
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyridinic and graphitic nitrogen-rich graphene for high-performance supercapacitors and metal-free bifunctional electrocatalysts for ORR and OER†
Shaikh Nayeem
Faisal
 *,
Enamul
Haque
,
Nikan
Noorbehesht
,
Weimin
Zhang
,
Andrew T.
Harris
,
Tamara L.
Church
and
Andrew I.
Minett
*
*,
Enamul
Haque
,
Nikan
Noorbehesht
,
Weimin
Zhang
,
Andrew T.
Harris
,
Tamara L.
Church
and
Andrew I.
Minett
*
Laboratory of Sustainable Technology, School of Chemical & Biomolecular Engineering, University of Sydney, NSW 2006, Australia. E-mail: shaikh.faisal@sydney.edu.au; andrew.minett@sydney.edu.au
First published on 23rd March 2017
Abstract
A facile synthesis method to produce nitrogen-doped graphene containing a high atomic percentage of pyridinic N and graphitic N via the thermal annealing of graphene oxide and uric acid is reported. The method yielded N-doped graphene nanosheets that had high surface areas and that contained 9.22 at% nitrogen, which was present in four bonding configurations with a high ratio of pyridinic N and graphitic N. The synthesized N-doped graphene showed excellent capacitance properties, which led to the fabrication of a stacked electrode supercapacitor cell of 2-electrode configuration with a specific capacitance of 230 F g−1 at a current density of 1 A g−1 and with a remarkably high energy density of 62.6 W h kg−1 in aqueous electrolyte. Additionally, this material showed superior properties as an electrocatalyst for both the oxygen-reduction reaction (ORR) and oxygen evolution reaction (OER), producing high current density primarily via the four-electron pathway for ORR at a current density of −4.3 mA cm−2 and OER activity of a generating current density of 10 mA cm−2 at 1.74 V vs. RHE in alkaline media. Hence, this dual-natured N-doped graphene could provide a platform for developing nanostructured graphene-based advanced energy storage and conversion devices.
1. Introduction
Carbon-based materials for clean and renewable energy are becoming promising candidates to help meet the global demand for sustainable energy development. In particular, nanostructured materials for efficient energy storage and conversion devices have been reported for a wide range of applications including as solar cells,1 fuel cells, lithium-ion batteries and electrochemical double-layer capacitors.2–4 Among these energy storage devices, supercapacitors have gained attention due to their orders-of-magnitude-higher power density, cycle efficiency and charge/discharge rates than batteries.5,6 Besides energy storage, energy conversion is also crucial to meeting global energy demands. Fuel cells are among the promising methods of delivering energy in the future, but their widespread implementation is hindered by the expense of the cathode material (platinum), as well as by their deactivation by the by-products of fuel decomposition (the fuel crossover effect), making the development of alternative, precious-metal-free electrode materials essential.7–9 Graphene, composed of a one-atom-thick honeycomb lattice of sp2-hybridized carbons, has shown great potential as an electrode material in energy storage and conversion due to its excellent electrical conductivity and high specific surface area.10–13Chemical dopants can alter the electronic properties of graphene.14 The introduction of dopants, either by the adsorption of molecules on the graphene surface or by the substitution of dopant atoms into the carbon lattice, can produce a band gap in graphene, and this strategy has led to the development of high-performance energy storage and conversion devices.15,16 Various heteroatoms, including N, B, and S, have imparted excellent electrochemical properties,17–21 and N-doped graphene in particular has shown capacitance suitable for high-performance supercapacitor applications,22–26 excellent electrocatalytic activity for oxygen reduction in fuel cells27,28 and oxygen evolution reaction for rechargeable metal–air batteries.29,30 The properties of nitrogen-doped graphene (NG) are conferred by nitrogen atoms within the carbon lattice, where they can exist as pyridinic, pyrrolic, graphitic, and oxidized N (Scheme 1).31
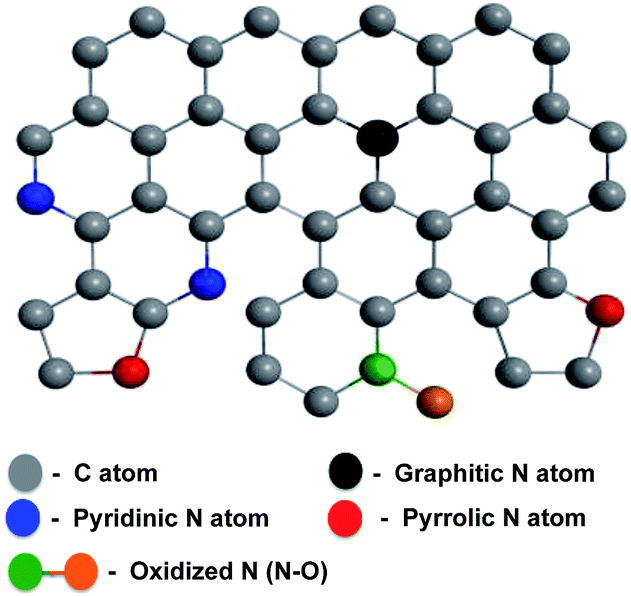 | ||
| Scheme 1 Schematic structure of NG. | ||
The roles of the differently configured nitrogen atoms in enhancing capacitance or generating catalytically active sites are still controversial,32,33 but both quantum mechanical calculations34–36 and experimental24,37 investigations indicate that graphitic, pyridinic, and pyrrolic N can enhance the capacitance, whereas pyridinic N and graphitic N can be essential to catalyzing oxygen reduction reaction by enhancing the adsorption of O2 on the adjacent carbon atoms and thus promotes the four-electron pathway.38–41 Besides the capacitance and oxygen reduction reaction, oxygen evolution reaction has gained a considerable attention due to the promising future of rechargeable metal–air batteries.42–44 N-doped graphene,29 NG/CNT composites,30 N-doped coaxial carbon nanotube,45 carbon nitrogen nanotubes46 and nitrogen and phosphorous co-doped nanocarbons47,48 have shown to be promising alternatives to noble and transition metal based OER catalysts.
The enhanced supercapacitive and electrocatalytic properties are highly dependent on the type and amount of nitrogen present. To obtain both the capacitance and electrocatalytic properties, high atomic concentrations of pyridinic, pyrrolic and graphitic nitrogen are required. The types and amounts of the different nitrogen bonding configurations in an N-doped graphene sample depend on the nitrogen source as well as the synthesis method. Nitrogen has been incorporated into the graphitic lattice from sources like acetonitrile,49 pyridine50 and ammonia51via chemical vapor deposition (CVD) or high-temperature treatment, and the resulting materials have been applied in lithium batteries,49 field effect transistors,50 and as catalysts for oxygen reduction.51 However, it is difficult to obtain materials with high levels of atomic nitrogen doping, and on a large scale, via CVD using gaseous or liquid sources of nitrogen, and the method also suffers from practical limitations due to possible contamination from the metal deposition catalyst as well as the toxicity of the precursors. On the other hand, the co-pyrolysis of graphene or a graphene precursor with a solid source of nitrogen, such as melamine51 or cyanamide,52 has produced NG with high levels of nitrogen (∼9 at%). Co-pyrolysis methods are attractive because they can be scaled up without any catalyst.53 However, the NG synthesized from melamine had a low surface area;51 whereas cyanamide, on the other hand, required a surfactant to bind to the graphene surface.52 Moreover, the NG synthesized from cyanamide did not contain pyrrolic nitrogen,52 which plays an important role in enhancing capacitance.34 In addition to melamine and cyanamide, urea has been employed as a solid source of nitrogen in the synthesis of N-doped graphene, however both the capacitance and electrocatalytic properties in the material were not investigated.53–56 In order to show both high capacitance and electrocatalytic activity, an NG sample must contain sufficient percentages of pyridinic, pyrrolic, and graphitic nitrogen atoms, and have large surface area and high pore volume.
Herein, we report the synthesis of NG via the thermal annealing of graphene oxide (GO) with uric acid at 800 °C under an argon atmosphere. Uric acid has a high nitrogen content (∼33 wt%), and its crystals can adhere to surfaces bearing various organic functionalities, and via multiple types of forces (electrostatic interactions, hydrogen bonding and/or van der Waals interactions), without the aid of surfactants.57 These properties allowed the co-pyrolysis of graphene oxide and uric acid to produce a nitrogen-doped reduced graphene oxide (rGO) with nitrogen atoms in all of the possible configurations in the graphitic lattice, a high total nitrogen content with a high ratio of pyridinic N and graphitic N and a high surface area after pyrolysis. As a result, the N-doped graphene had both high capacitance and bifunctional electrocatalytic activity towards oxygen reduction and evolution.
2. Experimental
Synthesis of nitrogen-doped graphene
GO was prepared from naturally expanded graphite (Ashbury Carbons, Grade-3772) using a reported procedure for the generation of ultra-large GO sheets.58 The GO sheets (approx. 20 mg) were dispersed in water by ultrasonication for 30 min at a frequency of 50 Hz, then mixed with uric acid in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:5 or 1:10 by mass. The mixtures were then stirred continuously and heated at 80 °C for 10–12 h to remove the water. The resulting solids were transferred to a tubular furnace and heated in an argon atmosphere at 5 °C min−1 to 800 °C, then annealed for 1 h. The synthesized nitrogen-doped graphenes produced with 1:1, 1:5 and 1:10 ratios of graphene oxide and uric acid were named NG1, NG5 and NG10, respectively. Pure GO was also thermally annealed to yield reduced graphene oxide (rGO) under the same conditions, and used as reference.
:1, 1:5 or 1:10 by mass. The mixtures were then stirred continuously and heated at 80 °C for 10–12 h to remove the water. The resulting solids were transferred to a tubular furnace and heated in an argon atmosphere at 5 °C min−1 to 800 °C, then annealed for 1 h. The synthesized nitrogen-doped graphenes produced with 1:1, 1:5 and 1:10 ratios of graphene oxide and uric acid were named NG1, NG5 and NG10, respectively. Pure GO was also thermally annealed to yield reduced graphene oxide (rGO) under the same conditions, and used as reference.
Characterization
The microstructures and morphology of the synthesized samples were investigated by Field Emission Scanning Electron Microscopy (FESEM, Zeiss ULTRA plus) and High Resolution Transmission Electron Microscopy (HRTEM, JEOL 2200FS) with an acceleration voltage of 200 kV. Thermogravimetric Analysis (TGA, TA Q500) was performed under Ar and at a heating rate of 5 °C min−1. X-ray diffractometry (XRD) was performed on a Shimadzu S6000 equipped with Cu Kα radiation (λ = 0.154 nm) with a resolution of 0.02°. Raman spectra were collected with Renishaw inVia Raman Spectrometer with laser wavelength of 514 nm. Nitrogen sorption isotherms and Brunauer–Emmett–Teller (BET) surface areas were measured over P/P0 = 0.05–0.3 at 77 K on an Autosorb IQ (Quantachrome, USA). X-ray photoelectron spectroscopy (XPS) analyses were performed by ESCALAB250Xi (Thermo Scientific, UK) with a monochromated Al Kα (energy 1486.68 eV) operating at 150 W (13 kV × 12 mA) under a vacuum of 2 × 10−9 mbar. The analysis spot was 500 µm in diameter. The binding energies were referenced to the adventitious hydrocarbon C 1s signal at 284.9 eV.Electrochemical measurement
For electrochemical characterization and measurement, the nitrogen-doped graphenes were deposited from a 1 mg mL−1 mixture, prepared by sonicating in degassed isopropanol, on a glassy-carbon electrode. The glassy-carbon working electrode was polished mechanically with a slurry of 0.05 µm alumina particles to obtain a mirror-like surface, washed with Milli-Q water and acetone, and allowed to dry. It was then tested in a three-electrode cell, using platinum wire as the counter electrode and a Ag/AgCl, KCl (3 M) electrode as the reference electrode. Measurements were taken using a Bio-logic SP300 potentiostat. The binding agents were 10 wt% PTFE for capacitance measurements and 5 wt% Nafion for electrocatalytic measurements. Capacitance was determined using a T-cell device in a two-electrode cell configuration, as described previously.59,60 The device was fabricated using a Swaglok system with two stainless steel current collector. The PVDF separator was placed between the current collectors where the material to be studied was sandwithed by pressing together at high pressure. 0.5 M H2SO4 was used as the electrolyte and the cell was airtight. 2.0 mg active materials were deposited on the diameter of an 8 mm stainless steel electrode surface using a 10-wt% PTFE binder. The gravimetric capacitance from galvanostatic charge/discharge was calculated using eqn (1).6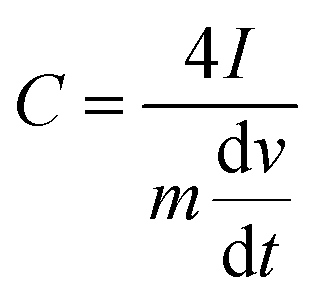 | (1) |
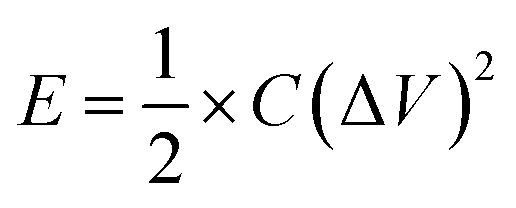 | (2) |
| P = E/t | (3) |
3. Results and discussion
In the synthesis of NG (Scheme 2), graphene oxide and uric acid were mixed together in water (in ratios of 1:1, 1:5 or 1:10 by mass in mg scale for NG1, NG5 and NG10, respectively), and then stirred continuously at 80 °C to remove the water. We expect that the uric acid molecules adsorbed on the graphene oxide surface during this time. The solid mixtures were then heated in a tubular furnace at a rate of 5 °C min−1 to 800 °C in an argon environment. The formation of N-doped graphenes from the pyrolysis of GO with melamine51 and cyanamide53 had been proposed to proceed in two steps via formation of carbon nitride first, which forms from the pyrolysis of cyanamide.62 For the present case, from the thermogravimetric-analysis curves examined under argon in a thermogravimetric analyser (TGA) (Fig. S1†), it is observed that the rapid weight losses started from 400 °C due to the decomposition of uric acid and the complete decomposition of the GO/uric acid mixture occurred around 750 °C. The thermogravimetric analysis of uric acid has also been performed to measure the possible amount of hard carbon transformed from the uric acid after carbonization. The TGA curve of uric acid showed the almost complete decomposition from 650 °C and only 0.38% of the initial mass of uric acid remained after the complete carbonization. This behaviour supports the mechanism of interaction of heterocyclic –NH groups of uric acid with the functional groups of GO, contributing to the incorporation of N into the graphene lattice rather than via formation of carbon nitride.53 Due to organic in nature and high surface attachment ability,57 uric acid can be attached on the graphene surface better than the inorganic solid precursor urea and thus higher atomic percentage of nitrogen with four possible bonding configurations has been achieved.
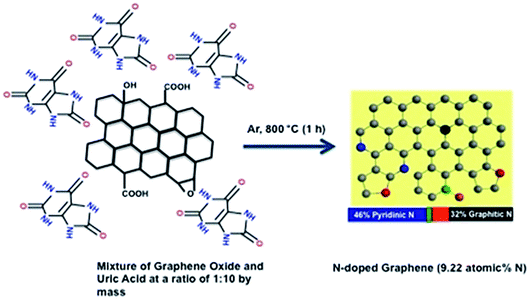 | ||
| Scheme 2 Schematic diagram of the formation of NG via the thermal annealing of graphene oxide with uric acid. | ||
The similar mechanism has been observed with organonitrogen compounds having high nitrogen contents26,71 as well as the high nitrogen containing liquid organic isomers of phenylenediamine.72 The advantages of uric acid are, the less expensive commercial availability compared to organonitrogen compounds like aminoterephthalic acid26 and methylimidazole71 as well as safe handling during synthesis due to its solid powder form rather than handling toxic liquids. The morphology, structure, and graphitic layers of the obtained NGs were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM images of NG10 showed a laminar morphology (Fig. 1a), and its low-resolution TEM image showed transparent stacked graphene sheets. The high-resolution transmission electron micrograph (HRTEM) and selected area electron diffraction (SAED) pattern revealed the highly crystalline nature of NG10 (Fig. 1b). The well-defined diffraction spots and rings in the SAED pattern confirmed that the sample was comprised of the hexagonal carbon lattice typical of graphene-like materials.63 The XRD patterns of NG1, NG5 and NG10 (Fig. 1c) showed broad peaks at around 26.0°, which corresponded to an interlayer spacing of 0.342 nm.
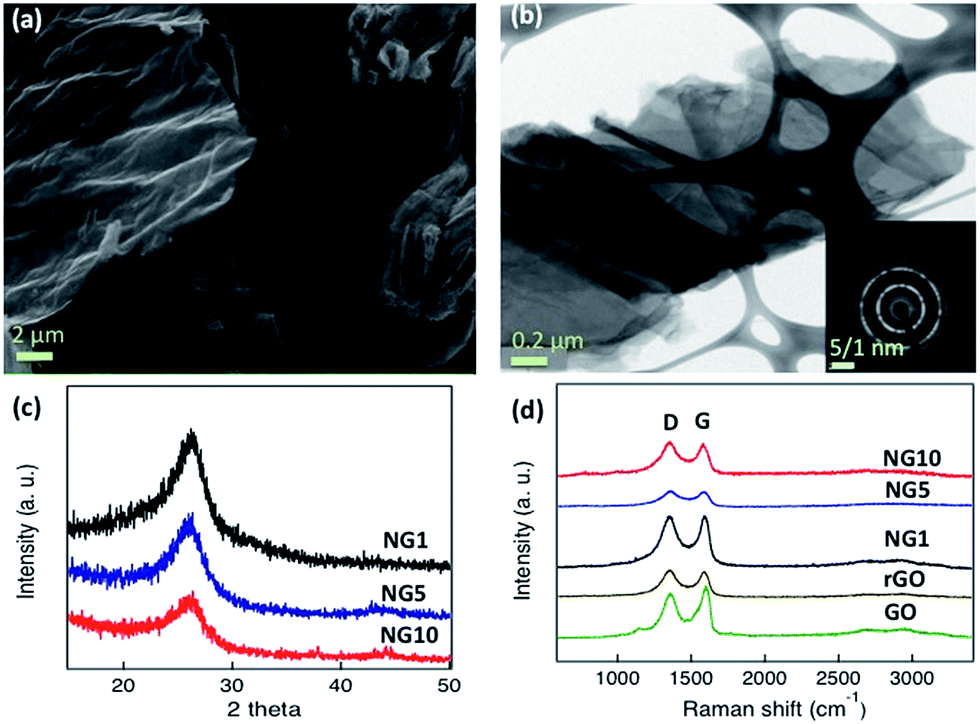 | ||
| Fig. 1 (a) SEM image of NG10, (b) TEM image and selected area electron diffraction (SAED) pattern (inset) of NG10, (c) XRD pattern of NG1 (black), NG5 (blue) and NG10 (red), (d) Raman spectra of NG10 (red), NG5 (blue), NG1 (black), reduced graphene oxide (brown) and graphene oxide (green). | ||
GO, rGO, and the N-doped materials NG1, NG5 and NG10, were characterized by Raman spectroscopy at an excitation wavelength of 514 nm under ambient conditions. As shown in Fig. 1d and Table 1, the G peaks of the GO, rGO, NG1, NG5 and NG10 occurred between 1582 and 1598 cm−1. The G peaks shifted to lower energy from GO to NG10, indicating that a conjugated sp2 structure was restored during pyrolysis.53 The ID/IG ratios of GO, rGO, NG1, NG5 and NG10 (Table 1) were used to evaluate the defects or in-plane crystallite size after pyrolysis, and the crystallite sizes, La, were calculated according to the Tuinstra–Koenig (TK) relation, eqn (4).38,64
| La = (2.4 × 10−10)λ4(ID/IG)−1 | (4) |
| ν(G)a (cm−1) | I D/IGa (−) | L a b (nm) | S BET c (m2 g−1) | V total d (cm3 g−1) | N sitese (% of total N) | Total Ne (at%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pyridinic | Pyrrolic | Graphitic | Oxide | |||||||
| a Position of the G peak (ν(G)) and ratio of D and G peak intensities (ID/IG) determined from Raman spectroscopy using λexcitation = 514 nm. b Crystallite size (La) calculated from Raman data according to the Tuinstra–Koenig relation.64 c BET surface area calculated over P/P0 = 0.05–0.30 from N2 adsorption at 77 K. d Total pore volume calculated from P/P0 = 0.995 from N2 adsorption at 77 K. e Calculated from X-ray photoelectron spectroscopy. Total nitrogen was calculated from detection-sensitivity-adjusted peak areas. f Not determined. | ||||||||||
| GO | 1598 | 0.87 | 19 | —f | —f | —f | —f | —f | —f | —f |
| rGO | 1594 | 1.03 | 16.3 | 75.68 | 0.07 | —f | —f | —f | —f | —f |
| NG1 | 1592 | 0.99 | 16.8 | 96.06 | 0.20 | 41.4 | 34.9 | 19.1 | 4.61 | 1.52 |
| NG5 | 1582 | 1.03 | 16.2 | 164.04 | 0.68 | 46.3 | 13.6 | 28.4 | 9.33 | 6.75 |
| NG10 | 1582 | 1.03 | 16.2 | 197.80 | 0.80 | 46.6 | 14.1 | 32.4 | 6.83 | 9.22 |
To characterize the elemental composition and nitrogen content in the NGs, X-ray photoelectron spectra (XPS) of the materials were measured. A comparison of the spectra of GO and rGO showed a decrease in oxygen content in rGO; neither spectrum showed obvious N 1s peaks (Fig. S3†). As shown in Fig. 2a, the survey spectra of NG samples revealed the presence of C, O and N. In NG1, NG5 and NG10, the high-resolution C 1s peak was centred at 284.4 eV and tailed at higher binding energies, indicating the connection of carbon atoms with N and O atoms. Though NG10 (Fig. 2b), NG5 (Fig. S4†) and NG1 (Fig. 2c) showed significant amounts of graphite-like C (284.40 eV),65 quantitative analysis of the C 1s region of the spectrum was impossible due to the likely presence of adventitious carbon in the samples. The deconvolutions of the N 1s peaks (Fig. 2d, S5 and S6,†Table 1) showed the presence of four nitrogen configurations. All the NG1, NG5 and NG10 contained primarily (>40%) pyridinic nitrogen and minor amounts (<10%) of N-oxides, but they differed in that NG1 contained much more pyrrolic N, whereas NG5 and NG10 was richer in graphitic N. The possible reason of high content of pyridinic N and graphitic N in NG5 and NG10 is due to interaction of higher amount of available nitrogen atoms with the active sites of created vacant carbon bonds from the functional groups during the doping process at high temperature. Due to the presence of higher ratio of functional groups on the basal planes and six members benzene rings, the high amount of nitrogen doping occurred in pyridinic and graphitic sites.31,34,73
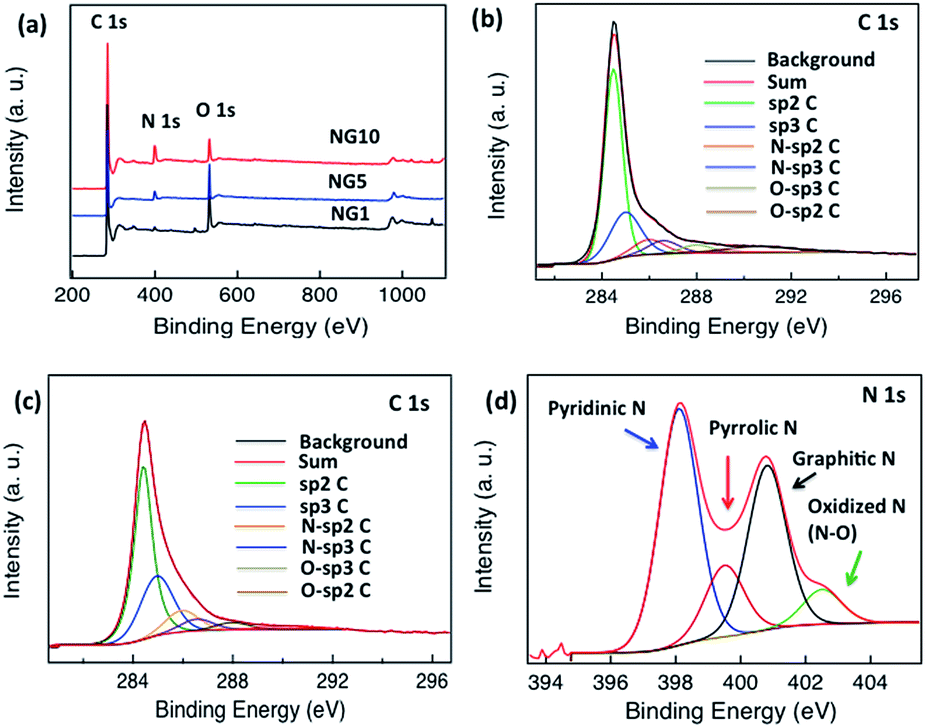 | ||
| Fig. 2 (a) XPS spectra of NG10 (red), NG5 (blue) and NG1 (black), (b) high-resolution C 1s spectrum of NG10, (c) high-resolution C 1s spectrum of NG1, (d) N 1s spectrum of NG10. | ||
Moreover, NG10 and NG5 had a much greater total nitrogen content (9.22 at% for NG10 and 6.75 at% for NG5, Table 1) and higher Brunauer–Emmett–Teller (BET) surface area (197.8 m2 g−1 measured over P/P0 = 0.05–0.30 for NG10 and 164.04 m2 g−1 measured over P/P0 = 0.05–0.30 for NG5, Fig. S7, S8† and Table 1) than did NG1 (1.52 at% N, Table 1; SBET = 96.06 m2 g−1 measured over P/P0 = 0.05–0.30, Fig. S9† and Table 1); the NG materials had greater surface area than rGO (75.68 m2 g−1 measured over P/P0 = 0.05–0.30, Fig. S10† and Table 1). NG10 also displayed high pore volume of 0.8 cm3 g−1 (as defined at P/P0 = 0.995), whereas the pore volumes of NG5, NG1 and rGO were 0.68, 0.2 and 0.07 cm3 g−1 (as defined at P/P0 = 0.995), respectively. Based on the above results, it is evident that the inclusion of N from uric acid contributed to raise the surface areas and pore volumes of the pyrolyzed N-doped graphene materials.
Cyclic voltammetry (CV) was used to investigate the electrochemical behavior of rGO, NG1, NG5 and NG10 in 0.5 M aqueous H2SO4 electrolyte deposited on a glassy carbon electrode at potential limits of 1.2 to −0.2 V vs. Ag/AgCl electrode in a three-electrode system at a scan rate of 100 mV s−1 (Fig. 3a), with 10 wt% PTFE in isopropanol being used as a binder. A comparison of the CVs for rGO, NG1, NG5 and NG10 revealed that the capacitive response became larger with a highly rectangular nature with increasing nitrogen content, and the higher doping percentage resulted in an enhancement of the double layer capacitance, indicating the more effective contribution of electrochemical double layer capacitance with pseudocapacitance for the sample with higher nitrogen content, NG10. Thus the electrochemical activity and reversibility of the capacitance of NG10 were recorded at different scan rates (Fig. S11†), and the capacitance increased at greater scan rates.
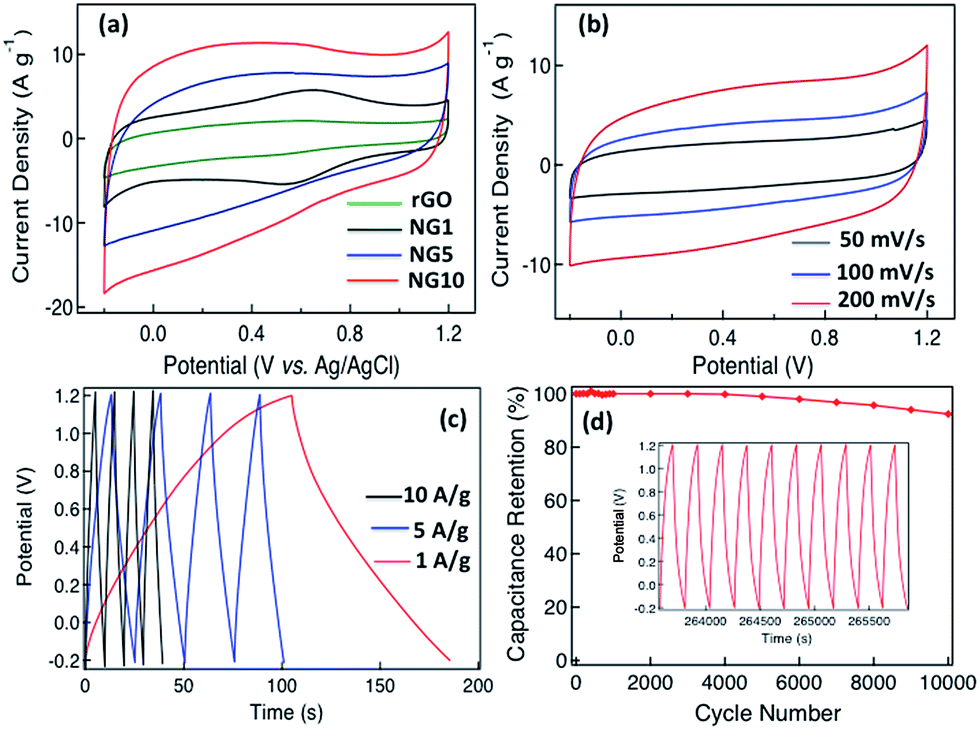 | ||
| Fig. 3 (a) Cyclic voltammograms of rGO, NG1, NG5 and NG10 using a three-electrode cell at a scan rate of 100 mV s−1 in 0.5 M H2SO4 solution, (b) cyclic voltammograms of NG10 at different scan rates (50, 100 and 200 mV s−1) in a two-electrode T-cell system, (c) galvanostatic charge/discharge curves for NG10 in a two-electrode T-cell system tested at current density of 1–10 A g−1, (d) cycling stability of NG10 measured at 5 A g−1 over the potential range of 1.2 to −0.2 V. The inset shows the charge–discharge curves over the last ten cycles. | ||
A T-cell device consisting of stainless steel electrodes with a Teflon cover was used to investigate the capacitive properties of NG10 for practical application (Fig. S12†).60 The active material was deposited on the electrodes with a PTFE binder and dried in an oven at 110 °C for 1 h. A PVDF membrane separator was inserted between the two electrodes, which were then pressed and sandwiched under high pressure. The H2SO4 electrolyte was poured from the top to completely fill the device before it was sealed air-tight. CVs recorded in the T-cell at different scan rates (50, 100 and 200 mV s−1) showed near-ideal rectangular voltammetric responses (Fig. 3b). The specific capacitance (Csp) of the NG10 electrode material was calculated from the galvanostatic discharge curves (Fig. 3c) to be 230 F g−1 in 0.5 M H2SO4 at a current density of 1 A g−1. At a high current density of 10 A g−1, the specific capacitance value remained as high as 137 F g−1. The RC constants at 1 and 10 A g−1 were 40.25 and 2.39 s, respectively. The energy density and power density of the device were calculated based on the discharge rates and plotted on a Ragone plot (Fig. S13†). The highest energy density, 62.61 W h kg−1 was achieved at the power density of 2.79 kW kg−1, and an energy density of 37.29 W h kg−1 was retained even at a high power density of 28.08 kW kg−1; this was superior than the values reported for chemical-doped graphene-based materials. (ESI Table S1†).
Besides the gravimetric capacitance, volumetric capacitance is also very important for fabricating devices for practical applications. The average density of the active material on the electrodes was calculated based on the thickness measured by SEM to be 0.5 g cm−3, meaning that the volumetric capacitance was 115 F cm−3, which is higher than that of nanostructured graphene-based supercapacitor materials such as activated graphene,6 graphene foam,66 graphene hydrogel67 and laser-scribed graphene.68 The superior performance of NG10 could be attributed to its high contents of pyridinic, pyrrolic and graphitic N, as well as to its high surface area with large pore volume, which provided excellent electrical conductivity and pseudocapacitance with favourable electrolyte penetration. The stability of the device was also investigated over 10000 cycles between 1.2 and −0.2 V at a high current density of 5 A g−1 (Fig. 3d). The material maintained almost 100% of its initial specific capacitance upto 4000 cycles and then gradually started decreasing and kept 92.4% of its initial specific capacitance after 10000 cycles. The charge/discharge curves for the last ten cycles demonstrate the stability of the device (inset of Fig. 3d).
NG10 was also tested as an electrocatalyst for the oxygen-reduction reaction (ORR) under alkaline conditions. Thus, NG10 was deposited on a glassy carbon electrode and its cyclic voltammetry was measured in both argon- and oxygen-saturated 0.1 M KOH(aq) at a scan rate of 50 mV s−1. As shown in Fig. 4a, a clean capacitive CV background was observed within a potential range of 1.0 to −0.05 V in the argon-saturated electrolyte. In contrast, in the oxygen-saturated electrolyte, a well-defined cathodic current was detected, and peaked at ≈+0.6 V. This indicated that NG10 was an active catalyst for O2 reduction. The electrocatalytic activities of NG10 and the related materials rGO, NG1 and NG5 were further investigated using rotating disk electrodes (RDE). Fig. 4b shows the linear sweep voltammograms of rGO, NG1, NG5 and NG10, all measured at a scan rate of 10 mV s−1 and rotation frequency of 1600 rpm in oxygen-saturated 0.1 M KOH(aq). To reveal the best performance in electrocatalysis, the effect of mass loading of the active material on the glassy carbon electrode has been investigated (Fig. S14†). Different masses of NG10 (4, 8, 12, 16 and 20 µg) have been deposited on a glassy carbon electrode having an area of 0.197 cm2. From the linear sweep voltammograms curves at 1600 rpm with the presence of oxygen, it was observed that the current density was increasing with the mass loading from 4 to 12 µg due to the increment of density of the active material (NG10). However, the current density was started to decrease even with the increasing mass loading from 16 to 20 µg. The possible reason for this behaviour is due to the restacking of graphene layers after covering the whole surface area which hampering the permeation of electrolyte between the graphene layers causing less current density.61 The electrochemical impedance spectroscopy (EIS) study (Fig. S15†) also showed the highest conductivity as well as very small inherent resistance from 4 to 12 µg mass loading and a formation of semi Nyquist plot for 20 µg mass loading indicated higher charge transfer resistance compared to the rest of the plots in higher frequencies. Moreover, all the spectra in low frequencies exhibit a Warburg angle higher than 45° indicating the electrodes were highly controlled by ion diffusion/transport process.
 | ||
| Fig. 4 (a) Cyclic voltammograms of NG10 at a scan rate of 50 mV s−1 in Ar-saturated (black) and O2-saturated (red) 0.1 M KOH(aq) solution, (b) linear sweep voltammograms (LSV) of rGO, NG1, NG5, NG10 and Pt/C at 1600 rpm and a scan rate of 10 mV s−1 in O2-saturated 0.1 M KOH(aq), (c) RDE measurements of NG10 in O2-saturated 0.1 M KOH(aq) at scan rates of 10 mV s−1, and (d) Koutecky–Levich plots of NG10 at electrode potentials of 0.6, 0.5, 0.4 and 0.3 V. | ||
The NG10 produced the most positive onset potential of +0.75 V and highest current density of −4.3 mA cm−2, and further RDE measurements (Fig. 4c) were thus used to calculate, via the Koutecky–Levich equation (eqn (5)),28 the number of electrons transferred to an oxygen molecule by the NG10 electrode.
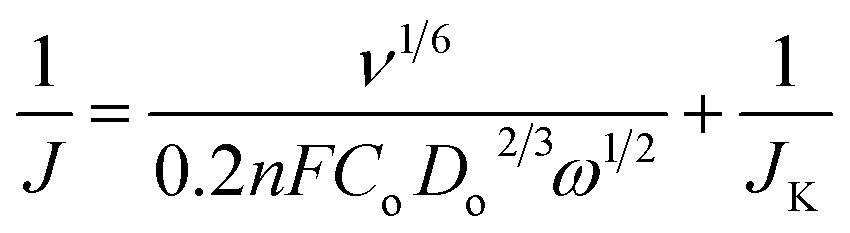 | (5) |
In the Koutecky–Levich equation, J, JL, JK are the measured current density, the diffusion-limiting current density and the kinetic-limiting current density, respectively; ω is the angular velocity of the disc, n is the overall number of electrons transferred in the oxygen reduction, F is the Faraday constant (96485 C mol−1), Co is the bulk concentration of oxygen (1.2 × 10−6 mol cm−3), Do is the diffusion coefficient of oxygen in 0.1 M KOH(aq) (1.9 × 10−5 cm2 s−1), v is the kinetic viscosity (0.01 cm2 s−1) and 0.2 is a constant that is valid when the rotation speed is expressed in rpm. The Koutecky–Levich plot of J−1vs. ω−1/2 at a potential of 0.6 V on the NG10 electrode (Fig. 4d) exhibited good linearity (R2 = 0.998). The average electron transfer number was 3.82 at different potentials of 0.6, 0.5, 0.4 and 0.3 V, which indicated that a four-electron process, with water as the product, was the preferred pathway. In addition, efficient electrocatalytic activity was observed from the cyclic voltammetry in the presence of 3 M methanol (Fig. S17†), indicating the stability of NG10 to the fuel crossover effect.54
To further investigate the potential application of NG10 enriched with pyridinic N and graphitic N, OER activity was assessed by sweeping the RDE potential from 1.0 to 2.0 V. From the LSV curves of NG10, NG5, NG1, Ru/C and Pt/C plotted in Fig. 5a, the onset potentials were measured 1.52, 1.56, 1.72, 1.46, and 1.68 V respectively. The rGO did not show any significant response towards OER activity. The potentials for a current density of 10 mA cm−2 for NG10, NG5, Ru/C and Pt/C were evaluated at 1.74, 1.76, 1.64 and 1.88 V respectively as the required potential to oxidize water at the current density of 10 mA cm−2 is used to judge the OER activity. Therefore, NG10 exhibited the lowest onset potential and large current density among the studied catalysts of NG5 and Pt/C as well as close to Ru/C (one of the best electrocatalysts for OER at present). The effect of mass loading was also investigated for OER activity and the similar behaviour was obtained showing 12 µg mass loading as the optimum amount for achieving the best electrocatalytic performance (Fig. S16†). The Tafel slope for the NG10 and NG5 were about 104 and 122 mV per decade respectively, much smaller than the Pt/C (208 mV per decade), which indicated the better OER activity of NG10 (Fig. 5b). The Tafel slope of NG10 was further analyzed in two regions (low overpotential and high overpotential with the values of 106 and 99 mV per decade respectively) according to the following reports.74,75 The relatively higher low potential value indicated the first electron transfer reaction rate for OER.76 The OER performance of NG10 was found to be superior to the commercial Pt/C as well as similar to transition metal oxides supported by carbon materials.45,69,70 Even though the exhibition of OER activities in metal-free nitrogen-doped few layers graphene is highly rare,30,47 the possible reason of the attribution of OER activity in NG10 even in two-dimensional nanostructure is due to the presence of high atomic percentages of N including high level of pyridinic N and graphitic N as well as the large surface area with high pore volume. The high percentage of pyridinic N and graphitic N on the edge of the surface creates the active sites for efficient bi-catalysis reaction towards oxygen reduction and evolution. The comparison of bifunctional electrocatalysis with Pt/C measuring by LSV plots at 1600 rpm in oxygen-saturated 0.1 M KOH, (Fig. 5c) the comparable ORR with four electrons transfer pathway and better OER activity of NG10 were observed. In ORR the onset potential and current density of NG10 were lower than the commercial Pt/C as it is the state-of-the-art ORR electrode, however in OER the good catalytic activity of NG10 was reflected for its lower onset potential and higher current density than the Pt/C. Even though the OER activity of Ru/C is better than NG10; however, the ORR activity of Ru/C is poor compared to Pt/C and nitrogen-doped carbons, 48 thus not suitable as a bifunctional electrocatalyst. The electrocatalytical stability of NG10 was also investigated by continuous chronoamperometric measurement at a constant potential of +0.6 V (Fig. 5d). The nitrogen and oxygen gas was added in a flow rate of 50 cm3 min−1 in 100 ml of electrolyte in a closed cell that saturated the electrolyte very quickly and helps to occur electrocatalysis instantly. The sharp increment of current density was observed after adding oxygen to the solution as well as keeping the rotation speed at 400 rpm. A nearly constant current density curve after stabilization was observed for a long cycle time and even at 25000 s around 92% of its initial current density had been retained. Thus this active material can be a potential platform for developing bifunctional electrocatalyst for metal–air batteries and rechargeable fuel cells as well as further modification of structure and/or incorporation of nanomaterials can enhance the electrocatalytical activity superior than the commercial noble metal based catalysts.
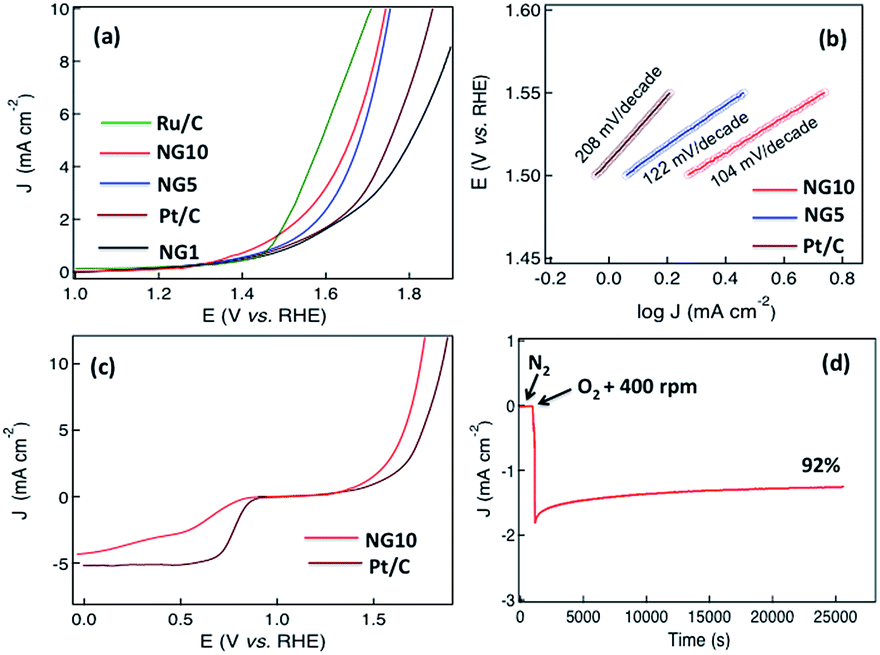 | ||
| Fig. 5 (a) Linear sweep voltammograms (LSV) of Ru/C, NG10, NG5, Pt/C and NG1 at a scan rate of 10 mV s−1 in 0.1 M KOH(aq); (b) shows the Tafel plots for Pt/C, NG5 and NG10 and (c) LSV curves of NG10 and Pt/C on an RDE (1600 rpm) in O2-saturated 0.1 M KOH(aq) at scan rates of 10 mV s−1 showing the electrocatalytic activities towards both ORR and OER, (d) chronoamperometric response of ORR activity in oxygen saturated electrolyte for stability test. | ||
4. Conclusions
In conclusion, we report a facile synthesis of N-doped graphenes with high surface areas as well as nitrogen atoms in all of the reported nitrogen configurations, using uric acid as a solid source of nitrogen. The synthesized material NG10, produced from the co-pyrolysis of graphene oxide and uric acid in a 1:10 mass ratio, showed excellent capacitive properties in acidic media and efficient catalytic activity for oxygen reduction and evolution reaction in an alkaline solution, and could therefore be applied as a promising material in devices for energy storage and conversion. The superb stability and efficient bifunctional electrocatalytic activity could make this material an ideal candidate not only in the field of supercapacitors, fuel cell and metal–air battery electrodes, but also in the area of sensors and transistors. The incorporation of nanoparticles, polymers and other carbon-based nanostructures could further enhance the electrochemical properties for a broader range of applications.
Acknowledgements
S. N. Faisal and E. Haque are grateful to the University of Sydney for USydIS postgraduate scholarships. N. Noorbehesht gratefully acknowledges financial support in the form of a Postgraduate Scholarship from the CSIRO-National Research Flagships Program (Future Manufacturing Flagship; No. FlagshipPhD12/03743). ATH and AIM thank the ARC and the University of Sydney for the ongoing support. The authors also thank Dr Dennis Antiohos for his assistance as well as Dr Bill Gong of the University of New South Wales for assistance with XPS.Notes and references
- M. P. Ramuz, M. Vosgueritchian, P. Wei, C. Wang, Y. Gao, Y. Wu, Y. Chen and Z. Bao, ACS Nano, 2012, 6, 10384–10395 CrossRef CAS PubMed
.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. V. Schalkwijk, Nat. Mater., 2005, 4, 36–377 CrossRef PubMed
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed
- Y. Shao, J. Liu, Y. Wang and Y. Lin, J. Mater. Chem., 2009, 19, 46–59 RSC
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed
- H.-J. Choi, S.-M. Jung, J. M. Seo, D. W. Chang, L. Dai and J.-B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS
- X. Zhang, B. Wang, J. Sunarso, S. Liu and L. Zhi, Wiley Interdiscip. Rev.: Energy Environ., 2012, 1, 317–336 CrossRef CAS
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 CAS
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC
- B. Guo, L. Fang, B. Zhang and J. R. Gong, Insci. J., 2011, 1, 80–89 CrossRef CAS
- R. Lv and M. Terrones, Mater. Lett., 2012, 78, 209–218 CrossRef CAS
- J. Han, L. L. Zhang, S. Lee, J. Oh, K.-S. Lee, J. R. Potts, J. Ji, X. Zhao, R. S. Ruoff and S. Park, ACS Nano, 2013, 7, 19–26 CrossRef CAS PubMed
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed
- Z.-H. Sheng, H.-L. Gao, W.-J. Bao, F.-B. Wang and X.-H. Xia, J. Mater. Chem., 2012, 22, 390–395 RSC
- S. Yang, L. Zhi, K. Tang, X. Fang, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS
- H.-L. Guo, P. Su, X. Kang and S.-K. Ning, J. Mater. Chem. A, 2013, 1, 2248–2255 CAS
- Y. Qiu, X. Zhang and S. Yang, Phys. Chem. Chem. Phys., 2011, 13, 12552–12558 RSC
- L. Sun, L. Wang, C. Tian, T. Tan, Y. Xie, K. Shi, M. Li and H. Fu, RSC Adv., 2012, 2, 4498–4506 RSC
- Z. Wen, X. Wang, S. Mao, Z. Bo, H. Kim, S. Cui, G. Lu, X. Feng and J. Chen, Adv. Mater., 2012, 24, 5610–5616 CrossRef CAS PubMed
- E. Haque, M. M. Islam, E. Pourazadi, M. Hasan, S. N. Faisal, A. K. Roy, K. Konstantinov, A. T. Harris, A. I. Minett and V. G. Gomes, RSC Adv., 2015, 5, 30679–30686 RSC
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed
- Z. Lin, G. H. Waller, Y. Liu, M. Liu and C.-P. Wong, Carbon, 2013, 53, 130–136 CrossRef CAS
- G.-L. Tian, M.-Q. Zhao, D. Yu, X.-Y. Kong, J.-Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 2251–2259 CrossRef CAS PubMed
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS
- M. S. Thorum, J. M. Hankett and A. A. Gewirth, J. Phys. Chem. Lett., 2011, 2, 295–298 CrossRef CAS
- H. M. Jeong, J. W. Lee, W.-H. Shin, Y. J. Choi, J. H. Shi, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed
- L. L. Zhang, X. Zhao, H. Ji, M. D. Stoller, L. Lai, S. Murali, S. Mcdonnell, B. Cleveger, R. M. Wallace and R. S. Ruoff, Energy Environ. Sci., 2008, 5, 9618–9625 Search PubMed
- T. Ikeda, M. Boero, S.-F. Huang, K. Terakura, M. Oshima and J. Ozaki, J. Phys. Chem. C, 2008, 112, 14706–14709 CAS
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337–6342 CrossRef CAS PubMed
- E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. C, 2010, 114, 15306–15314 CAS
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, ACS Nano, 2014, 8, 6856–6862 CrossRef CAS PubMed
- J. Wu, L. Ma, R. M. Yadav, Y. Yang, X. Zhang, R. Vajtai, J. Lou and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2015, 7, 14763–14769 CAS
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed
- M. Park, H. Sun, H. Lee, J. Lee and J. Cho, Adv. Energy Mater., 2012, 2, 780–800 CrossRef CAS
- A. Kraytsberg and Y. Ein-Eli, Nano Energy, 2013, 2, 468–480 CrossRef CAS
- G.-L. Tian, T. Q. Zhang, B. Zhang, Y.-G. Jin, J.-Q. Huang, D. S. Su and F. Wei, Adv. Funct. Mater., 2014, 24, 5956–5961 CrossRef CAS
- R. M. Yadav, J. Wu, R. Kochandra, L. Ma, C. S. Tiwari, L. Ge, G. Ye, R. Vajtai, J. Lou and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2015, 7, 11991–12000 CAS
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed
- R. Li, Z. Wei and X. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS
- Z. Jin, J. Yao, C. Kittrell and J. M. Tour, ACS Nano, 2011, 5, 4112–4117 CrossRef CAS PubMed
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 CAS
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed
- H. Wang, C. Zhang, Z. Liu, L. Wang, P. Han, H. Xu, S. Dong, J. Yao and G. Cui, J. Mater. Chem., 2011, 21, 5430–5434 RSC
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Müllen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed
- Z. Lin, G. Waller, Y. Liu, M. Liu and C.-P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS
- Z. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 CAS
- Z. Mou, X. Chen, Y. Du, X. Wang, P. Yang and S. Wang, Appl. Surf. Sci., 2011, 258, 1704–1710 CrossRef CAS
- J. B. Presores and J. A. Swift, Langmuir, 2012, 28, 7401–7406 CrossRef CAS PubMed
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, R. L. Shepherd, J. Chen, S. A. Yamini, K. Konstantinov, A. I. Minett, J. M. Razal and G. G. Wallace, Adv. Funct. Mater., 2013, 23, 5345–5355 CrossRef CAS
- M. S. Romano, N. Li, D. Antiohos, J. M. Razal, A. Nattestad, S. Beirne, S. Fang, Y. Chen, R. Jalili, G. G. Wallace, R. Baughman and J. Chen, Adv. Mater., 2013, 25, 6602–6606 CrossRef CAS PubMed
- D. Antiohos, M. S. Romano, J. M. Razal, S. Beirne, P. Aitchison, A. I. Minett, G. G. Wallace and J. Chen, J. Mater. Chem. A, 2014, 2, 14835–14843 CAS
- N. Jung, S. Kwon, D. Lee, D. Yoon, Y. M. Park, A. Benayad, J. Choi and J. S. Park, Adv. Mater., 2013, 25, 6854–6859 CrossRef CAS PubMed
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS
- C. J. Powell, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 1–3 CrossRef CAS
- Y. Zhao, J. Liu, Y. Hu, H. Cheng, C. Hu, C. Jiang, L. Jiang, A. Cao and L. Qu, Adv. Mater., 2013, 25, 591–596 CrossRef CAS PubMed
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed
- M. F. Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef PubMed
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed
- Y. Wang, W. Ding, S. Chen, Y. Nie, K. Xiong and Z. Wei, Chem. Commun., 2014, 50, 15529–15532 RSC
- M. M. Islam, S. N. Faisal, A. K. Roy, S. Ansari, D. Cardillo, K. Konstantinov and E. Haque, J. Nanotechnol. Mater. Sci., 2015, 2(1), 1–5 Search PubMed
- Y. Zou, W. Zhong, S. Li, J. Luo, C. Xiong and W. Yang, Electrochim. Acta, 2016, 212, 828–838 CrossRef CAS
- X.-F. Li, K.-Y. Lian, L. Liu, Y. Wu, Q. Qui, J. Liang and M. Deng, Sci. Rep., 2016, 6, 23495 CrossRef CAS PubMed
- L. Wang, Z. Tang, W. Yan, H. Yang, Q. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 20635–20641 CAS
- L. Wang, Z. Tang, W. Yan, Q. Wang, H. Yang and S. Chen, J. Power Sources, 2017, 343, 458–466 CrossRef CAS
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01355h |
| This journal is © The Royal Society of Chemistry 2017 |