Charrier-El Bouhtoury, Wood Sci. Technol., 2014, 49, 205–221 CrossRef.
- R. Soto, J. Freer and J. Baeza, Bioresour. Technol., 2005, 96, 95–101 CrossRef CAS PubMed.
- L. Chupin, C. Motillon, F. C.-E. Bouhtoury, A. Pizzi and B. Charrier, Ind. Crops Prod., 2013, 49, 897–903 CrossRef CAS.
- Z. H. Huang, B. Zhang and G. Z. Fang, BioResources, 2013, 8, 4593–4608 Search PubMed.
- C. W. Oo, M. J. Kassim and A. Pizzi, Ind. Crops Prod., 2009, 30, 152–161 CrossRef CAS.
- L. Y. Foo, Phytochemistry, 1981, 20, 1397–1402 CrossRef CAS.
- R. C. Zhou, S. X. Si and Q. Y. Zhang, Appl. Surf. Sci., 2012, 258, 3578–3583 CrossRef CAS.
- P. Navarrete, A. Pizzi, H. Pasch, K. Rode and L. Delmotte, Ind. Crops Prod., 2010, 32, 105–110 CrossRef CAS.
- M. B. Ucar, G. Ucar, A. Pizzi and O. Gonultas, Ind. Crops Prod., 2013, 49, 697–704 CrossRef CAS.
- H. Saad, A. Khoukh, N. Ayed, B. Charrier and F. C.-E. Bouhtoury, Ind. Crops Prod., 2014, 61, 517–525 CrossRef CAS.
- S. Bianchi, A. N. Gloess, I. Kroslakova, I. Mayer and F. Pichelin, Ind. Crops Prod., 2014, 61, 430–437 CrossRef CAS.
- S. Bianchi, I. Kroslakova, R. Janzon, I. Mayer, B. Saake and F. Pichelin, Phytochemistry, 2015, 120, 53–61 CrossRef CAS PubMed.
- A. Duval and L. Averous, J. Agric. Food Chem., 2016, 64, 1751–1760 CrossRef CAS PubMed.
- C. H. Luo, W. Grigsby, N. Edmonds, A. Easteal and J. Al-Hakkak, J. Appl. Polym. Sci., 2010, 117, 352–360 CAS.
- Y. Sert, A. A. El-Emam, O. A. Al-Deeb, A. A. Al-Turkistani, F. Ucun and C. Cirak, Spectrochim. Acta, Part A, 2014, 126, 86–97 CrossRef CAS PubMed.
- F. Hassan, A. A. Hameed, A. Alshanon, B. M. Abdullah, H. Z. Huri, N. Hairunisa and E. Yousif, Asian J. Biochem., 2015, 10, 252–266 CrossRef.
- S. Sivakumaran, A. L. Molan, L. P. Meagher, B. Kolb, L. Y. Foo, G. A. Lane, G. A. Attwood, K. Fraser and M. Tavendale, Phytochemistry, 2004, 65, 2485–2497 CrossRef CAS PubMed.
- K. Kamiya, C. Watanabe, H. Endang, M. Umar and T. Satake, Chem. Pharm. Bull., 2001, 49, 551–557 CrossRef CAS PubMed.
- Y. Matsuo, Y. Fujita, S. Ohnishi, T. Tanaka, H. Hirabaru, T. Kai, H. Sakaida, S. Nishizono and I. Kouno, Food Chem., 2010, 121, 1073–1079 CrossRef CAS.
- R. Kusano, S. Ogawa, Y. Matsuo, T. Tanaka, Y. Yazaki and I. Kouno, J. Nat. Prod., 2010, 74, 119–128 CrossRef PubMed.
- A. Arbenz and L. Avérous, Green Chem., 2015, 17, 2626–2646 RSC.
- M. Gaugler and W. J. Grigsby, J. Wood Chem. Technol., 2009, 29, 305–321 CrossRef CAS.
- A. Vernhet, S. Dubascoux, B. Cabane, H. Fulcrand, E. Dubreucq and C. Poncet-Legrand, Anal. Bioanal. Chem., 2011, 401, 1559–1569 CrossRef PubMed.
- L. Mouls and H. Fulcrand, J. Mass Spectrom., 2012, 47, 1450–1457 CrossRef CAS PubMed.
- H. Fulcrand, M. Dueñas, E. Salas and V. Cheynier, Am. J. Enol. Vitic., 2006, 57, 289–297 CAS.
- H. Fulcrand, V. Atanasova, E. Salas and V. Cheynier, The Fate of Anthocyanins in Wine: Are There Determining Factors? In Red Wine: Color-Revealing the Mysteries, ed. A. L. Waterhouse and J. A. Kennedy, American Chemical Society, Washington, DC, 2004, vol. 6, pp. 68–88 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |