
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and investigation on liquid crystal and optical properties of dyads based on triphenylene and perylene†
Xiangfei Konga,
Liting Xiaa,
Haifeng Zhanga,
Shengping Daia,
Caili Yua,
Zheng Liua,
Linping Mub,
Guixia Wang *a and
Zhiqun He*c
*a and
Zhiqun He*c
aCollege of Chemistry and Bioengineering, Guangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials, Guilin University of Technology, Guilin 541004, China. E-mail: 2010033@glut.edu.cn
bSchool of Physics and Information Engineering, Shanxi Normal University, Linfen 041004, China
cKey Laboratory of Luminescence and Optical Information, Ministry of Education, Institute of Optoelectronic Technology, Beijing Jiaotong University, Beijing 100044, China. E-mail: zhqhe@bjtu.edu.cn
First published on 17th March 2017
Abstract
Four novel mesogenic dyads consisting of a hexa(alkoxy)triphenylene donor that was linked to a perylene tetracarboxylic esters acceptor by the bridges (O–(CH2)2–O), (O–(CH2)6–O), (O–(CH2)10–O) and (O–(CH2)12–O), had been synthesized. Their structures were characterized by 13C and 1H nuclear magnetic resonance (NMR), infrared spectroscopy (IR) and elemental analysis (EA). The experimental results of cyclic voltammetry and UV/Vis showed the aliphatic linkage of the donor–acceptor dyads preserved the genuine electrochemical behaviors and light absorption properties of the donor and acceptor units, and the energy level difference between the HOMO of the donor and the LUMO of the acceptor is about 1.94 eV. The differential scanning calorimetry (DSC) traces and polarizing optical microscopy (POM) textures confirmed that all dyads had mesophase. When excited at 443 nm, fluorescence quenching process of the acceptor unit was ascribed to a intramolecular ground-state charge transfer from the donor to it. And the fluorescence quenching varying with the bridge lengths were understood by Dexter-type energy transfer theory. These behaviors of photoinduced intramolecular charge transfer and forming columnar liquid crystal phase made these dyads candidates of new active materials in organic solar cells.
Introduction
Since the first discovery of mesogenic benzene-hexa-n-alkanoate discotic organics by Chandrasekhar in 1977,1 discotic liquid crystals (DLCs) have received much attention due to their attractive potentials in organic electronic devices. A DLC molecule is normally composed of two components, a planar rigid core and several flexible alkyl chains as substitutes peripherally connected to the core.2 Discotic mesogens tend to self-organize into columns due to π–π interactions between the neighbouring cores, as well the van der Waals attraction involving alkyl chains.3 This offers DLCs good electron and/or hole transporting properties along the column direction.4 The exchange of charge carriers between adjacent columns is strongly hindered due to the insulation surrounding shell forming by alkyl chains.5 The anisotropy in conductivity of a triphenylene (TP) mesogen in its columnar phase was found to be typically about σ‖/σ⊥ = 1000 (σ‖ and σ⊥ denoting conductivity parallel and perpendicular to the column, respectively).6 These materials are therefore considered as quasi one-dimensional semiconductors.DLC materials have already demonstrated applications in optoelectronic devices, such as organic photovoltaics (OPV) and light emitting devices (OLEDs).7 In these applications, tripheny-lene-based DLCs are normally electron-rich and used as p-type semiconductor,8 while DLCs based on perylene bisimides and tetracarboxylic esters (PTE) are deficient in electrons and often used as n-type semiconductors.9 Recently DLCs dyads, triads and oligomers having electron donor (D) units and electron acceptor (A) units connected by covalent bond bridges, have been synthesized.10–15 Due to intramolecular charge transfer states, a large stokes shift had been observed.15 Efficient light-induced electron and energy transfer processes also occurred in these systems when excited in the visible region.14 However the mechanism of the light-induced charge and energy transfer processes in these DLCs dyads, triads and oligomers has not been fully understood.13
When the D–A typed mesogenic molecules underwent face-on arrangement on the substrates, a configuration for one-dimensional hole and electron transport pathways will be realized. This will be an optimized layout for OPV applications.16 For in this device, the electrons and holes are effectively formed by intramolecular light-induced charge transfer process, and will be transported to the cathode and anode along electron and hole transport channels, respectively. But so far, the controllable alignment of these D–A typed DLCs on a substrate has still been a considerable challenge.
The studying of bulk heterojunction (BHJ) solar cells reveals that the open-circuit voltage (Voc) is (almost linearly) proportional to the energy level difference between the highest occupied molecular orbital (HOMO) level of the electron donors and the lowest occupied molecular orbital (LUMO) level of the acceptors.17 Xiao et al. reported the BHJ solar cells based on blending RR-P3HT with perylene diimides (PDI) or PTE as acceptors, and the Voc value of the latter was 0.3 V higher than that of the former, since the LUMO level of PTE is 0.3 eV higher than that of PDI.18
Previously we have studied the liquid crystal and optical properties of alkoxy-bridged triphenylene and perylene monoimide diesters dyads (TP6-Cn-PMIBE, n = 2, 6, 10 and 12).14 In order to improve the energy level difference between the HOMO of the donor and the LUMO of the acceptor, four novel dyads DC2A, DC6A, DC10A and DC12A, composing of a TP moiety as donor and a PTE moiety as acceptor, linked through (O–(CH2)2–O), (O–(CH2)6–O), (O–(CH2)10–O) and (O–(CH2)12–O) bridges, respectively, were synthesized in this work (Fig. 1). Molecular structures in relation to electrochemical behaviours, the fluorescent emission/quenching and the energy transfer, as well as liquid crystalline phase transitions, were studied. The experimental results reveal that the energy level difference between the HOMO of the donor and the LUMO of the acceptor is about 1.94 eV, and the dyads have mesogenic phase even at room temperature.
 | ||
| Fig. 1 Chemical structures of the synthesized D–A dyads, labelled according to the length of linking groups as indicated. As well as that of reference systems hexa(n-hexyloxy)triphenylene (HAT6, donor monomer) and 3,4,9,10-tetra-(n-hexyloxy-carbonyl)-pery-lene (PTE, acceptor monomer). | ||
Experimental
Materials
All chemicals and solvents are from Aladdin Industrial Corporation, XiLong Chemical, and FuYu Chemical. All were analytical-grade reagents and used as received unless otherwise noted.Synthesis of 2-(6-bromohexyloxy)-3,6,7,10,11-penta(hexyloxy)-triphenylene (3)
2-Hydroxy-3,6,7,10,11-penta(hexyloxy)-triphenylene (compound 1 (1.00 g, 1.34 mmol)), 1,6-dibromohexane (1.96 g, 8.03 mmol), tetrabutylammonium bromide (0.13 g, 0.40 mmol) and potassium hydroxide (0.30 g, 5.36 mmol) were added to a dichloromethane (8 mL) and water (5 mL) mixed solution. The mixture was stirred at room temperature for 24 hours under N2 atmosphere. Then the solution was extracted with dichloromethane (10 mL × 3) before the organic phase was washed with brine and dried over anhydrous sodium sulfate. After evaporation of the solvent in vacuo, the crude product was purified by chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/50) to yield 3 as a colourless solid (1.13 g, 93%). 1H NMR (400 MHz, CDCl3) δ: 7.83 (s, 6H), 4.23 (t, J = 6.8 Hz, 12H), 3.38 (t, J = 6.8 Hz, 2H), 1.98–1.92 (m, 14H), 1.60–1.51 (m, 14H), 1.41–1.39 (m, 20H), 0.93 (t, J = 6.8 Hz, 15H). FT-IR (KBr) νmax (cm−1): 3450, 2930 (C–H), 2840 (C–H), 1620, 1520, 1430, 1390, 1260 (C–O), 1170, 1070, 1050.Synthesis of 2-(10-bromodecyloxy)-3,6,7,10,11-penta(hexyloxy)-triphenylene (4)
Under the reaction condition of compound 3, compound 4 was obtained as a colourless solid (89%). 1H NMR (400 MHz, CDCl3) δ: 7.84 (s, 6H), 4.23 (t, J = 6.8 Hz, 12H), 3.40 (t, J = 6.8 Hz, 2H), 1.98–1.91 (m, 12H), 1.85 (t, J = 6.8 Hz, 2H), 1.62–1.54 (m, 12H), 1.42–1.35 (m, 30H), 0.94 (t, J = 6.8 Hz, 15H). FT-IR (KBr) νmax (cm−1): 3440, 2930 (C–H), 2850 (C–H), 1620, 1520, 1440, 1390, 1260 (C–O), 1170, 1050, 837.The general procedure for DCnA (n = 2, 6, 10 and 12)
ω-Bromo-alkoxy-triphenylenes (1.22 mmol) and KI (2.21 g, 13.31 mmol) were dissolved in acetone (30 mL). The mixture was refluxing for 24 hours under N2 atmosphere to give ω-iodo-alkoxy-triphenylenes. Then the mixture was cooled to room temperature, compound 10 (0.92 g, 1.59 mmol), 1-hexanol (16 mL) and anhydrous potassium carbonate (2.74 g, 20.15 mmol) were added, the mixture was stirred at 50 °C for 72 hours under N2 atmosphere. Then water (300 mL) was added, and the mixture was extracted by ethyl acetate (20 mL × 3). Then the organic phase was dried over anhydrous sodium sulfate, and the solvents removed under reduced pressure. The crude product was purified by chromatography (silica gel, eluted with ethyl acetate/petroleum ether = 1/10) to give an orange solid.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1610, 1510, 1430, 1390, 1270 (C–O), 1170, 1040, 749. Elemental analysis calcd for C92H122O14 (1452): C, 76.10, H, 8.47, found: C, 76.05, H, 8.54. MS (ESI): m/z (% relative intensity) = 1473.9 (100) (M + Na)+.O), 1620, 1520, 1400, 1390, 1270 (C–O), 1160, 1100, 1030, 839, 748. Elemental analysis calcd for C96H130O14 (1508): C, 76.46, H, 8.69, found: C, 76.31, H, 8.75. MS (ESI): m/z (% relative intensity) = 1530.9 (100) (M + Na)+.O), 1620, 1520, 1440, 1390, 1270 (C–O), 1160, 1040, 839, 747. Elemental analysis calcd for C100H138O14 (1564): C, 76.79, H, 8.89, found: C, 76.77, H, 8.92. MS (ESI): m/z (% relative intensity) = 1587.0 (100) (M + Na)+.O), 1620, 1520, 1430, 1390, 1270 (C–O), 1170, 1040, 845, 748. Elemental analysis calcd for C102H142O14 (1592): C, 76.94, H, 8.99, found: C, 76.88, H, 9.01. MS (ESI): m/z (% relative intensity) = 1615.0 (100) (M + Na)+.
O), 1610, 1510, 1430, 1390, 1270 (C–O), 1170, 1040, 749. Elemental analysis calcd for C92H122O14 (1452): C, 76.10, H, 8.47, found: C, 76.05, H, 8.54. MS (ESI): m/z (% relative intensity) = 1473.9 (100) (M + Na)+.O), 1620, 1520, 1400, 1390, 1270 (C–O), 1160, 1100, 1030, 839, 748. Elemental analysis calcd for C96H130O14 (1508): C, 76.46, H, 8.69, found: C, 76.31, H, 8.75. MS (ESI): m/z (% relative intensity) = 1530.9 (100) (M + Na)+.O), 1620, 1520, 1440, 1390, 1270 (C–O), 1160, 1040, 839, 747. Elemental analysis calcd for C100H138O14 (1564): C, 76.79, H, 8.89, found: C, 76.77, H, 8.92. MS (ESI): m/z (% relative intensity) = 1587.0 (100) (M + Na)+.O), 1620, 1520, 1430, 1390, 1270 (C–O), 1170, 1040, 845, 748. Elemental analysis calcd for C102H142O14 (1592): C, 76.94, H, 8.99, found: C, 76.88, H, 9.01. MS (ESI): m/z (% relative intensity) = 1615.0 (100) (M + Na)+.Characterisation and instrumentation
FT-IR was performed on a VECTOR 22 FT-IR spectrometer (KBr tablet). UV/Vis was recorded on a Shimadzu UV-2450 spectrometer. Fluorescent emissions were performed on a HITACHI F-4600 instrument. 1H NMR spectra were recorded on a Bruker spectrometer (300 MHZ, 400 MHz or 500 MHz). EA was recorded on a Thermo Flash EA-1112 instrument. DSC experiments were carried out on a Thermal Analysis DSC-Q100 instrument. The mesomorphic properties were evaluated by a polarized optical microscopy (Olympus THMS600) instrument provided with a heating stage (Linkam THMSE 600). Electronic energy levels were performed on a Shanghai Chen Hua CHI760E electrochemical workstation. Mass spectra were obtained using an Agilent 6495 in the ESI mode.Results and discussion
Synthesis
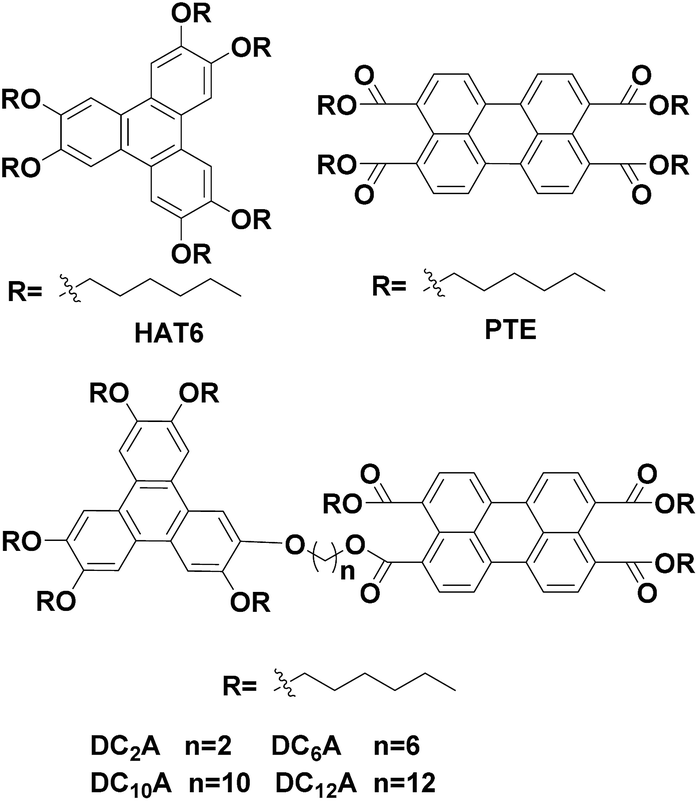
The D–A dyads in this work were synthesized from a triphenylene derivative as a donor and a perylene derivative as an acceptor. The preparation of monohydroxypentahexyloxy-triphenylene (compound 1) was one of the key steps towards asymmetric triphenylene derivatives synthesis, which followed the procedures reported previously.19 The perylene monoanhydride diester (compound 10) were prepared according to the report in literature.14 The dyads were denoted as DC2A, DC6A, DC10A and DC12A according to the length of the linkage chain (Scheme 1).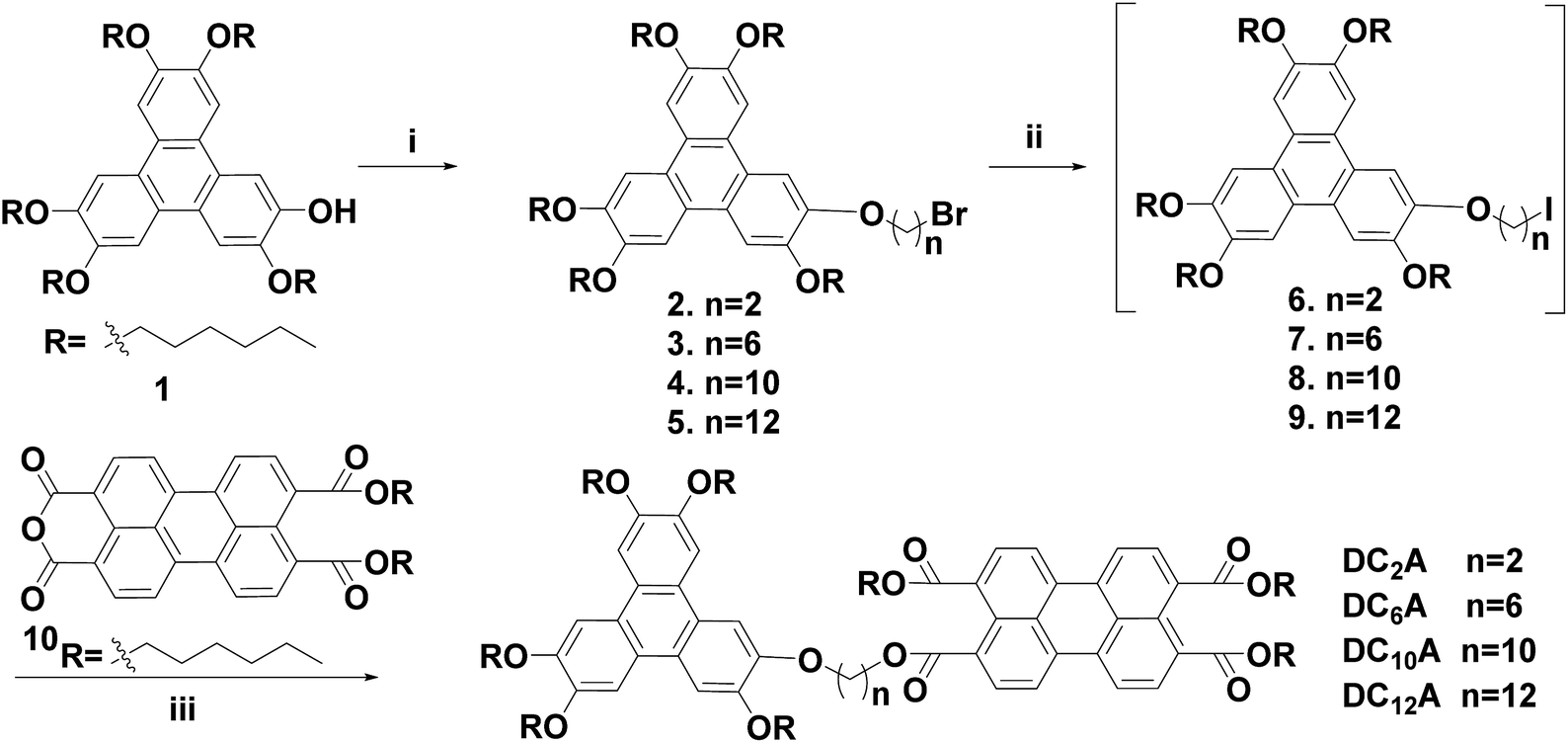 | ||
| Scheme 1 Synthesis of D–A dyads based on triphenylene and perylene cores. Reagents and conditions: (i) KOH, H2O, 1, ω-dibromoalkanes, tetrabutylammonium bromide, dichloromethane, rt, 24 h, 68–93%; (ii) KI, acetone, refluxing, 24 h; (iii) K2CO3, 1-hexanol, 50 °C, 72 h, 28–40% (two steps). | ||
ω-Bromo-alkoxy-triphenylenes (compounds 2–5) can be synthesized via alkylation of 1 with dibromoalkanes (Br(CH2)nBr, n = 2, 6, 10 and 12) under phase transfer conditions in high yields. Among these, compounds 2 and 5 have been reported in previous work.10,14 The intermediates ω-iodo-alkoxy-triphenylenes (compounds 6–9) were obtained by reacting compounds 2–5 with excessive potassium iodide in acetone under refluxing according to Finkelstein reaction.20 The raw ω-iodo-alkoxy-triphenylene products without further purification were then reacted with perylene monoanhydride diesters in the presence of 1-hexanol and anhydrous potassium carbonate at 50 °C to give the target dyads. Since the perylene monoanhydride diesters were not soluble well in these reactions, as well as the reaction solutions were highly viscous, it took about 72 hours to obtain the dyads in the yields of 28–40% for the last two steps.
Phase behaviours and potential in self-organization
Phase behaviours of the dyads were investigated by POM (optical textures see Fig. 2) and DSC (with a scan rate of 10 °C per minute). Peak transition temperatures along with their associated enthalpies (ΔH) were listed in Table 1. | ||
| Fig. 2 Optical textures of DC10A at 68 °C (a) and DC12A at 48 °C (b) (on cooling from the isotropic liquid, crossed polarizers, scale bar 100 μm). | ||
| Compounds | First heating scan T/°C (ΔH/kJ g−1) | First cooling scan T/°C (ΔH/kJ g−1) |
|---|---|---|
| a Dh, hexagonal discotic; Dx, unidentified discotic; I, isotropic phase. | ||
| DC2A | Dx 56 (10.58) I | I 6 (0.60) Dx |
| DC6A | Dx 55 (0.3) I | I −13 (0.58) Dx |
| DC10A | Dx 21 (0.44) Dx 48 (15.33) Dh 97 (1.48) I | I 89 (1.99) Dh |
| DC12A | Dx 43 (1.49) Dh 80 (2.19) I | I 53 (0.37) Dh 11 (2.00) Dx |
As shown in Fig. 3, DC2A showed one endothermic transition and peaked at 56 °C in the first heating cycle, while a broad and weak exothermic transition peaked about 6 °C during cooling. Under microscopic observation, it was found to be a birefringent and soft solid at −20 °C, clearly deformable under physical stress. Upon heating, it gradually turned to fluid, and the birefringent disappeared at 67 °C in an isotropic state. This isotropic liquid remained without birefringent even cooled to −20 °C, but the liquid became very viscous. The birefringence slowly occurred after two days at room temperature, but maintained the liquid state.
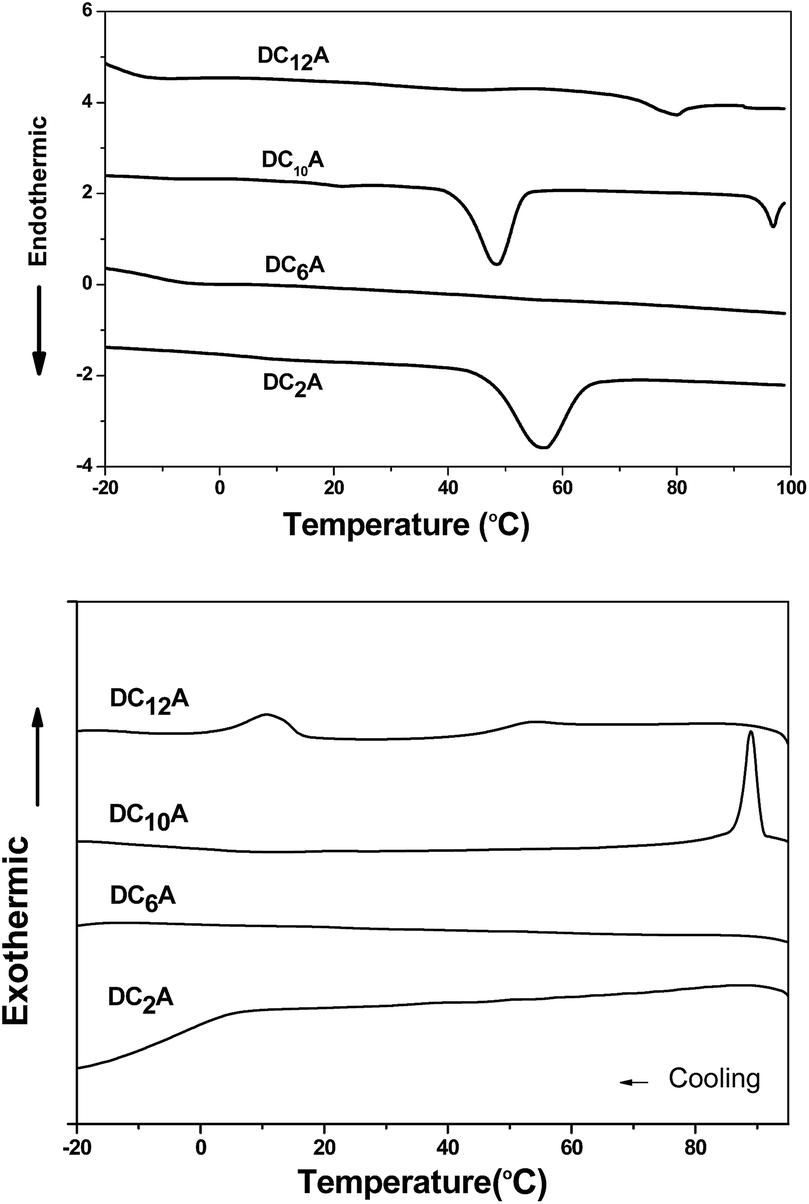 | ||
| Fig. 3 The DSC traces (10 °C min−1, first heating–cooling cycle) for DCnA, (n = 2, 6, 10 and 12). | ||
Under microscopic observation, DC6A was a birefringent and deformable solid at −20 °C. If heating up, it turned to fluid gradually. The birefringence disappeared around 59 °C. However, cooling down the isotropic liquid, the viscosity increased but no birefringence can be observed even at −20 °C. From DSC trace, it can be seen that in the heating circle an imperceptible peak at 55 °C. When cooled it has a broad phase transfer at −13 °C.
During heating run, DC10A showed three endothermic transitions which peaked at 21 °C, 48 °C and 97 °C, respectively. During cooling, a sharp exothermic transition occurred at 89 °C with an 8 °C supercooling. Under microscopic observation, it was birefringent and deformable at −20 °C. When heating up it started to flow at −16 °C with birefringence, which maintained to 98 °C and then disappeared. Upon cooling the birefringence returned around 94 °C. The birefringence fluid remained to −20 °C.
Two endothermic transitions at 43 °C and 80 °C were found for DC12A in the first DSC heating scan. During the cooling scan, two exothermic peaks at 53 °C and 11 °C. Under microscopic observation, on heating crystal DC12A melted at −5 °C, the birefringent fluid maintain till 85 °C where the birefringent disappeared. The birefringent fluid resorted at 74 °C during cooling and solidified at −18 °C.
The liquid crystal optical textures of dyads DC10A and DC12A, bearing longer spacers, are shown in Fig. 2. These textures are similar to the straight linear defects and focal conic textures for hexagonal columnar phase, respectively.12,21 For dyads DC2A and DC6A, when slowly cooled from the isotropic phase to room temperature, the birefringent phenomena was not resorted, the optical textures were not clear yet.
Electrochemical behaviour and HOMO/LUMO energy levels
The electrochemical behaviours of HAT6, PTE, and DC6A were investigated by cyclic voltammetry (CV) in dry acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. The experiments were performed at room temperature with a scan rate of 50 mV s−1, using a standard three-electrode cell with a platinum wire as the counter electrode, a AgCl/Ag electrode as the reference electrode, and a glass carbon electrode as the working electrode. The samples were solution-cast directly onto the working electrode. The cyclic voltammograms of these compounds were shown in Fig. 4, and all potentials were recorded versus AgCl/Ag reference. All these compounds showed one or two reversible redox waves. HAT6 exhibited one quasi-reversible redox waves with the half-wave potentials (Eox1/2) being 0.98 V, PTE exhibited one quasi-reversible redox waves (Eox1/2) being 1.49 V, and DC6A with the first and second half-wave potentials (Eox(1)1/2 and Eox(2)1/2, respectively) being 0.96 V and 1.44 V. Analysis of these data showed that the first redox wave of DC6A can be assigned to the oxidation reaction of the triphenylene unit, and the second redox wave can be correlated to the oxidation reaction of the perylene unit.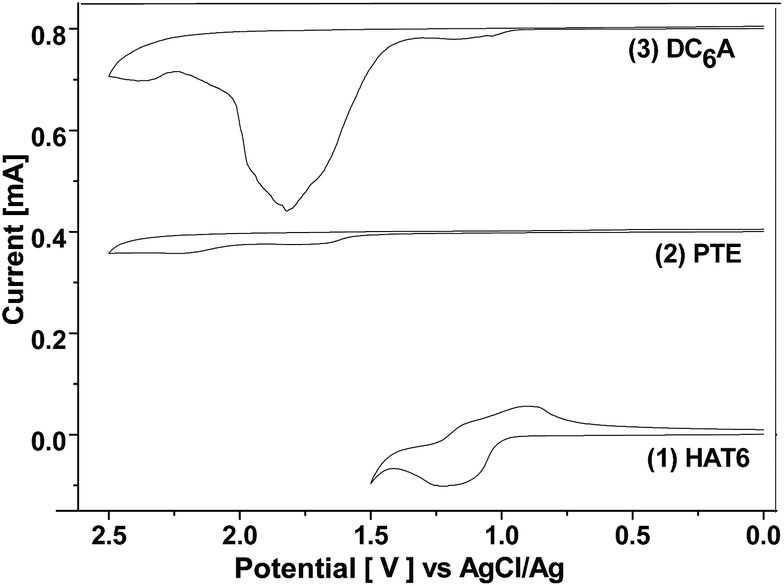 | ||
| Fig. 4 Cyclic voltammogram of HAT6, PTE and DC6A at room temperature (casted film in 0.1 M TBAPF6, rate 50 mV s−1). The curves of PTE and DC6A moved up 0.0004 and 0.0008 unit than that of HAT6, respectively. | ||
For all CV measurements, the ferrocene/ferrocenium (Fc/Fc+) redox couple was used for calibration.22 The energy levels of the HOMO of these electroactive compounds can be derived from the onset of the first oxidation wave, using the following equation: HOMO = −(Eox + 4.8 eV), where Eox is the onset potential of the first oxidation wave versus EFc/Fc+, and the data are summarized in Table 2. The HOMO energy levels of HAT6, PTE and DC6A were then estimated to −5.23, −5.74, and −5.21 eV, respectively. This indicates that the HOMO values of the dyads are determined mainly by that of the triphenylene moiety, which having higher HOMO energy than that of PTE. And the LUMO energy levels can be estimated from the optical energy gaps (Eg, see S21†) and the HOMO energy levels by LUMO = HOMO + Eg. It is obvious that for the dyad DC6A the perylene moiety is responsible for the red edge of the longest wavelength absorption in dichloromethane solution (see Fig. 5). The HOMO value used in this equation should be corresponding to that of perylene unit. So the estimated LUMO energy level of DC6A is −3.29 eV, indicating for the dyad the LUMO energy is governed by that of the perylene moiety, which has lower LUMO energy than that of HAT6. As a consequence, the HOMO energy of a D–A dyad connected by an aliphatic spacer is almost equal to that of the donor; and the LUMO energy of it is nearly the same as that of the acceptor. And the aliphatic linkage of the D–A dyads preserves the genuine electrochemical behaviours of the donor and acceptor units. It is consistent with the conclusion of Xiao Y., and et al.23
| Eox(1)1/2 | Eox(2)1/2/V | HOMO/eV | Eg/eV | LUMO/eV | |
|---|---|---|---|---|---|
| HAT6 | 0.98 | — | −5.23 | 3.35 | −1.88 |
| PTE | 1.49 | — | −5.74 | 2.40 | −3.34 |
| DC6A | 0.96 | 1.44 | −5.21 | 2.40 | −3.29 |
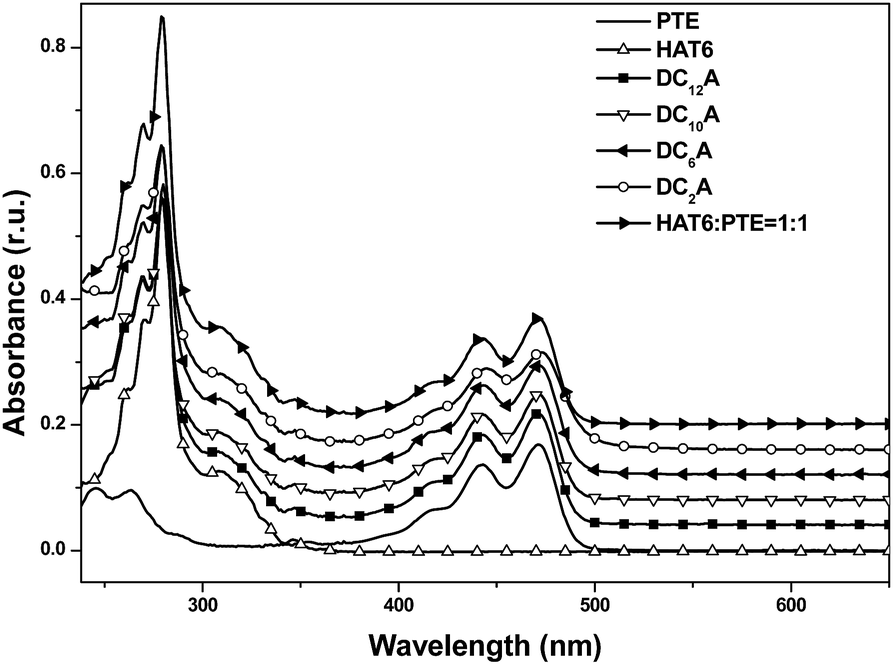 | ||
Fig. 5 UV-Vis absorption spectra of HAT6, PTE, DC2A, DC6A, DC10A and DC12A in dichloromethane, concn. = 5 × 10−6 mol L−1. In order to facilitate the observation, the spectral lines of DC12A, DC10A, DC6A, DC2A and HAT6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PTE = 1:1 successively moved up 0.04 units than HAT6. :PTE = 1:1 successively moved up 0.04 units than HAT6. | ||
Steady-state spectroscopy
:1 molar ratio was also recorded for comparison. The absorption of the mixture features essentially the sum spectrum of the isolated components. The absorption spectra of the dyads DC2A, DC6A, DC10A and DC12A are all similar to that of the former mixture. The absence of characteristic charge-transfer bands demonstrates that the charge transfer complex does not form between the molecules of a dyad. In summary, the lengths of the flexible bridges do not significantly affect the absorption spectra properties of the dyads in dilute solution, and which features the sum spectrum of their donor and acceptor units.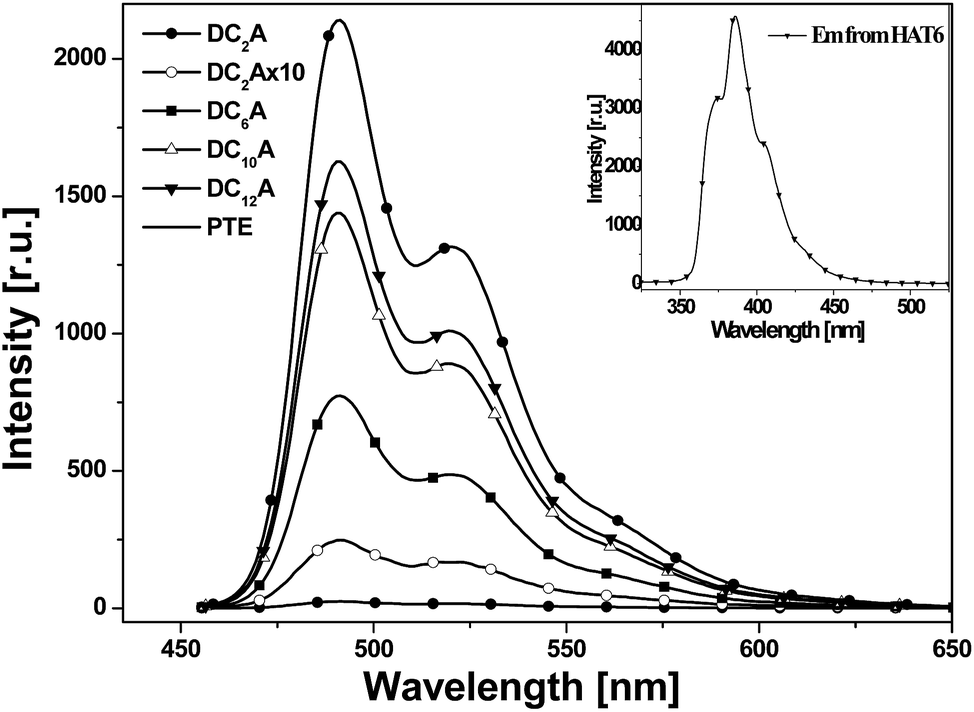 | ||
| Fig. 6 Photoluminescent emission spectra of PTE, DC2A, DC6A, DC10A and DC12A, λex = 443 nm. Inset: photoluminescent emission spectra of HAT6 excited at 280 nm (all in dichloromethane solutions at concn. = 5 × 10−6 mol L−1). | ||
It can be seen that emission spectra from the four dyads and PTE are extremely similar in spectral profile in range of 460 to 650 nm. They are multi-peak emissions around 491, 521 and 566 nm. It also can be seen from the inset that HAT6 emits in the range of 350–470 nm, which is very different from that of PTE. This implies that when excited at 443 nm the photoluminescence emissions of the dyads are mainly from the perylene units. However, the emission intensities of the dyads weakened as compared with that of PTE and further decreasing was found as the flexible linking bridges got shorter. The emission of DC2A was extremely weak, but when the spectrum was amplified 10 times the curve has the same structure as that of the other dyads. When the length of flexible bridges increasing, the emission from the dyads gradually strengthened. Although the DC12A has the longest bridge among the four dyads, its emission intensity is still lower than that of PTE. It is obvious that the general reduction of photoluminescence indicates the presence of photoluminescent quenching processes to the excitation of D–A dyads. The shorter the bridge, the stronger the fluorescence quenching effect of the dyads.
In order to explain the experimental observation described in Fig. 6, we treat the donor and the acceptor units of the dyads as entities that can be studied separately. This approximation is reasonable because the absorption spectra of the dyads correspond to the sum spectra of the individual molecular units. The normalized fluorescence excitation spectra of the dyads are almost completely coincident in the range of 360 to 510 nm (see Fig. 7). It is therefore reasonable to describe the molecular orbital of the donor and the acceptor units in a single-particle approximation.
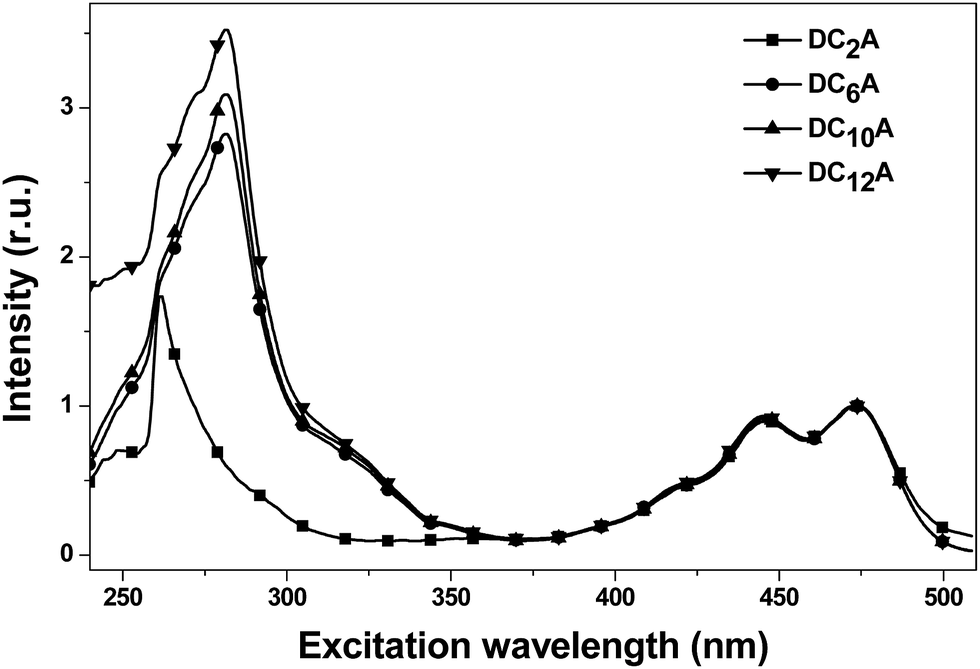 | ||
| Fig. 7 The normalized fluorescence excitation spectra of DC2A, DC6A, DC10A and DC12A in dichloromethane, concn. = 5 × 10−6 mol L−1. The emission was monitored around 520 nm. | ||
We deduced that the intramolecular charge transfer processes were responsible for the fluorescence quenching of the dyads, and shown schematically in Fig. 8. The energetic positions of the HOMOs and LUMOs corresponded to results from cyclo-voltammetric experiments of the HOMO and LUMO levels of DC6A. The progress starts from the left hand, and the dotted arrow indicates the direction of an electron being about to move.
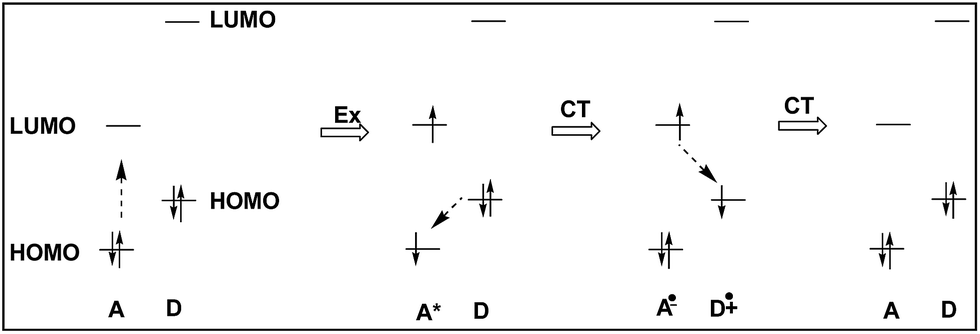 | ||
| Fig. 8 Schematic representation of the ground-state charge-transfer (CT) of dyads DCnA (n = 2, 6, 10 and 12). The energetic positions of the HOMOs and LUMOs correspond to results from cyclovoltammetric experiments of the HOMO and LUMO levels of DC6A, respectively. | ||
When the dyad is excited at 443 nm, due to the extinction coefficient of the donor is negligible at this wavelength and only A unit is excited, electron transits from its HOMO to its LUMO. The excitation state generated is indicated by symbol A*, and this excitation process is shown by the Ex sign. The pair of electrons in the HOMO of donor unit has higher energy than the HOMO energy level of A*, so donor unit transfers one electron to it. Thus donor unit loses one electron and becomes a radical cation (D+˙), and A* gets one electron and becomes radical anion (A−˙). This is a ground-state charge-transfer process, denoted as CT. Finally, the electron in the LUMO of A−˙ has higher energy than the HOMO of D+˙, and transfers to it, resulting in the dyad returning to the electronic ground state. This is another process of charge transfer, represented by CT. These charge transfer processes can be explained by Dexter-type energy transfer theory. It shows that the energy transfer rate constant decays exponentially with the distance.26 Thus the longer the distance between D and A units, the weaker the excitation energy decay, and the stronger the fluorescence emission. For DC2A, the bridge length is the shortest in the dyads, so its fluorescence emission is almost absolutely quenched. In the case of compound DC12A, the spacer is the longest, and its fluorescence intensity is the strongest of all.
In summary, when excited at 443 nm, the intramolecular photoinduced charge-transfer processes occur to the four dyads, and which are responsible for the fluorescent quenching of the dyads in dilute solution.
Conclusions
Four novel dyads DCnA (n = 2, 6, 10 and 12) were synthesized and fully characterized. The experimental results of CV and UV/Vis showed the aliphatic linkage of the D–A dyads preserved the genuine electrochemical behaviours of the D and A units. When the four dyads in dilute dichloromethane solution are excited at 443 nm, the fluorescence quenching of the A unit varies with the bridge length. This is ascribed to a ground-state charge transfer from the D unit to the A unit. For compound of DC2A, the bridge length is the shortest and energy transfer rate is the fastest, so its fluorescence is almost absolutely quenched. In the case of compound DC12A, the bridge length is the longest and energy transfer rate is the slowest, and its fluorescence intensity is the strongest. Considering the mesogenic behaviours of DC10A and DC12A, these dyads are potential active materials for fabricating OPV with ideal double channels for charge carriers transporting.Acknowledgements
We gratefully acknowledge financial support from National Natural Science Foundation of China (11364013, 11474017) and the Education Department of Guangxi Province (No. KY2015YB129). We would like to thank Collaborative Innovation Center for Exploration of Hidden Nonferrous Metal Deposits and Development of New Materials in Guangxi and Guang Xi Key Laboratory of Hidden Metallic Ore Deposits Exploration for the POM service.References
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Pramana, 1977, 9, 471–480 CrossRef CAS.
- R. J. Bushby and O. R. Lozman, Curr. Opin. Colloid Interface Sci., 2002, 7, 343–354 CrossRef CAS.
- A. Benito-Hernandez, U. K. Pandey, E. Cavero, R. Termine, E. M. Garcia-Frutos, J. L. Serrano, A. Golemme and B. Gomez-Lor, Chem. Mater., 2013, 25, 117–121 CrossRef CAS.
- D. Adam, F. Closs, T. Frey, D. Funhoff, D. Haarer, H. Ringsdorf, P. Schuhmacher and K. Siemensmeyer, Phys. Rev. Lett., 1993, 70, 457–460 CrossRef CAS PubMed; D. Adam, P. Schuhmacher, J. Simmerer, L. Haussling, K. Siemensmeyer, K. H. Etzbach, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef; A. M. van de Craats, J. M. Warman, A. Fechtenkotter, J. D. Brand, M. A. Harbison and K. Mullen, Adv. Mater., 1999, 11, 1469–1472 CrossRef; H. Zhao, Z. He, M. Xu, C. Liang and S. Kuma, Phys. Chem. Chem. Phys., 2016, 18, 8554–8560 RSC; K. Zhao, M. Jing, L. An, J. Du, Y. Wang, P. Hu, B. Wang, H. Monobe, B. Heinrichc and B. Donnio, J. Mater. Chem. C, 2017, 5, 669–682 RSC.
- D. Markovitsi, Mol. Cryst. Liq. Cryst., 2003, 397, 25–45 CrossRef CAS; K. Ohta, K. Hatsusaka, M. Sugibayashi, M. Ariyoshi, K. Ban, F. Maeda, R. Naito, K. Nishizawa, A. M. Van de Craats and J. M. Warman, Mol. Cryst. Liq. Cryst., 2003, 397, 89–98 CrossRef; C. F. van Nostrum, Adv. Mater., 1996, 8, 1027–1030 CrossRef.
- E. O. Arikainen, N. Boden, R. J. Bushby, J. Clements, B. Movaghar and A. Wood, J. Mater. Chem., 1995, 5, 2161–2165 RSC; N. Boden, R. J. Bushby and J. Clements, J. Chem. Phys., 1993, 98, 5920–5931 CrossRef CAS.
- R. Freudenmann, B. Behnisch and M. Hanack, J. Mater. Chem., 2001, 11, 1618–1624 RSC; L. Schmidt-Mende, A. Fechtenkotter, K. Mullen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed; W. Pisula, A. Menon, M. Stepputat, I. Lieberwirth, A. Kolbe, A. Tracz, H. Sirringhaus, T. Pakula and K. Mullen, Adv. Mater., 2005, 17, 684–689 CrossRef; M. Bajpai, N. Yadav, S. Kumar, R. Srivastava and R. Dhar, Liq. Cryst., 2016, 43, 928–936 CrossRef.
- I. Seguy, P. Destruel and H. Bock, Synth. Met., 2000, 111–112, 15–18 CrossRef CAS; C. Ruiz, U. K. Pandey, R. Termine, E. M. Garcia-Frutos, G. Lopez-Espejo, R. P. Ortiz, W. Huang, T. J. Marks, A. Facchetti, M. C. R. Delgado, A. Golemme and B. Gomez-Lor, ACS Appl. Mater. Interfaces, 2016, 8, 26964–26971 Search PubMed.
- Z. An, J. Yu, S. C. Jones, S. Barlow, S. Yoo, B. Domercq, P. Prins, L. D. A. Siebbeles, B. Kippelen and S. R. Marder, Adv. Mater., 2005, 17, 2580–2583 CrossRef CAS; F. Wurthner, C. Thalacker, S. Diele and C. Tschierske, Chem.–Eur. J., 2001, 7, 2245–2253 CrossRef; J. van Herrikhuyzen, A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 10021–10027 CrossRef PubMed; M. Oukachmiha, P. Destruela, I. Seguya, G. Ablarta, P. Jolinata, S. Archambeaua, M. Mabialaa, S. Fouetb and H. Bock, Sol. Energy Mater. Sol. Cells, 2005, 85, 535–543 Search PubMed.
- M. Bagui, T. Dutta, S. Chakraborty, J. S. Melinger, H. Zhong, A. Keightley and Z. Peng, J. Phys. Chem. A, 2011, 115, 1579–1592 CrossRef CAS PubMed; S. K. Varshney, H. Monobe, Y. Shimizu, H. Takezoe and V. Prasad, Liq. Cryst., 2010, 37, 607–615 CrossRef; X. Kong, Z. He, Y. Zhang, L. Mu, C. Liang, B. Chen, X. Jing and A. N. Cammidge, Org. Lett., 2011, 13, 764–767 CrossRef PubMed.
- K. Zhao, L. An, X. Zhang, W. Yu, P. Hu, B. Wang, J. Xu, Q. Zeng, H. Monobe, Y. Shimizu, B. Heinrich and B. Donnio, Chem.–Eur. J., 2015, 21, 10379–10390 CrossRef CAS PubMed; M. Bagui, T. Dutta, H. Zhong, S. Li, S. Chakraborty, A. Keightley and Z. Peng, Tetrahedron, 2012, 68, 2806–2818 CrossRef.
- S. K. Gupta, S. Setia, S. Sidiq, M. Gupta, S. Kumar and S. K. Pal, RSC Adv., 2013, 3, 12060–12065 RSC.
- K. J. Lee, J. H. Woo, E. Kim, Y. Xiao, X. Su, L. M. Mazur, A. Attias, F. Fages, O. Cregut, A. Barsella, F. Mathevet, L. Mager, J. W. Wu, A. DEleo and J. Ribierre, Phys. Chem. Chem. Phys., 2016, 18, 7875–7887 RSC.
- X. Kong, P. Liu, G. Wang, L. Xia, S. Dai, J. Su, P. Liao, Z. Liu and L. Mu, Chin. J. Org. Chem., 2016, 36, 1325–1334 CrossRef CAS.
- W. Xiao, Z. He, S. Remiro-Buenamanana, R. J. Turner, M. Xu, X. Yang, X. Jing and A. N. Cammidge, Org. Lett., 2015, 17, 3286–3289 CrossRef CAS PubMed.
- L. Schmidt-Mende, M. Watson, K. Mullen and R. H. Friend, Mol. Cryst. Liq. Cryst., 2003, 396, 73–90 CrossRef CAS.
- M. C. Scharber, D. Muhlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS; H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 Search PubMed.
- Y. Jiang, L. Lu, M. Yang, C. Zhan, Z. Xie, F. Verpoortacd and S. Xiao, Polym. Chem., 2013, 4, 5612–5620 RSC.
- X. Kong, Z. He, H. Gopee, X. Jing and A. N. Cammidge, Tetrahedron Lett., 2011, 52, 77–79 CrossRef CAS; W. Xiao, Z. He, M. Xu, N. Wu, X. Kong and X. Jing, Tetrahedron Lett., 2015, 56, 700–705 CrossRef.
- T. Zoller, D. Uguen, A. De Clan and J. Flsche, Tetrahedron Lett., 1998, 39, 8089–8092 CrossRef CAS.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hagele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS PubMed.
- M. Thelakkat and H. Schmidt, Adv. Mater., 1998, 10, 219–223 CrossRef CAS.
- Y. Xiao, X. Su, L. Sosa-Vargas, E. Lacaze, B. Heinrich, B. Donnio, D. Kreher, F. Mathevet and A. Attias, CrystEngComm, 2016, 18, 4787–4798 RSC.
- D. Markovitsi, A. Germain, P. Millie, P. Lecuyert, L. K. Gallos, P. Argyrakis, H. Bengss and H. Ringsdorf, J. Phys. Chem., 1995, 99, 1005–1017 CrossRef CAS.
- R. Hertmanowski, A. Biadasz, T. Martynski and D. Bauman, J. Mol. Struct., 2003, 646, 25–33 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, Nanodevices for the Life Sciences, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2nd edn, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01320e |
| This journal is © The Royal Society of Chemistry 2017 |