
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothermally prepared nanosized and mesoporous Ce0.4Zr0.6O2 solid solutions with shape dependence in photocatalysis for the degradation of methylene blue†
Wei Tiana,
Jun Yinb,
Lingfei Weia,
Quanhao Shena,
Rehana Bibia,
Ming Liua,
Bin Yanga,
Naixu Li*a and
Jiancheng Zhou *acd
*acd
aSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, Jiangsu, PR China. E-mail: naixuli@seu.edu.cn; jczhou@seu.edu.cn
bDepartment of Biological and Chemical Engineering, Jingdezhen College, Jingdezhen 333000, Jiangxi, PR China
cDepartment of Chemical and Pharmaceutical Engineering, Southeast University Chengxian College, Nanjing 210088, Jiangsu, PR China
dJiangsu Province Hi-Tech Key Laboratory for Bio-medical Research, Southeast University, Nanjing 211189, PR China
First published on 17th March 2017
Abstract
Mesoporous Ce0.4Zr0.6O2 solid solutions with four different shapes, including mixed nanorod and nanoparticle (RP, cubic), nanopolyhedron (PH, tetragonal), nanosphere (SP, cubic) and nanoegg (EG, tetragonal), were hydrothermally fabricated. Various physical and chemical techniques were used to reveal shape-dependent structural, optical, electronic and surface properties, providing a powerful insight into the morphological dependence of photocatalytic activity towards methylene blue (MB) degradation. Among these samples, RP, consisting of a cubic Ce-rich phase (Ce0.6Zr0.4O2 nanorods) and a tetragonal Zr-rich phase (Ce0.2Zr0.8O2 nanoparticles), affords the optimal functionality, with a top degradation rate (D = 70.5%) and a peak rate constant (k = 0.0102 min−1). This occurrence results mainly from the prompt separation of excitons and suitable textural properties for MB absorption and activation. A brief introduction to the reaction path when using RP is proposed.
Introduction
Semiconductor-based photocatalytic technology is highly appreciated as a promising and powerful response to human concerns over the energy crisis and environmental contamination, by virtue of the use of inexhaustible and renewable solar radiation as a substitute for non-renewable resources, such as coal and petroleum.1–4 This appealing feature considerably prompts its versatile application in H2 evolution,5–9 CO2 utilization10–12 and environmental cleaning.13–17 It is unquestionable that the exploration of photocatalytic materials of high activity and stability is an indispensable part of its development. Over the past few years, the focusing of great attention on the advanced structural, optical and electronic properties of semiconductors has remarkably inspired the design and fabrication of exciting light-responsive materials based on TiO2 (ref. 18–20) and titanates,14,21,22 metal sulfides,23–27 g-C3N4 (ref. 28–31) and WO3 (ref. 13 and 32) from abundantly available feedstocks, leading to an incredible expansion in their application to areas involving photocatalysis and photothermalcatalysis.Apart from the aforementioned catalytic materials, there are other first-rank analogues of substantial interest. In particular, CeO2, a well-documented typical pale yellow/white rare earth oxide with non-stoichiometric properties and a face-centered cubic (f.c.c.) fluorite structure, is endowed with special characteristics including but not limited to (a) outstanding (photo)chemical and thermal stability, low toxicity and cost,33 (b) redox, acid–base bifunctional and semiconducting features that are hardly accessible in other materials34,35 and (c) facile fabrication using various existing protocols from earth-abundant starting materials.36–38 However, the material hardly escapes its intrinsic defects, such as weak surface acid strength and the low separation efficiency of charge barriers, which are detrimental for practical applications. Great efforts connected with countering these issues, mainly including structural and chemical modifications, have accelerated a collection of amazing catalytic breakthroughs in a broad array of diverse applications ranging from the oxidation of pollutants to H2 evolution and fuel cells to photo(thermal)catalysis.34,39
The structurally functionalized design of CeO2 is mainly classified into two groups: the construction of a specific morphology (e.g. nanorod, nanocube, nanopolyhedron or nanotube)40 and the introduction of porous behavior (e.g. mesoporous or macroporous) through template-assisted methods41,42. The morphology dependence of (photo)catalytic performance is more widely observed and well-rationalized using tunable active atomic modes on preferentially exposed reactive planes through the precise control of thermodynamics and kinetics in material preparation.43–45 For example, Wang et al.46 developed CeO2 nanorods, nanocubes, octahedrons and spindles through careful command of the hydrothermal process, including hydrothermal temperature and time, and the category and concentration of mineralizers for dimethyl carbonate (DMC) synthesis from CO2 and methanol. The author confirmed shape-dependent exposed facets with designated acid and basic strength through TEM, SEM and TPD results. On the other hand, spindle-like CeO2 enclosed by a (111) crystal plane turn out to be most catalytically active among the four shapes possibly associated with the exposed (111) plane, defect sites, and acidic-basic sites. Notably, from a kinetic viewpoint, weak acid sites played a major role in the elevated catalysis due to the appropriate strength available for reactant adsorption and resultant desorption, as compared to moderate and strong sites. In other words, in this system, CeO2 acted as acid–base bifunctional promoter, which offered shape-controlled catalytic performance. Lu et al.47 correlated shapes with photocatalysis (redox behavior) by using hexagonal CeO2 nanorods as a catalyst for photocatalytic hydrogen evolution. Rod-shaped CeO2 provided an elevated hydrogen evolution rate (HER) relative to CeO2 nanoparticles. The occurrence was closely associated with Ce3+ and oxygen vacancies. CeO2 rods enclosed by exposed less stable {110} planes accommodated more Ce3+ and oxygen vacancies than CeO2 nanoparticles with the most stable {111} facets predominantly exposed. These two factors respectively encouraged a boost in light absorption and the separation efficiency of excitons,34,48 resulting in a remarkable increase in the HER. To conclude, morphology has an evident effect on photocatalysis.
Chemically modified CeO2, through doping with foreign elements or the constructing a CeO2-based hybrid system, has been a research hotspot both in academia and the industrial community. Chemical doping proves an effective strategy to tailor the physical and chemical properties of catalytic materials to improve their performances.49–51 The first transition series metals, Zr and rare earth elements are preferentially selected for CeO2 modification. Liyanage et al.48 made a detailed comparison of the catalytic performance towards the photothermalcatalytic degradation of indigo carmine and rhodamine B over hydrothermally prepared Y doped CeO2 rods (Ce–Y–O solid solutions, YOC). Compared with the pristine analogue, YOC displayed higher photocatalytic activity below 50% Y doping. Among doped samples, YOC with 10% loading was above others when ranked in terms of catalysis. The observation was intimated to be related to the bandgap (optical harvesting capacity) and oxygen vacancies (separation efficiency of excitons, as set forth) as a function of Y concentration. Thus, the optimal light-driven catalytic activity was obtained through setting a reasonable Y concentration (10% loading). The conclusion is also applied to CeO2 doping with other metals, such as Mn,52,53 Zr54 and Sn.55 Among these elements, Zr draws extensive attention among scholars. Zhang et al.54 prepared tetragonal CexZr1−xO2 (x = 0.1, 0.2 and 0.3) solid solutions via a citrate complexation route and evaluated their catalytic function in the photocatalytic decolourization of rhodamine B (RhB) and 2,4-dichorophenol (2,4-DCP). They proved more catalytically active than pure CeO2, partly due to the former amplified visible-light response resulting from the visible-light-induced metal-to-metal charge transfer (MMCT) transition (“Zr(IV)–O–Ce(III)” → “Zr(III)–O–Ce(IV)”) in a specific oxo-bridged bimetallic Zr(IV)–O–Ce(III) linkage between two neighboring flattened tetrahedrons of the structural framework. This occurrence distinguishes them from their analogues, including Y doped CeO2 nanorods48 and ZnxCd1−xS,23,25 ignoring a conventional concept that the optical absorption of a solid solution is supposed to fall in between those of its parent compounds.
In addition to chemical doping, coupling plain or doped CeO2 with some useful materials to construct a series of CeO2-based functionalized assemblies has enjoyed tremendous publicity in the past few decades. Among these, plain or doped CeO2 (dominated by CeO2 and CexZr1−xO2) serves as an active support for noble metals (e.g. Pt56,57 and Au58,59) and metal oxides (e.g. NiOx (ref. 60 and 61)) or essential components of supporting materials.62,63 Although they show striking advances in thermal catalysis, they fail to receive due attention in photo(thermal)catalysis.
As can be seen from the aforementioned introduction, CeO2 of various shapes and CexZr1−xO2 solid solutions have been well explored, but investigating the morphological effects of CexZr1−xO2 solid solutions on photocatalytic activity remains an untouched area to date. Herein, we hydrothermally access nanosized Ce0.4Zr0.6O2 with four different shapes, RP, PH, SP and EG. To begin with, a comparative study on the photocatalytic removal of methylene blue (MB) over CexZr1−xO2 (0 ≤ x ≤ 1) obtained using the synthetic strategy for RP indicates Ce0.4Zr0.6O2 RP as the optimal catalyst (Table S1†). Therefore, x = 0.4 was fixed in following experiments. Next, as-prepared samples were characterized using XRD, Raman, EDS-mapping, SEM, TEM, XPS, DRS, photocurrent response, N2 sorption, and NH3- and CO2-TPD. Different acid–base behaviors from TPD analyses not only correspond to various separation efficiencies for photogenerated charges, but also state the preferentially exposed reactive facets of samples with various shapes. Furthermore, photocatalytic activity tests confirm Ce0.4Zr0.6O2 RP as the best of the four catalysts, which derives mainly from the prompt separation of excitons and suitable textural properties that favor MB adsorption and activation. Finally, based on analytical results, a possible reaction mechanism was proposed.
Results and discussion
XRD and Raman analysis
Structural characteristics of various samples are revealed unambiguously through XRD and Raman studies in Fig. S1† and Fig. 1. Fig. S1a† indicates that CeO2 and ZrO2 samples respectively crystallize in face-centered cubic fluorite and tetragonal structures, from consulting their individual PDF cards (PDF#34-0394 and PDF#50-1089). Besides, a successive shift in all patterns to higher diffraction angles with increasing Zr content is clearly observed, indicative of CexZr1−xO2 as a typical solid solution.54 The idea is also highly approved upon seeing that the evidently depressed peak intensities of each CeO2–ZrO2 sample compared with pristine oxides discloses the introduction of Ce4+ (Zr4+) into the ZrO2 (CeO2) framework, which is similar to previous results15,16. Interestingly, unlike homogeneous CexZr1−xO2 (x = 0.2, 0.6 and 0.8), Ce0.4Zr0.6O2 RP is identified as a diphasic solid due to peak splitting. In addition, by virtue of cubic CeO2 and tetragonal ZrO2, as well as biphasic Ce0.4Zr0.6O2, CexZr1−xO2 samples are reasonably assumed to be cubic (x ≥ 0.4) and tetragonal (x < 0.4) systems. Therefore, an RP-shaped sample is composed of a CeO2-rich phase (cubic Ce0.6Zr0.4O2) and a ZrO2-rich phase (tetragonal Ce0.2Zr0.8O2) from a comparative analysis of their XRD patterns.34 The results are made clearer in the locally magnified Fig. S1a† (Fig.S1b†). | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of Ce0.4Zr0.6O2 solid solutions with different shapes (“Raman shift/cm−1” and “Raman intensity/a.u.” are applied to the longitudinal and horizontal coordinates of the insets, the Raman spectra of CeO2R and ZrO2 P). | ||
Fig. 1a provides solid evidence that PH, SP and EG-shaped Ce0.4Zr0.6O2 are characterized as uniphase solid solutions by virtue of (a) the respective appearance of peaks around 29.8°, 34.8°, 49.6° and 58.3° between (111), (200), (220) and (311) in cubic CeO2 and (011), (002), (112) and (013) in tetragonal ZrO2 (marked with dashed lines) and (b) the absence of a combined XRD pattern for pure CeO2 and ZrO2, excluding the formation of CeO2–ZrO2 composites.50 However, it is very difficult to make accurate judgments regarding the crystalline phase of each solid solution using XRD alone. The issue has been well addressed with the aid of Raman analysis.54 Fig. 1b presents Raman spectra of samples with four shapes, along with those of pristine CeO2 R and ZrO2 P as reference samples, provided as insets. Obviously, in the left inset, a steep peak centered at 461 cm−1, ascribed to the Raman active F2g vibration mode of the cubic phase, suggests pure CeO2 with a cubic syngony,48,54,64 which is consistent with the XRD results. In addition, the other peak at ca. 593 cm−1 in the inset reveals a small amount of Ce3+ cations in the CeO2 network.48 The two observations equally happen in RP and SP-shaped materials, implying their cubic-fluorite structures with a small amount of Ce3+ ions, similar to pure CeO2. However, with respect to the two other samples, the bands around 140, 266, 311, 471 and 626 cm−1 stem from Raman active models (A1g + 2B1g + 3Eg) of tetragonal ZrO2 (the right inset), denoting their tetragonal nature.54 Combining XRD and Raman results, lattice spacings (d) and relevant planes are certainly determined and summarized in Table S2.†
Electron microscopy analysis
Fig. S2–S5† provide STEM images and corresponding mappings of Ce0.4Zr0.6O2 with various morphologies defining well the element components and distributions. To begin with, Fig. S2e† recognizes markedly Ce0.4Zr0.6O2 RP, which can be divided into two parts: Group R and Group P (Fig. S2d and f†). Each group is composed of three homogeneously distributed elements: Ce, Zr and O (Fig. S2a–c and g–i†). Notably, Group R embraces a Ce-rich and Zr-lean phase (Ce0.59Zr0.41O2, from the EDS results, as below), which is opposite to that of Group P (Ce0.21Zr0.79O2). To conclude, when integrated with XRD and Raman analyses, cubic Ce0.4Zr0.6O2 RP is well-defined as a solid solution assembly of cubic Ce0.6Zr0.4O2 R and tetragonal Ce0.2Zr0.8O2 P. Judging from the evenly dispersed Ce, Zr and O in Fig. S3–S5,† three other samples are equally identified as Ce0.4Zr0.6O2 solid solutions, which is in line with the respective XRD results. Nevertheless, these shaped bodies are too small to discern only via STEM micrographs. Thus, SEM and HRTEM are of exceptional importance for this purpose.Fig. 2 allows the analysis of the morphology and grain size of each Ce0.4Zr0.6O2 material. As seen from Fig. 2a, Ce0.4Zr0.6O2 embodies rod-like bodies, with lengths of up to ca. 400–500 nm and a peak diameter of up to ca.100 nm, and morphologically irregular particulate matter on a nanometer scale. In Fig. 2b, an irregular shaped particle with a tetragonal structure is intimately attached to the R-shaped matrix. In addition, RP displays four sets of lattice fringes with interplanar crystal spacing of 0.293, 0.293, 0.254 and 0.18 nm, respectively corresponding to the (111), (011), (200) and (220) planes. Finally, the lattice spacing of 0.543 nm is indexed as the lattice constant (a) of the Ce0.6Zr0.4O2 solid solution.65
 | ||
| Fig. 2 SEM and HRTEM images of Ce0.4Zr0.6O2 RP (a and b), PH (c and d), SP (e and f) and EG (g and h). | ||
With the exception of RP, another three samples are difficult to identify in shape from their SEM graphs (Fig. 2c, e and g) due to relatively smaller particles (ca.10–45 nm in size). HRTEM images completely differentiate their nanoscale morphologies. In Fig. 2d, tetragonal Ce0.4Zr0.6O2 PH, with clear-cut edges and corners (highlighted with various coloured polygons), shows a narrow edge length distribution of 4–15 nm and a (011) facet with lattice spacing of 0.312 nm. As shown in Fig. 2f, the cubic SP-like solid solution is well constructed with a diameter range of 8–16 nm, and is enclosed by (111) and (200) planes with interplanar distances of 0.312 and 0.273 nm, respectively. Fig. 2h unequivocally elucidates an interesting tetragonal EG-shaped sample of (5 × 7)–(17 × 33) nm2, long × short axis, with a lattice spacing of 0.306 nm, ascribed to (011) facets. In all cases, the perfect agreement of the lattice spacings from HRTEM with corresponding data from XRD analysis (Table S2,† no. 4, 7–9) again substantiates the formation of solid solutions.
XPS analysis
To fully investigate surface components and the chemical state of samples, XPS spectra are provided in Fig. 3, along with related important data on binding energies (Table S3†) and on the content of designated species (Table S4†) for four different shaped samples. The survey spectra in Fig. 3a offer solid evidence of Ce, Zr and O as the basic elements of all shape-dependent samples, which is in good agreement with the mapping results. In addition, the real components of these samples (Table S4†) are calculated from the corresponding peak areas provided by CasaXPS software. Obviously, these XPS data are roughly consistent with those obtained using EDS, with the exception of RP. However, both display some deviation from the theoretical values, possibly due to the irregular surfaces of the determinant or other technical limitations in both. Fig. 3b depicts the intricate Ce 3d spectrum associated with a tunable valence state and a particular 4f configuration.66 It is well-decomposed into Series A (900.6–917.0 eV) and B (882.3–898.5 eV), which are assigned to the spin–orbit coupling Ce 3d3/2 and Ce 3d5/2 states, respectively.65 Among the two, A1 and B1 are indexed as Ce3+, and others as Ce4+.66 The binding energies of 882.3–882.6 eV in Series B being below that of bulk CeO2 (882.9 eV) support the evaluation of all samples as solid solutions67 as the XRD and mapping results also do. Besides, Ce3+ content is obtained from a peak area percentage of A1 and B1 in Series A and Series B. As seen from Table S4,† it follows the rank: PH > SP > EG ≈ RP, which coincides with that for oxygen defect concentration, which is directly proportional to the former. In Fig. 3c, the characteristic doublets positioned at ca. 182 and ca. 184 eV respectively, related to core-level Zr 3d5/2 and 3d3/2, support Zrn+ (4 > n > 0) and Zr4+ species, the former providing second convincing evidence for well-constructed solid solutions with adjustable shapes.54,67 Fig. 3d elaborates the deconvolution of the high resolution O 1s profile into three peaks centered at 529.4–533.4 eV, which are respectively ascribed to Ce–O, Zr–O and H–O bonds.66,67 Each O species content is sensitive to sample shape, especially for those in the same crystal system (e.g. for A1s of RP and SP, a marked margin of up to 8.5%, Table S4†). O species content is expected to impact the surface acid–base nature (Table 1) in close relation with the separation efficiency of light-induced excitons.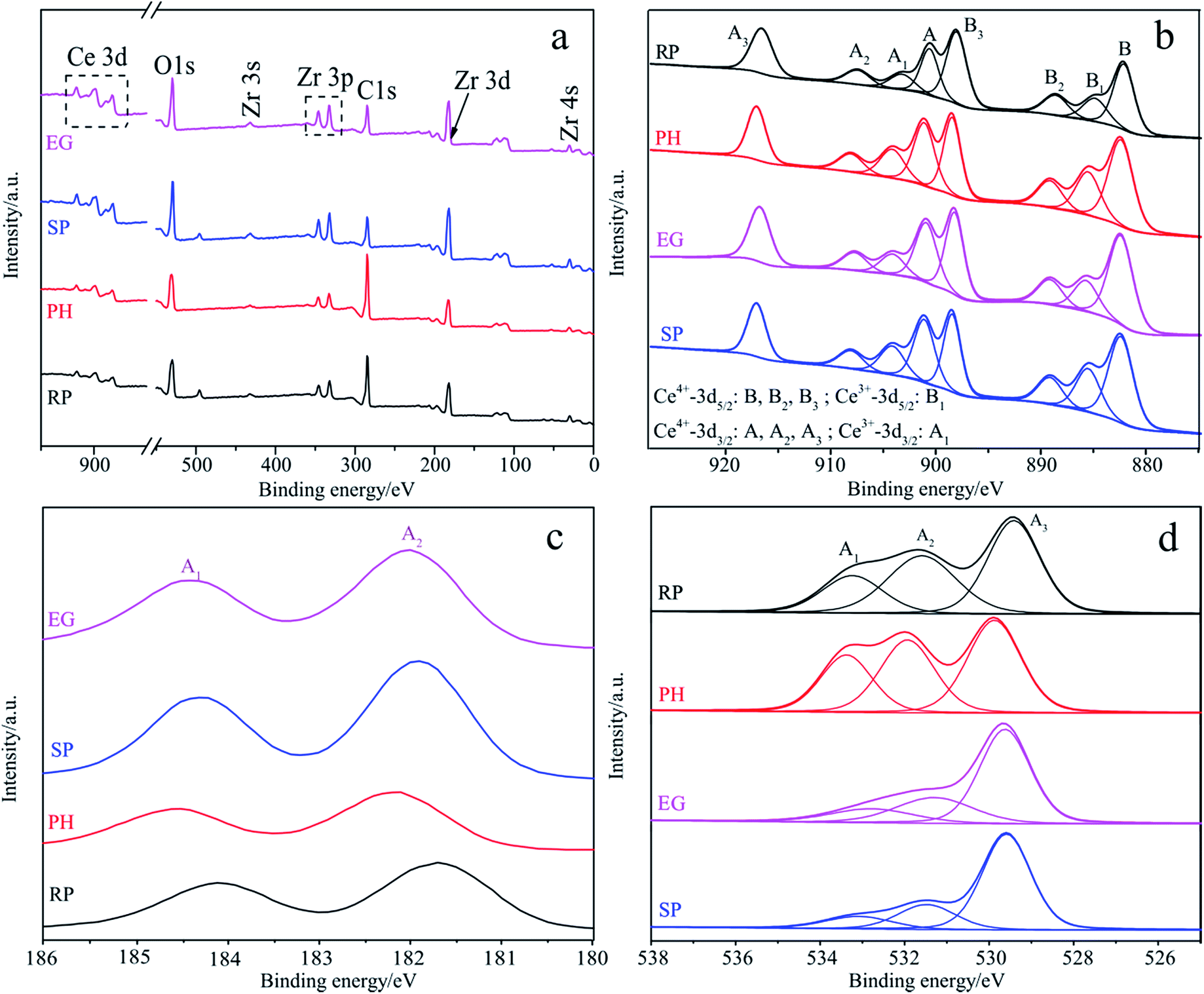 | ||
| Fig. 3 Survey spectra (a) and high-resolution XPS spectra of Ce 3d (b), Zr 3d (c) and O 1s (d) for four samples with various shapes. | ||
| Samples | NH3 absorption (mmol g−1)a | CO2 absorption (mmol g−1)b | ||||||
|---|---|---|---|---|---|---|---|---|
| Weak (<200 °C) | Moderate (200–400 °C) | Strong (>600 °C) | Total | Weak (<200 °C) | Moderate (200–400 °C) | Strong (>600 °C) | Total | |
| a NH3-TPD results.b CO2-TPD results.c nd = not detected. | ||||||||
| RP | 0.184 | 0.349 | 0.148 | 0.681 | 0.155 | 0.324 | Trace | 0.479 |
| PH | 0.245 | 0.258 | ndc | 0.503 | 0.184 | 0.149 | nd | 0.333 |
| SP | 0.320 | 0.452 | nd | 0.772 | 0.288 | 0.317 | nd | 0.605 |
| EG | 0.281 | 0.379 | nd | 0.660 | 0.257 | 0.257 | nd | 0.514 |
DRS, VBXPS and I–V analyses
UV-Vis diffuse reflection spectroscopy (DRS), valence band XPS (VBXPS) and photocurrent–voltage (I–V) curves are collected in Fig. 4 for a comparison of the optical absorption capacity, the construction of the energy band structure and the evaluation of separation probability of light-driven excitons. Fig. 4a displays typical semiconducting and shape-dependent optical absorption with a sole and steep absorption band in the ultraviolet and visible window of the solar spectrum for each sample. RP, SP, PH and EG shaped materials display absorption bands near 431, 458, 480 and 497 nm, respectively. Cubic SP is higher in rank, possibly due to enhanced Ce3+ content relative to that of cubic RP48 (Table S4†). With regard to tetragonal CeO2–ZrO2, such as EG and PH-shaped bodies (Fig. 1b), a robust visible-light response benefits partially from a metal-to-metal charge transfer (MMCT) transition in the oxo-bridged Zr(IV)–O–Ce(III) linkage.54 The former is slightly more light-responsive than the latter, primarily on account of having more access to the transition. A comparative analysis of light harvesting between the two phases deals with the two effects above. In the case of EG and SP-like bodies, the MMCT transition plays the main part in the former optical excellence. In the advantageous optical absorption see in PH over that of SP, both may work together. | ||
| Fig. 4 DRS and Tauc plots (inset) (a), VBXPS profiles (b) and I–V curves (c) of samples with different shapes. | ||
The inset in Fig. 4a offers bandgap (Eg) values (2.87–3.14 eV) from a Tauc plot interpretation based on the following equation:13,21,68
| αhν = A(hν − Eg)n/2 | (1) |
| ECB = EVB − Eg | (2) |
Therefore, their ECB values are determined to be −0.58 (RP), 0.25 (PH), −0.32 (SP) and 0.29 eV (EG), respectively.
Comparative resolution of the separation efficiencies of light-induced charge carriers is conducted by means of an I–V curve under Xe lamp irradiation (Fig. 4c). The four differently shaped bodies exhibit significant differences in net photocurrent response as an indicator of separation efficiency. These efficiencies follow the order: RP ≈ SP > EG > PH, which is inconsistent with the optical responses (Fig. 4a) but accords well with that of the surface acid–base amounts (Table 1). In a word, the optical properties, band structures and excitonic separations of a semiconductor are largely dependent on its morphology.
N2 sorption analysis
N2 sorption profiles in Fig. 5 are used to shed light on the textural properties of specific morphological samples. All sorption curves in Fig. 5a display an adsorption jump in the P/P0 range of ca. 0.4–1.0. They are classified into (a) typical type-II isotherm curves with a clear H3-type hysteresis loop, which applies to the cubic RP and SP-shaped samples21,69 and (b) type-IV isothermal absorption models of a typical mesoporous material with a H2 hysteresis loop, suitable for tetragonal Ce0.4Zr0.6O2 PHs and EGs.70 Fig. 5b describes shape-related BJH pore width distributions centered at 9.51, 11.5, 13.6 and 3.61 nm for RP, PH, SP and EG-shaped materials, indicating their mesoporous structures.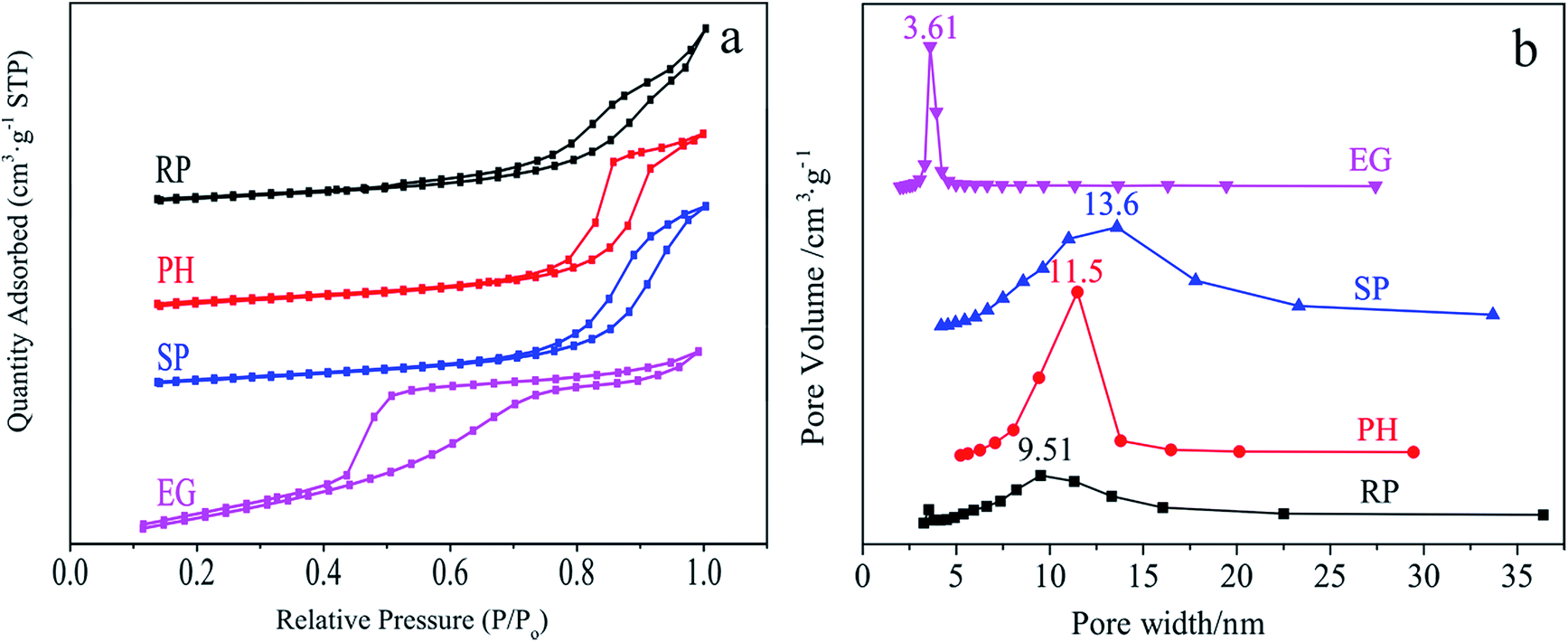 | ||
| Fig. 5 N2 sorption (a) and BJH pore size distribution (b) of RP, PH, SP and EG-shaped samples. | ||
It is well documented that the textural properties of a catalytic material have a remarkable effect on the adsorption and activation of reagents such as methylene blue (MB) on its surface. Table S5† collects important data comprising of BET surface areas (SBET), pore volumes (PV), average pore sizes (APS) and methylene blue absorbances (MBA) associated with this behaviour. Obviously, the SP-shaped body with enhanced textural properties (SBET = 63.0 m2 g−1 and PV = 0.237 cm3 g−1) relative to those of the RP analogue (SBET = 39.6 m2 g−1 and PV = 0.150 cm3 g−1) provides elevated MBA (41.1% vs. 29.6%). Nevertheless, the substitution of PH (SBET = 47.0 m2 g−1 and PV = 0.164 cm3 g−1) with identical superiority for SP in the aforementioned discussion enables a completely opposite result (1.56% vs. 29.6%). The two occurrences are rationalized by the N2 sorption profiles (Fig. 5). With respect to samples such as SP and RP with identical types of isotherm curves and hysteresis loops, their textural properties feature as the basis for an estimation of their adsorption capacity for a given adsorbate. So do those of EG and PH (a small pore size in the tetragonal phase represents superiority from the viewpoint of MB adsorption). If not, the relationship is hardly well established and thus analysed experimentally. Such is the case for EG and RP. As indicated from Table S5,† tetragonal materials prove inefficient in MB absorption and activation, which is hazardous for its removal.
NH3-and CO2-TPD analysis
Enhanced surface acid–base characteristics of samples are reported to allow the suppressed recombination of light-induced excitons, leading to improved photocatalytic performance.71–73 Therefore, this is well-examined by means of NH3- and CO2-TPD techniques. Fig. S7† and Table 1 summarize three types of acid–base sites and their amounts in the four shaped samples, respectively. Fig. S7† indicates the classification of these sites into weak (<200 °C), moderate (200–400 °C) and strong (>600 °C) ones. PH, EG and SP-like bodies merely cover the first two. Both numbers follow the order: SP > EG > PH (Table 1), which is in good agreement with the separation efficiencies of charge carriers (Fig. 4c), implying that increased acidity and alkalinity favour excitonic splitting. Unlike the three other materials, the RP-shaped sample embraces all three sites possibly associated with its phase separation. Notably, its exclusive strong acidic sites endow it with as good separation effects for electron–hole pairs as that of an SP-like analogue (Fig. 4c). In addition, the marked variation in the amounts of acid–base sites represents different anion–cation layouts on the exposed surfaces of variously shaped bodies. That is to say, the samples with various shapes tune their surface acid–base properties for the control of excitonic separation efficiencies by selectively exposing reactive facets to obtain desirable photocatalytic performances.Photocatalytic performance evaluation
MB photocatalytic removal serves as a target reaction to test the catalytic functionality of four samples with different shapes. Fig. 6 illustrates the rank of their activities: RP > EG > SP > PH (Fig. 6a), along with the fact that the reaction is a first-order kinetic process (Fig. 6b). A comprehensive analysis of the aforementioned characterization conclusions, especially those from Fig. 4 and 5 and Table 1, sheds light on the sequence. Of the four, the RP-like solid is selected as the undisputed champion by virtue of its advantages, including (a) the desirable separation probability of the photogenerated charges (Fig. 4c) resulting from the optimal acid–base behavior (Fig. S7† and Table 1) by selectively exposing the designated reactive planes, and (b) the suitable textural properties for efficient MB adsorption and activation (Fig. 5 and Table S5†), over those of the three other samples in the dominant factors that influence photocatalytic behavior. Next, compared with the RP-like solid, the elevated light harvesting capacity (Fig. 4a) of the EG-like material among the four is not sufficient to rectify the undesirable situation caused by the aforementioned factors. In addition, SP loses its predomination, that equals or surpasses the first two, in that it suffers from excessively large SBET and PV values (Table S5†) and imperfect optical behavior (Fig. 4a), despite its superior separation efficiency. The former is adverse to the resultant desorption and light input of the reaction system by virtue of exorbitantly adsorbed MB. The latter enables the obstruction of photogenerated electron–hole pairs. Finally, the most catalytically inactive PH-shaped body suffers mainly from exceptionally low MB absorbance (Table S5†), hardly absorbing and activating MB, and unsatisfactory splitting attributes toward excitons (Fig. 4c). | ||
| Fig. 6 (a) Degradation efficiency (D) and (b) reaction rate constant (k) of samples with different shapes. | ||
Conclusions
RP, PH, SP and EG-like Ce0.4Zr0.6O2 materials were synthesized via a technically viable hydrothermal route. These materials are endowed with unique physical and chemical behaviors as a function of shape that are well-determined by means of various tools including XRD, Raman, EDS-mapping, SEM, TEM, XPS, N2 sorption, NH3- and CO2-TPD, DRS and photocurrent response. Therefore, the morphological dependence of light-induced catalytic performance is well-established. Among the aforementioned samples, RP stands out from its photocatalytically active competition with the three other samples by means of its excellence in excitonic splitting and textural properties. A possible reaction mechanism is proposed based on the well-identified main reactive species.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities of China (No. 3207045403 and 3207045409), the National Natural Science Foundation of China (No. 21576050 and No. 51602052), the Jiangsu Provincial Natural Science Foundation of China (BK20150604) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- Y. J. Zhang, T. Mori and J. H. Ye, Sci. Adv. Mater., 2012, 4, 282–291 CrossRef CAS.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B. Chen, H. X. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- H. Xu, S. X. Ouyang, L. Q. Liu, P. Reunchan, N. Umezawa and J. H. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 CAS.
- H. J. Li, Y. Zhou, W. G. Tu, J. H. Ye and Z. G. Zou, Adv. Funct. Mater., 2015, 25, 998–1013 CrossRef CAS.
- M. C. Liu, L. Z. Wang, G. Q. Lu, X. D. Yao and L. J. Guo, Energy Environ. Sci., 2011, 4, 1372–1378 CAS.
- M. Liu, Y. Chen, J. Su, J. Shi, X. Wang and L. Guo, Nat. Energy, 2016, 1, 16151 CrossRef CAS.
- N. X. Li, M. C. Liu, Z. H. Zhou, J. C. Zhou, Y. M. Sun and L. J. Guo, Nanoscale, 2014, 6, 9695–9702 RSC.
- N. X. Li, L. Z. Zhang, J. C. Zhou, D. W. Jing and Y. M. Sun, Dalton Trans., 2014, 43, 11533–11541 RSC.
- Q. W. Tian, L. Zhang, J. H. Liu, N. X. Li, Q. H. Ma, J. C. Zhou and Y. M. Sun, RSC Adv., 2015, 5, 734–739 RSC.
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed.
- T. Wang, X. G. Meng, P. Li, S. X. Ouyang, K. Chang, G. G. Liu, Z. W. Mei and J. H. Ye, Nano Energy, 2014, 9, 50–60 CrossRef CAS.
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517–11519 RSC.
- N. Li, H. Teng, L. Zhang, J. Zhou and M. Liu, RSC Adv., 2015, 5, 95394–95400 RSC.
- J. H. Liu, L. Zhang, N. X. Li, Q. W. Tian, J. C. Zhou and Y. M. Sun, J. Mater. Chem. A, 2015, 3, 706–712 CAS.
- W. Tian, N. Li and J. Zhou, Appl. Surf. Sci., 2016, 361, 251–258 CrossRef CAS.
- W. Tian, Q. Shen, N. Li and J. Zhou, RSC Adv., 2016, 6, 25568–25576 RSC.
- J. Wang, Y. Shen, Y. Li, S. Liu and Y. Zhang, Chem.–Eur. J., 2016, 22, 12449–12454 CrossRef CAS PubMed.
- Q. Kang, T. Wang, P. Li, L. Q. Liu, K. Chang, M. Li and J. H. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS PubMed.
- H. Xu, P. Reunchan, S. Ouyang, H. Tong, N. Umezawa, T. Kako and J. Ye, Chem. Mater., 2013, 25, 405–411 CrossRef CAS.
- N. Li, M. Liu, B. Yang, W. Shu, Q. Shen, M. Liu and J. Zhou, J. Phys. Chem. C, 2017, 121, 2923–2932 CAS.
- L. Zhang, W. Tian, Y. Chen, J. Chen, H. Teng, J. Zhou, J. Shi and Y. Sun, RSC Adv., 2016, 6, 83471–83481 RSC.
- C. J. Xing, D. W. Jing, M. C. Liu and L. J. Guo, Mater. Res. Bull., 2009, 44, 442–445 CrossRef CAS.
- M. C. Liu, D. W. Jing, Z. H. Zhou and L. J. Guo, Nat. Commun., 2013, 4, 1–8 Search PubMed.
- M. Liu and X. Wang, SPIE Newsroom, 2016 DOI:10.1117/2.1201604.006486.
- N. X. Li, B. Y. Zhou, P. H. Guo, J. C. Zhou and D. W. Jing, Int. J. Hydrogen Energy, 2013, 38, 11268–11277 CrossRef CAS.
- M. C. Liu, B. Wang, Y. Q. Zheng, F. Xue, Y. B. Chen and L. J. Guo, Catal. Sci. Technol., 2016, 6, 3371–3377 CAS.
- M. C. Liu, D. W. Jing, L. A. Zhao and L. J. Guo, Int. J. Hydrogen Energy, 2010, 35, 7058–7064 CrossRef CAS.
- Z. X. Zhou, J. H. Wang, J. C. Yu, Y. F. Shen, Y. Li, A. R. Liu, S. Q. Liu and Y. J. Zhang, J. Am. Chem. Soc., 2015, 137, 2179–2182 CrossRef CAS PubMed.
- Z. Zhou, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, ACS Nano, 2015, 9, 12480–12487 CrossRef CAS PubMed.
- Z. Zhou, Q. Shang, Y. Shen, L. Zhang, Y. Zhang, Y. Lv, Y. Li, S. Liu and Y. Zhang, Anal. Chem., 2016, 88, 6004–6010 CrossRef CAS PubMed.
- J. Shi, C. Cheng, Y. Hu, M. Liu and L. Guo, Int. J. Hydrogen Energy, 2017, 42, 4651–4659 CrossRef CAS.
- N. Li, Y. Zheng, L. Wei, H. Teng and J. Zhou, Green Chem., 2017, 19, 682–691 Search PubMed.
- K. Wu, L. D. Sun and C. H. Yan, Adv. Energy Mater., 2016, 6, 1600501 CrossRef.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- L. Vivier and D. Duprez, Chemsuschem, 2010, 3, 654–678 CrossRef CAS PubMed.
- Y. D. Liu, J. Goebl and Y. D. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- R. I. Walton, Prog. Cryst. Growth Charact. Mater., 2011, 57, 93–108 CrossRef CAS.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- G. Nahar and V. Dupont, Renewable Sustainable Energy Rev., 2014, 32, 777–796 CrossRef CAS.
- C. W. Sun, H. Li and L. Q. Chen, Energy Environ. Sci., 2012, 5, 8475–8505 CAS.
- D. Terribile, A. Trovarelli, J. Llorca, C. de Leitenburg and G. Dolcetti, J. Catal., 1998, 178, 299–308 CrossRef CAS.
- Y. Wei, Z. Zhao, J. Liu, C. Xu, G. Jiang and A. Duan, Small, 2013, 9, 3957–3963 Search PubMed.
- Y. Li and W. J. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- M. C. Liu, Y. Q. Zheng, S. F. Xie, N. X. Li, N. Lu, J. G. Wang, M. J. Kim, L. J. Guo and Y. N. Xia, Phys. Chem. Chem. Phys., 2013, 15, 11822–11829 RSC.
- M. C. Liu, Y. Q. Zheng, L. Zhang, L. J. Guo and Y. N. Xia, J. Am. Chem. Soc., 2013, 135, 11752–11755 CrossRef CAS PubMed.
- S. P. Wang, L. F. Zhao, W. Wang, Y. J. Zhao, G. L. Zhang, X. B. Ma and J. L. Gong, Nanoscale, 2013, 5, 5582–5588 RSC.
- X. H. Lu, T. Zhai, H. N. Cui, J. Y. Shi, S. L. Xie, Y. Y. Huang, C. L. Liang and Y. X. Tong, J. Mater. Chem., 2011, 21, 5569–5572 RSC.
- A. D. Liyanage, S. D. Perera, K. Tan, Y. Chabal and K. J. Balkus, ACS Catal., 2014, 4, 577–584 CrossRef CAS.
- Y. J. Zhang, T. Mori, J. H. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS PubMed.
- X. H. Zhang, D. W. Jing, M. C. Liu and L. J. Guo, Catal. Commun., 2008, 9, 1720–1724 CrossRef CAS.
- Y. J. Zhang, T. Mori, L. Niu and J. H. Ye, Energy Environ. Sci., 2011, 4, 4517–4521 CAS.
- L. Yue and X.-M. Zhang, J. Alloys Compd., 2009, 475, 702–705 CrossRef CAS.
- P. Borker and A. V. Salker, Mater. Chem. Phys., 2007, 103, 366–370 CrossRef CAS.
- C. C. Zhang and J. Lin, Phys. Chem. Chem. Phys., 2011, 13, 3896–3905 RSC.
- H. R. Pouretedal, Z. Tofangsazi and M. H. Keshavarz, J. Alloys Compd., 2012, 513, 359–364 CrossRef CAS.
- J. C. Serrano-Ruiz, J. Luettich, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, J. Catal., 2006, 241, 45–55 CrossRef CAS.
- Q. Yuan, Q. Liu, W.-G. Song, W. Feng, W.-L. Pu, L.-D. Sun, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2007, 129, 6698–6699 CrossRef CAS PubMed.
- A. Primo, T. Marino, A. Corma, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 6930–6933 CrossRef CAS PubMed.
- Q. Xu, W. Lei, X. Li, X. Qi, J. Yu, G. Liu, J. Wang and P. Zhang, Environ. Sci. Technol., 2014, 48, 9702–9708 CrossRef CAS PubMed.
- G. Nahar, V. Dupont, M. V. Twigg and E. Dvininov, Appl. Catal., B, 2015, 168–169, 228–242 CrossRef CAS.
- T. Mondal, K. K. Pant and A. K. Dalai, Int. J. Hydrogen Energy, 2015, 40, 2529–2544 CrossRef CAS.
- N. S. Arul, D. Mangalaraj, R. Ramachandran, A. N. Grace and J. I. Han, J. Mater. Chem. A, 2015, 3, 15248–15258 CAS.
- M. J. Muñoz-Batista, M. N. Gómez-Cerezo, A. Kubacka, D. Tudela and M. Fernández-García, ACS Catal., 2014, 4, 63–72 CrossRef.
- Y. Wei, Z. Zhao, J. Liu, S. Liu, C. Xu, A. Duan and G. Jiang, J. Catal., 2014, 317, 62–74 CrossRef CAS.
- Y. Zhang, J. Wen, J. Wang, D. Pan, M. Shen and Y. Lu, Nano Res., 2011, 4, 494–504 CrossRef CAS.
- P. Ji, J. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2008, 112, 17809–17813 CAS.
- G. Postole, B. Chowdhury, B. Karmakar, K. Pinki, J. Banerji and A. Auroux, J. Catal., 2010, 269, 110–121 CrossRef CAS.
- S. Ouyang, P. Li, H. Xu, H. Tong, L. Liu and J. Ye, ACS Appl. Mater. Interfaces, 2014, 6, 22726–22732 CAS.
- N. Li, L. Wei, R. bibi, L. Chen, J. Liu, L. Zhang, Y. Zheng and J. Zhou, Fuel, 2016, 185, 532–540 CrossRef CAS.
- S. Zeng, X. Zhang, X. Fu, L. Zhang, H. Su and H. Pan, Appl. Catal., B, 2013, 136–137, 308–316 CrossRef CAS.
- X. C. Wang, J. C. Yu, Y. D. Hou and X. Z. Fu, Adv. Mater., 2005, 17, 99–102 CrossRef CAS.
- Y. H. Zhang, G. X. Xiong, N. Yao, W. S. Yang and X. Z. Fu, Catal. Today, 2001, 68, 89–95 CrossRef CAS.
- Z. Z. Lin and X. C. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterization details and some related results from characterization and photocatalytic performance evaluation. See DOI: 10.1039/c7ra01201b |
| This journal is © The Royal Society of Chemistry 2017 |