
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward a detoxification fabric against nerve gas agents: guanidine-functionalized poly[2-(3-butenyl)-2-oxazoline]/Nylon-6,6 nanofibers†
Wu Bin Ying‡
 a,
Sohee Kim‡a,
Min Woo Leea,
Na Yeong Goa,
Hyunsook Jungb,
Sam Gon Ryub,
Bumjae Lee*a and
Kyung Jin Lee*a
a,
Sohee Kim‡a,
Min Woo Leea,
Na Yeong Goa,
Hyunsook Jungb,
Sam Gon Ryub,
Bumjae Lee*a and
Kyung Jin Lee*a
aDepartment of Applied Chemical Engineering, College of Engineering, Chungnam National University, Daejeon 305–764, Korea. E-mail: yingwubin@cnu.ac.kr; kjlee@cnu.ac.kr
bAgency for Defense Development (ADD), Yuseong-Gu, Daejeon, Korea
First published on 7th March 2017
Abstract
A novel guanidine-functionalized polymer, poly[2-(3-butenyl)-2-oxazoline] (PBuOxz), has been co-electrospun with Nylon-6,6 to form fibers that could be used for the decontamination of chemical warfare agents (CWAs). PBuOxz was obtained from the living cationic polymerization of 2-(3-butenyl)-2-oxazoline, which was synthesized starting from 4-pentenoic acid. This clickable PBuOxz polymer could be easily functionalized through the introduction of amine groups via thiol–ene click chemistry, followed by guanidinylation to form guanidine-functionalized PBuOxz (G-PBuOxz). The synthesized G-PBuOxz/Nylon-6,6 fibers are an active hydrolysis species for diisopropyl fluorophosphate (DFP), an organophosphate mimic of nerve agents. The decontamination efficiency for DFP could be as high as 100% under trace aqueous solution conditions using this G-PBuOxz/Nylon-6,6 fiber as the decontamination agent at the ratio of [guanidine]/[DFP] = 100/1 in 2 hours. The hydrolysis occurs via a general SN2 mechanism for this G-PBuOxz-based catalysis. Furthermore, the kinetics of decontamination were also investigated based on time and guanidine concentration. Hence, this G-PBuOxz/Nylon-6,6 fiber is effective as a hydrolyzing agent against toxic organophosphates, and thus exhibits potential as a material for protecting against chemical warfare agents.
1. Introduction
Protective clothing is widely used to protect military personnel when they are exposed to toxic chemical warfare agents (CWAs).1–10 It consists of multiple layers, which can physically or chemically absorb or block toxic compounds.11–15 However, full protection brings the problems of undesirable weight and heat burdens for the wearers. Furthermore, the relatively low decontamination efficiency is another potential threat. These challenges motivate the development of a new protective clothing system that is lightweight with a relatively high decontamination efficiency.Nylon is a versatile engineering material, and is used in stockings and clothing of all types. This fabric is recognized for being thin and flexible, but is also valued for its durability and toughness. However, the difficulty in introducing functional groups into the nylon polymer chain has limited its application in various specialized fields that require functionalized polyamides. Polyoxazolines can be considered as a structural variant of nylon with amide functional groups in the side chain.16 Therefore, it is expected that polyoxazolines will have a similar solubility parameter (δ) to nylon, which will make it possible to form homogeneous blends of these two polymers.17–19 In addition, the functionality of poly(2-substituted-2-oxazoline)s can be tuned simply by varying the 2-substituted functional group of the 2-oxazoline monomer.20–28 The living cationic ring-opening polymerization of 2-substituted-2-oxazolines provides easy and direct access to a wide variety of well-defined polymers with large numbers of functional groups.
The guanidine group has been applied in molecular recognition devices, disinfectants and antiseptics for clinical use.29–31 It is capable of forming both electrostatic and hydrogen bond interactions with polar molecules. Due to its strong basicity (pKa = 13.6), it can be used as a catalyst to decompose phosphates in water, leading to activation of the guest toward nucleophilic substitution.32
Herein, due to the versatility of poly(2-substituted-2-oxazoline)s and their ability to form functional materials, poly[2-(3-butenyl)-2-oxazoline] (PBuOxz) was synthesized and fibrous materials have been prepared via co-electrospinning with nylon. In the previous literature, diverse methods have been reported concerning the preparation of functional polymers containing large amounts of functional groups. For example, various functional groups can be easily introduced onto the pedant vinyl groups of PBuOxz to form functionalized polyoxazoline via thiol–ene chemistry.33–39 However, most of the polymers reported in previous studies are very difficult to process, which will be a crucial drawback for industrial application. The polymer presented here will provide a new breakthrough to prepare functional polymeric materials with easy processability. Here, as a representative example of the easy processability of the polymer, an electrospun nanofiber web was prepared. Electrospun webs have attracted large scientific interest due to their high specific surface area, high porosity and low fabric weight, which are perfect properties for application in catalytic decomposition fabrics.40–50 In this study, the electrospun web prepared from a blend of guanidine functionalized PBuOxz/nylon was used as a detoxification fabric against CWAs.
2. Experimental
2.1 Materials
The solvents dichloromethane, diethyl ether, methanol, acetonitrile and N,N-dimethylformamide (Samchun Chem., extra pure grade, 99.5%) were stored over freshly ground calcium hydride (Sigma-Aldrich Chem., reagent grade, 95%) with vigorous stirring for one day, followed by distillation into a two-neck reactor fitted with a silicone rubber seal and a high-vacuum line (10−6 Torr) with a Rotaflo® stopcock. Sigma-Aldrich reagents: 4-pentenoic acid (98%), 2-chloroethylamine hydrochloride (99%), N-hydroxysuccinimide (98%), methyl-3-mercaptopropionate (98%), methyl triflate (99%), 2-methyl-2-oxazoline (99%), cysteamine (98%), cysteamine hydrochloride (98%), 2,2′-azobis(2-methylpropionitrile) (98%), and cyanamide (99%). TCI reagents: piperidine (99%). These reagents were used without further purification.2.2 Monomer synthesis
The synthesis of 2-(3-butenyl)-2-oxazoline (BuOxz) involved three steps starting from 4-pentenoic acid and proceeding through N-succinimidyl-4-pentenate and N-(2-chlorethyl)-4-pentenamide, to finally give (2-(3-butenyl)-2-oxazoline), as described by Schlaad.51 (1) N-Succinimidyl-4-pentenate: to a solution of 25.0 g (0.25 mol) of 4-pentenoic acid in 700 mL of dry dichloromethane (distilled from CaH2) were added 46.12 g (0.40 mol) of N-hydroxysuccinimide and 52.54 g (0.275 mol) of 1-(3-dimethylpropyl)-3-ethylcarbodiimide hydrochloride (EDAC). The mixture was then stirred for 12 h at 40 °C. After evaporation of the solvent, the residue was dissolved in a mixture of diethyl ether and water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), and subsequently extracted five times. After the combined organic layers were dried with sodium sulfate, the solvent was removed to yield N-succinimidyl-4-pentenate as a colorless solid. (2) N-(2-Chloroethyl)-4-pentenamide: a solution of 37.5 g (0.19 mol) of N-succinimidyl-4-pentenate in 500 mL of dry dichloromethane was added dropwise under vigorous stirring to a solution of 44.25 g (0.38 mol) of 2-chloroethylamine hydrochloride and 15.26 g (0.38 mol) of NaOH in 200 mL of deionized water. After being stirred overnight at 50 °C, the organic layer was separated and washed twice with 200 mL of deionized water. The combined fractions were dried over sodium sulfate and the solvent evaporated to give N-(2-chloroethyl)-4-pentenamide as a yellowish oil. (3) 2-(3-Butenyl)-2-oxazoline: in a flame-dried flask, 24.6 g (0.153 mol) of N-(2-chloroethyl)-4-pentenamide was dissolved in 100 mL of dry methanol. To this solution, a freshly ground solution of 12.85 g (0.23 mol) of KOH in 100 mL of dry methanol was added dropwise using a cannula. The reaction mixture was stirred for 12 h at 70 °C under an argon atmosphere (precipitation of KCl occurred after about 10 min). The salt was filtered off and the remaining solution was concentrated in an evaporator. Additionally, the precipitated salt was removed by passing the solution through 0.45 μm filter paper. For purification, the yellow solution was fractionally distilled at 60 °C via a high vacuum line to give 2-(3-butenyl)-2-oxazoline as a colourless liquid.
:1 v/v), and subsequently extracted five times. After the combined organic layers were dried with sodium sulfate, the solvent was removed to yield N-succinimidyl-4-pentenate as a colorless solid. (2) N-(2-Chloroethyl)-4-pentenamide: a solution of 37.5 g (0.19 mol) of N-succinimidyl-4-pentenate in 500 mL of dry dichloromethane was added dropwise under vigorous stirring to a solution of 44.25 g (0.38 mol) of 2-chloroethylamine hydrochloride and 15.26 g (0.38 mol) of NaOH in 200 mL of deionized water. After being stirred overnight at 50 °C, the organic layer was separated and washed twice with 200 mL of deionized water. The combined fractions were dried over sodium sulfate and the solvent evaporated to give N-(2-chloroethyl)-4-pentenamide as a yellowish oil. (3) 2-(3-Butenyl)-2-oxazoline: in a flame-dried flask, 24.6 g (0.153 mol) of N-(2-chloroethyl)-4-pentenamide was dissolved in 100 mL of dry methanol. To this solution, a freshly ground solution of 12.85 g (0.23 mol) of KOH in 100 mL of dry methanol was added dropwise using a cannula. The reaction mixture was stirred for 12 h at 70 °C under an argon atmosphere (precipitation of KCl occurred after about 10 min). The salt was filtered off and the remaining solution was concentrated in an evaporator. Additionally, the precipitated salt was removed by passing the solution through 0.45 μm filter paper. For purification, the yellow solution was fractionally distilled at 60 °C via a high vacuum line to give 2-(3-butenyl)-2-oxazoline as a colourless liquid.
2.3 Polymerization of 2-(3-butenyl)-2-oxazoline
Poly[2-(3-butenyl)-2-oxazoline] (PBuOxz) with a target Mw of 10000 g mol−1 was synthesized as follows.51 To a solution of 7.03 g (56.2 mmol) of 2-(3-butenyl)-2-oxazoline in 10 mL of dry acetonitrile (freshly distilled from CaH2) was added 0.08 mL (0.7 mmol) of methyl triflate under a dry argon atmosphere. The mixture was stirred for 24 h at 70 °C, followed by the addition of 1 mL (10.4 mmol) of piperidine at room temperature. After being stirred for another 5 h at 70 °C, the reaction mixture was precipitated in diethyl ether for 6 h. The resulting PBuOxz was purified by repeated precipitation from diethyl ether three times to remove any unreacted 2-(3-butenyl)-2-oxazoline monomer, and then dried in a vacuum oven at 60 °C to constant weight.
2.4 Guanidine-functionalization of poly[2-(3-butenyl)-2-oxazoline]
All of the experiments were carried out in N,N-dimethylformamide (DMF) at 70 °C by using 2,2′-azobis(2-methylpropionitrile) (AIBN) as a radical source via a thiol–ene reaction. The PBuOxz (3.04 g, vinyl moles: 24.32 mmol) was dissolved in 30 mL of dried N,N-dimethylformamide under a nitrogen atmosphere, followed by the addition of cysteamine (CA) or cysteamine hydrochloride (CA·HCl) (8.5 g, 72.96 mmol) and AIBN (0.82 g, 4.87 mmol) at a mole ratio of [CA]/[vinyl]/[AIBN] = 3/1/0.2. The functionalization was carried out at 70 °C for 24 hours, and then the reaction mixture was cooled down at 0 °C. The resulting amine functionalized poly[2-(3-butenyl)-2-oxazoline] (A-PBuOxz) was purified by repeated precipitation from THF three times to remove any unreacted CA or CA·HCl, residual radical source or unfunctionalized PBuOxz, and then dried in a vacuum oven at 60 °C to constant weight. Further functionalization via guanidinylation was carried out using A-PBuOxz as the base polymer. A-PBuOxz (6.36 g, amine moles: 25 mmol) was dissolved in 60 mL of 0.35 M HCl aqueous solution, followed by the addition of cyanamide (2.1 g, 50 mmol). Guanidine functionalization was carried out at 90 °C for 24 hours. After cooling to room temperature, the resulting polymer was precipitated three times in acetone to remove any unfunctionalized A-PBuOxz or cyanamide, and then dried in a vacuum oven at 60 °C to constant weight. Because the guanidinylation was carried out in HCl aqueous solution, HCl would be complexed with the guanidine group, stabilizing it and decreasing its reactivity. Therefore, HCl was removed by treating the reaction mixture with 10 wt% KHCO3 aqueous solution three times, followed by washing with deionized water three times to obtain a neutral solution. The resulting guanidine functionalized poly[2-(3-butenyl)-2-oxazoline] (G-PBuOxz) was dried in a vacuum oven at 60 °C to constant weight.2.5 Co-electrospinning of G-PBuOxz/Nylon-6,6
A 10 wt% solution of G-PBuOxz/Nylon-6,6 (5/5, wt/wt) was prepared in trifluoroethanol (CF3CH2OH) or formic acid (HCOOH) as the solvent. Before the co-electrospinning process, the prepared polymer solution was stirred well to ensure uniform mixing. The polymer solution was then transferred into a 1 mL syringe fitted with a metallic needle of 0.4 mm inner diameter. The syringe was horizontally fixed on a syringe stand, and the positive electrode of a high voltage power supply (CPS-60K02vl, Chungpa EMT Co., South Korea), capable of generating voltages up to 30 kV, was clamped to the metal needle tip. The applied voltage was fixed at 15 kV and the tip-to-collector distance was kept at 15 cm. All the experiments were conducted at room temperature. The developed nanofiber mats were collected on a rotating drum covered with aluminum foil. The co-electrospun sample of G-PBuOxz/Nylon-6,6 fibers was dried in a vacuum oven at room temperature for 24 h to remove the residual solvent.2.6 Decontamination of diisopropyl fluorophosphates
The desired amount of G-PBuOxz/Nylon-6,6 fibers was placed into a 5 mL vial purged with nitrogen. 0.5 μL of DFP and 25 μL of H2O were introduced into the system and the decontamination reaction was performed at 32 °C for 2 hours. Then, 1 mL of D2O was injected into the vial to extract the hydrolysis products, followed by the collection of the resulting D2O solution for 31P-NMR analysis.2.7 Characterization
IR absorption spectra were measured on a Nicolet iS5 FT-IR spectrometer with the iD5 ZnSe ATR accessory (Thermo Scientific). 1H-NMR spectra were recorded on a Unity Inova400 MHz spectrometer with tetramethylsilane as the internal standard. The concentration of the samples was in the range of 1–5 wt%. Gel permeation chromatography (GPC) analysis was performed using a waters 2690 separations module equipped with three Styragel columns and a refractive index detector, with refractive index detection at a flow rate of 1.0 mL min−1 in THF, after calibration using polystyrene standards. The morphology of the fibers obtained from electrospinning was observed using an S-4800 (HITACHI) scanning electron microscope (SEM) after platinum sputtering (50 seconds) onto the fibers. The kinetics of decontamination were determined by 31P-NMR spectrometry using a Bruker AvanceIII 600 spectrometer, with triphenyl phosphate (TPP) in deuterated acetone used as an external reference (0 ppm).3. Results and discussion
Scheme 1 contains a schematic diagram of the synthetic procedure for G-PBuOxz, the co-electrospinning process to produce G-PBuOxz/Nylon-6,6 fibers and the decontamination process of the detoxification fabric. Initially, 2-(3-butenyl)-2-oxazoline (BuOxz) was chosen as the functional monomer, not only because it is a structural isomer of nylon, but also because it has two reactive sites that exhibit orthogonal reactivity. This monomer can undergo ring-opening living cationic polymerization while maintaining the double bond. The BuOxz monomer was synthesized in three steps from commercially available 4-pentenoic acid27 (Scheme S1†) at a mild temperature, as described in the ESI.† The successful synthesis of the monomer is demonstrated in Fig. S1.†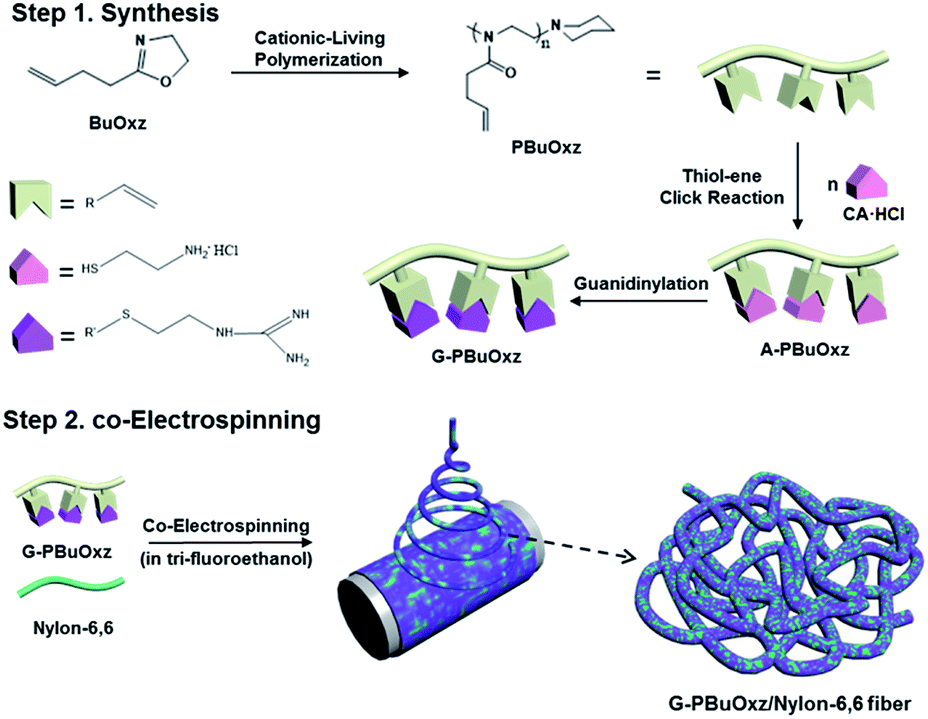 | ||
| Scheme 1 Synthesis of G-PBuOxz and the co-electrospinning of G-PBuOxz/Nylon-6,6 fibers. | ||
Homopolymerization of BuOxz was performed in acetonitrile solution using methyl triflate as an initiator, followed by the termination of the piperidine (Scheme 1). The chemical structure and Mw of PBuOxz were confirmed by 1H-NMR spectroscopy (Fig. 1(a)). The backbone intensity ratio was consistent with the theoretical value of 4:1:2. By comparing the integrals of the vinyl proton of P6 with the initiator end-group of P1 or the terminator end-group of P11, the Mw could be calculated to be 13400 g mol−1. The molecular weight of PBuOxz was also analysed by GPC (9600 g mol−1) and it was found to have a polydispersity index of 1.21, which is relatively well matched with that from the 1H-NMR calculation (Fig. 1(a)). From the FT-IR spectrum (Fig. 1(b)), the νC–H (2600–3200 cm−1), νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (∼1700 cm−1) and νC–N (∼1400 cm−1) stretching of the PBuOxz backbone could be easily confirmed, but the νCC signal was obscured by the νCO signal.
O (∼1700 cm−1) and νC–N (∼1400 cm−1) stretching of the PBuOxz backbone could be easily confirmed, but the νCC signal was obscured by the νCO signal.
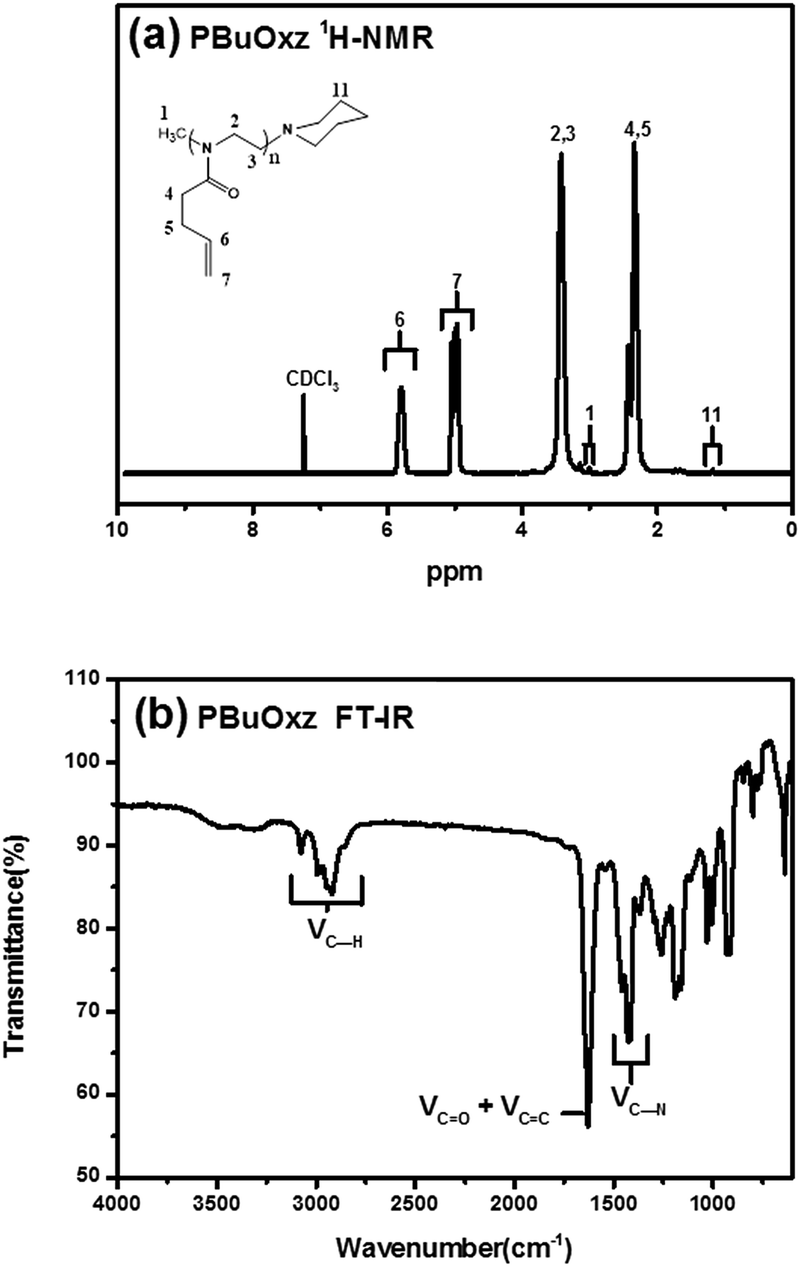 | ||
| Fig. 1 (a) 1H-NMR spectrum of PBuOxz; (b) FT-IR spectrum of PBuOxz. | ||
This functional PBuOxz monomer could be easily functionalized to introduce amine groups via thiol–ene click chemistry, followed by guanidinylation to form guanidine-functionalized PBuOxz (G-PBuOxz). Firstly, amine functionalization of PBuOxz was carried out using cysteamine (CA) or cysteamine hydrochloride (CA·HCl) as the functional agent via a thiol–ene reaction, as shown in Scheme 2.
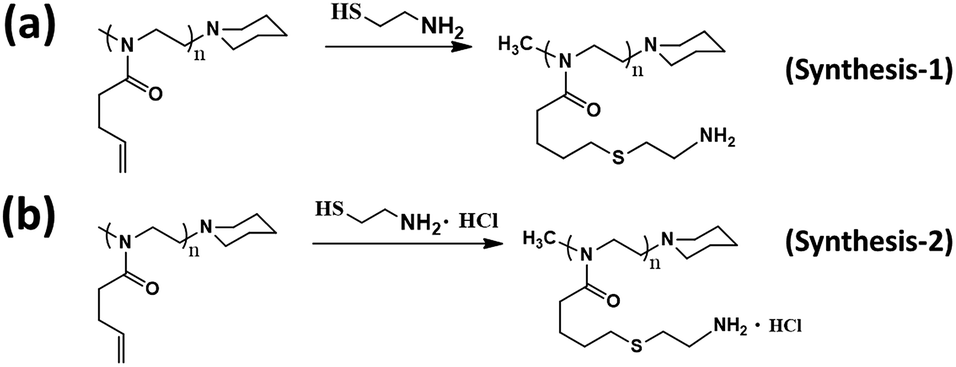 | ||
| Scheme 2 Amination routes of PBuOxz: (a) synthesis-1 (cysteamine) and (b) synthesis-2 (cysteamine hydrochloride). | ||
In the case of the CA click reaction, the conversion yield is only 7–14 mol%, even when the ratio of [CA]/[CC] is increased (PBuOxz-Syn-1(1) and PBuOxz-Syn-1(2) as listed in Table 1, obtained from the 1H-NMR spectra in Fig. 2(a)), mainly due to the basicity of the amine group. The basic amine prevents the formation of stable thiyl radicals during the procedure. As a result, we introduced the amine salt CA·HCl in order to increase the conversion yield of the thiol–ene click reaction. After analysis by 1H-NMR (Fig. 2(b)), almost 100% conversion for the click chemistry reaction can be obtained, as listed in Table 1 (PBuOxz-Syn-2), judging from the disappearance of the vinyl protons at 5–6 ppm. This CA·HCl aminated polyoxazoline was defined as A-PBuOxz, and was further guanidinylated to enhance the basicity of the polymer for the detoxification of nerve gases. From the FT-IR spectrum (Fig. 2(c)), the appearance of ν–[NH3]+Cl− at 2600–3200 cm−1 also proved that –NH2·HCl groups had been introduced.
| No. | Thiol–ene reaction | Amine-PBuOxz | ||
|---|---|---|---|---|
| [Amine]/[vinyl]/[AIBN] | Time (h) | Temp (°C) | Functionality | |
| PBuOxz-Syn-1(1) | CA: 3/1/0.2 | 24 | 70 | 7.0 |
| PBuOxz-Syn-1(2) | CA: 5/1/0.2 | 14.0 | ||
| PBuOxz-Syn-2 | CA·HCl: 3/1/0.2 | 100 | ||
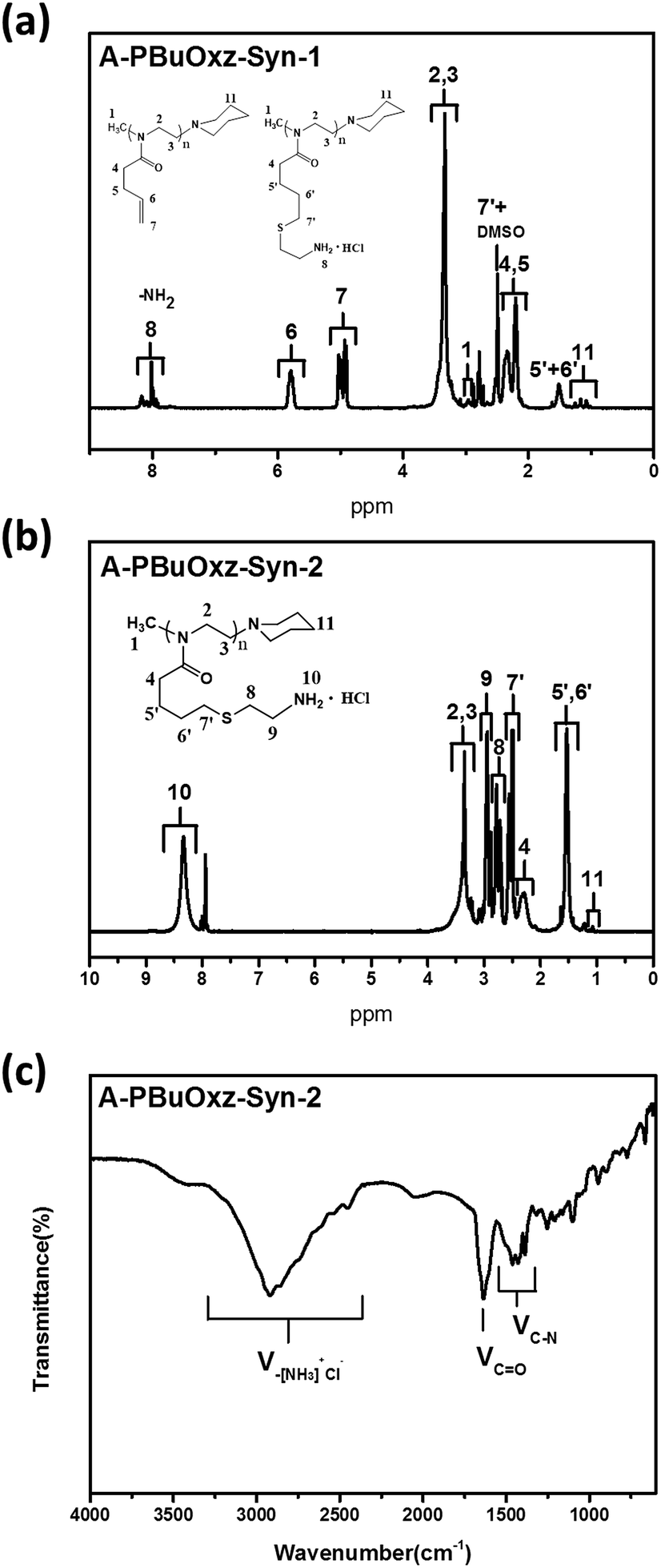 | ||
| Fig. 2 (a) 1H-NMR spectrum of A-PBuOxz-Syn-1 aminated by cysteamine; (b) 1H-NMR spectrum of A-PBuOxz-Syn-2 aminated by cysteamine hydrochloride; (c) FT-IR spectrum of A-PBuOxz-Syn-2. | ||
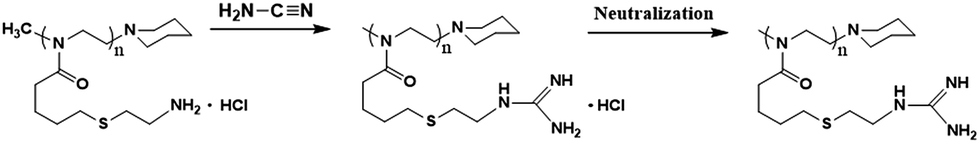
Guanidine functionalization was performed using cyanamide as a functional agent to form G-PBuOxz as a weakly acidic aqueous solution, as shown in Scheme 3. The chemical structure of G-PBuOxz was determined by 1H-NMR spectroscopy, as shown in Fig. 3(a). Compared to A-PBuOxz, the signal from the amine protons centered at 8.2 ppm diminished with the substitution of the guanidine protons at 5.5 ppm and 6.2 ppm in G-PBuOxz. When the signal area integral of the guanidine protons is compared with that of the backbone, it can be considered that almost 100% of the amine groups have been functionalized into guanidine groups. From the FTIR spectrum of G-PBuOxz shown in Fig. 3(b), the appearance of N–H stretching at 3320 cm−1 could be ascribed to the additional amine groups of guanidine. A decrease in the peak at 2600–3200 cm−1 was further evidence of guanidine formation, which indicates that the νN–H of –NH3+Cl− has been changed to guanidine groups. The appearance of CN stretching at 1580 cm−1 also indicates the formation of guanidine groups.
 | ||
| Scheme 3 Guanidinylation of A-PBuOxz. | ||
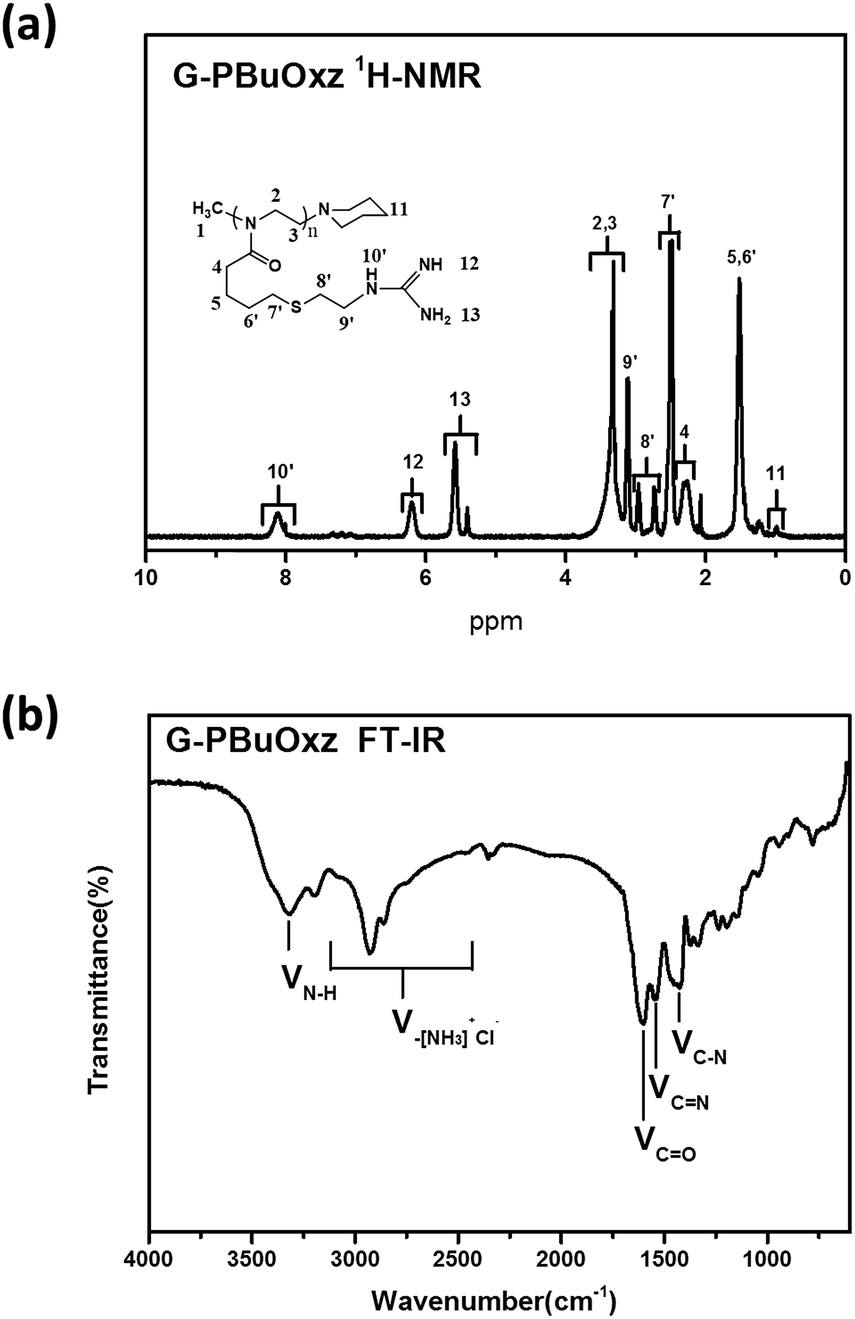 | ||
| Fig. 3 (a) 1H-NMR spectrum of G-PBuOxz; (b) FT-IR spectrum of G-PBuOxz. | ||
The molecular weights of the resulting G-PBuOxz and A-PBuOxz polymers were increased to 18900 g mol−1 and 15400 g mol−1 from the Mw of 9600 g mol−1 for PBuOxz, and the molecular weight distributions (PDIs) changed little (PBuOxz: 1.21, A-PBuOxz: 1.22, G-PBuOxz: 1.22) as shown in Fig. 4, which is strong evidence for successful functionalization.
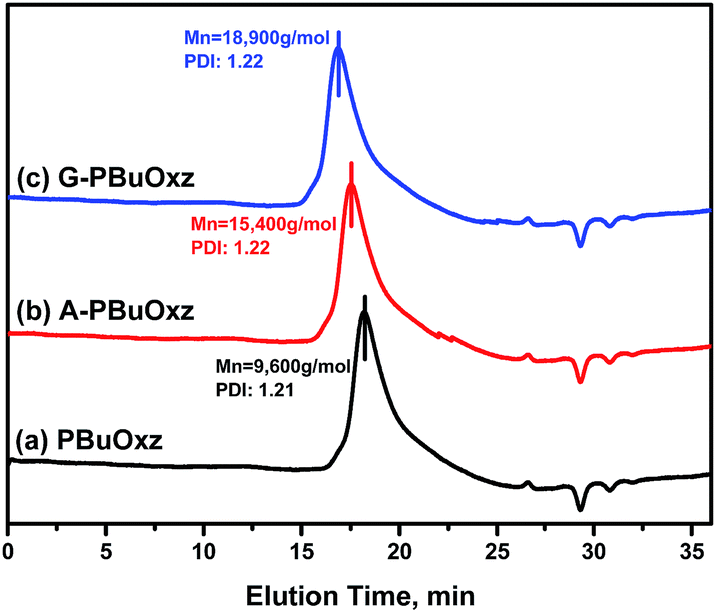 | ||
| Fig. 4 GPC curves of (a) the PBuOxz base polymer (Mn: 9600 g mol−1, PDI: 1.21), (b) A-PBuOxz (Mn: 15400 g mol−1, PDI: 1.22), and (c) G-PBuOxz (Mn: 18900 g mol−1, PDI: 1.22). | ||
Unfortunately, the as-prepared G-PBuOxz could not be electrospun to form nanofibers by itself because of its poor physical properties (gel-formation was observed, as shown in Fig. 5(a)). However, when this polymer is blended with nylon, its physical properties are enhanced enough to form nanofiber structures. Because of the molecular backbone similarity between PBuOxz and nylon, the polymeric blend was successfully fabricated without any notable phase separation, suggesting that an inter-penetrating network was formed, which is the main driving force for the enhancement of the physical properties. Fig. 5(b)–(d) show SEM images of electrospun nanofibers with different weight ratios of G-PBuOxz and Nylon-6,6, i.e. 7/3, 5/5 and 3/7, respectively. In terms of functionality, a higher amount of G-PBuOxz is preferred for further application, but a nanofiber web with the desired porosity can only be obtained when over 50% of nylon is introduced into the system (Fig. 5(c)). Another SEM image with low magnification (inset of Fig. 5(c)) illustrates the uniformity of the resulting G-PBuOxz/Nylon-6,6 fibers. Judging from the SEM images, the excellent processability of this G-PBuOxz/Nylon-6,6 fiber can be confirmed. By simply blending G-PBuOxz with Nylon-6,6, which is an economical and commercially available polyamide, followed by co-electrospinning, protective clothing materials can be easily produced. In addition, no allograft rejection occurs due to the similar solubility parameters (δ) of these two polyamides (Nylon-6,6: 23.37 MPa1/2, poly[2-(3-butenyl)-2-oxazoline]: 26 MPa1/2), so homogeneous nanofibers could be formed.17,18 This excellent processability provides great convenience for future industry applications, not just for military uniforms but also for conventional fiber industries to produce functional fabrics.
 | ||
| Fig. 5 SEM images of (a) G-PBuOxz fibers; (b) G-PBuOxz/Nylon-6,6 (7/3, wt/wt) fibers; (c) G-PBuOxz/Nylon-6,6 (5/5, wt/wt) fibers and (d) G-PBuOxz/Nylon-6,6 (3/7, wt/wt) fibers. | ||
Here, in order to maintain the catalytic activity of the guanidine functional groups toward organic phosphates, trifluoroethanol was selected as the electrospinning solvent, because formic acid (the solvent generally used for electrospinning nylon in other studies) can affect the basicity of the guanidine groups by leading to salt formation or urea formation, as shown in Fig. 6, resulting in a decrease in the detoxification yield (Fig. S2†).
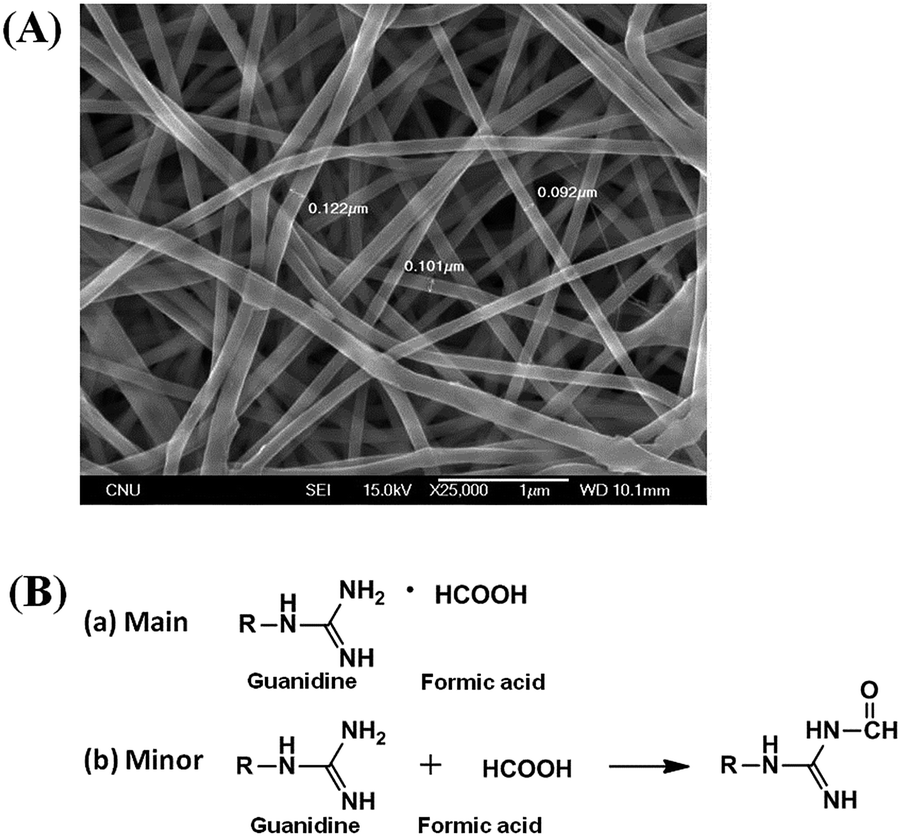 | ||
| Fig. 6 (A) SEM image of G-PBuOxz/Nylon-6,6 (5/5, wt/wt) fibers co-electrospun in formic acid. (B) Side reactions of guanidine groups with formic acid: (a) salt formation (complexing with formic acid); (b) urea formation (reacting with formic acid). | ||
Since guanidine is the strongest base (pKa = 13.6) among the neutral organic compounds, we expected it to exhibit excellent reactivity toward DFP – a simulant for combat organophosphates such as the nerve agents sarin and soman, which are inhibitors of acetylcholine esterase.52–54 In fact, DFP is known to be phosphorylated by guanidine at elevated temperatures via a hydrolysis mechanism.55 The guanidine-catalyzed hydrolysis of organophosphates such as DFP proceeds through the three amine groups in the guanidine groups via a general base-catalyzed bimolecular substitution mechanism (SN2), involving P–O bond cleavage by the solvated amino groups, RNH2⋯H–OH.
As shown in Fig. 7, the doublet at 5.0 and 9.0 ppm coupled to the P–F bond (JP–F = 970 Hz) is assigned to the reactant (DFP) in the 31P-NMR spectrum.56,57 A broad singlet signal centered at 16.7 ppm is attributed to the hydrolysis product, DHP, formed by the G-PBuOxz/Nylon-6,6 fibers co-electrospun in trifluoroethanol. Furthermore, increasing the [guanidine]/[DFP] ratio increased the decontamination efficiency massively, with 100% removal obtained at a ratio of 100/1. This can be regarded as great evidence of the successful preparation of fabric materials with decontamination properties. However, when the fibers were obtained using formic acid as the co-electrospinning solvent (Fig. 6(A)), the decontamination efficiency was 0%, as shown in Fig. S2,† which might be caused by the side reactions that occur between the guanidine groups and the carboxyl groups in formic acid, as shown in Fig. 6(B).
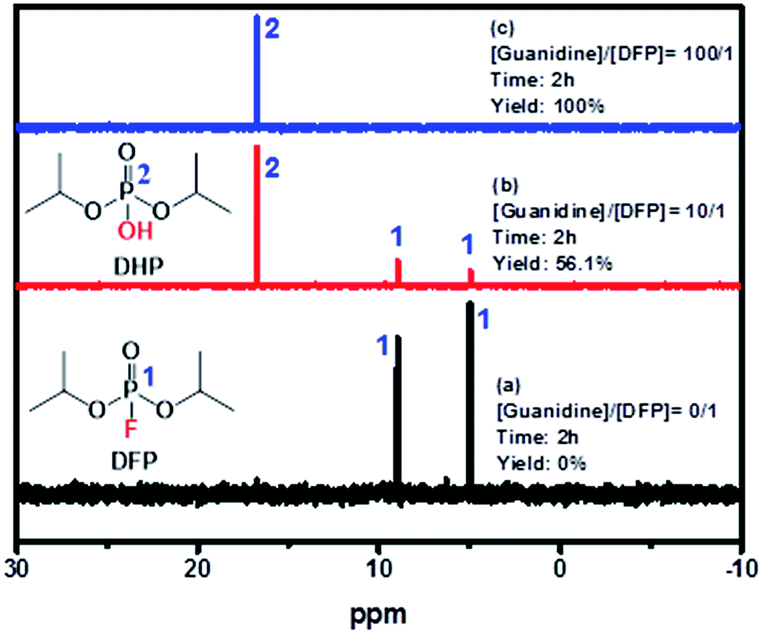 | ||
| Fig. 7 31P-NMR spectra of (a) pure DFP, (b) [guanidine]/[DFP] = 10/1, and (c) [guanidine]/[DFP] = 100/1 showing DFP decomposition by the catalyst. | ||
Under the conditions in this study, the degree of DFP hydrolysis will be related to the concentration of the substrate ([DFP]) and the reaction time. In this case, the decontamination process is governed not by guanidine itself, but by guanidine–H2O complexes as shown in Scheme 4.
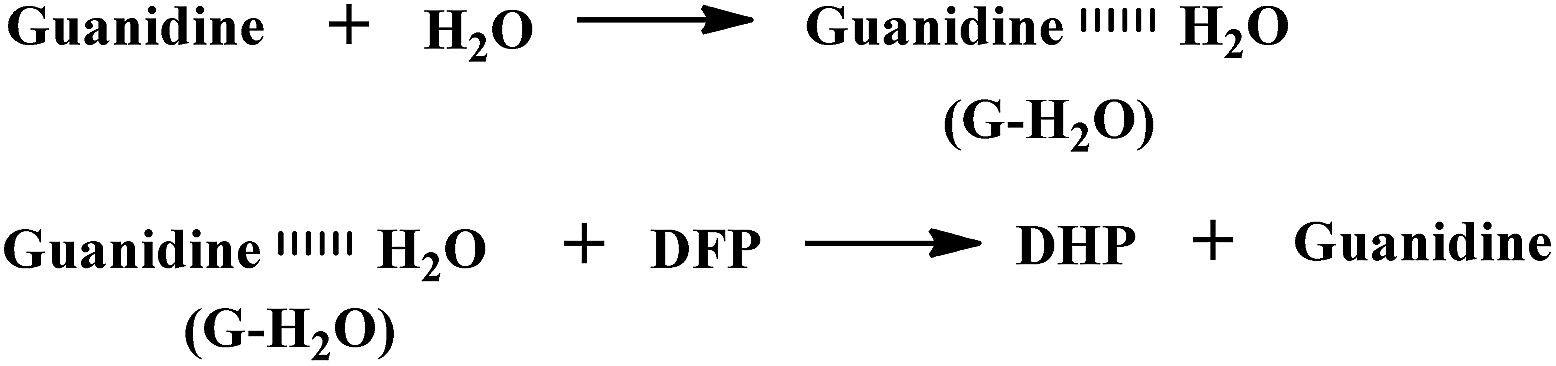 | ||
| Scheme 4 Decontamination of DFP by guanidine in the presence of H2O. | ||
Therefore, the governing kinetics equation can be written as follows:
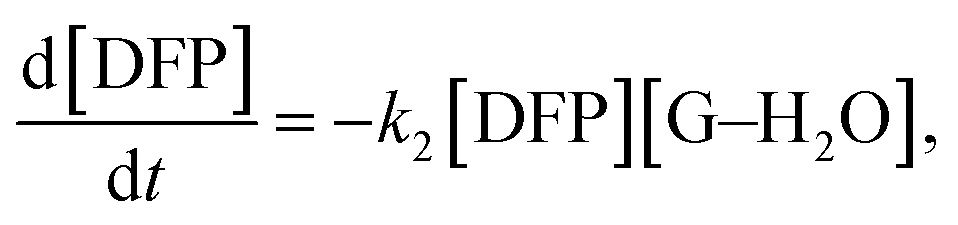 | (1) |
Kow ∼ −2.64).58 Therefore, guanidine combines with water molecules just for an instant.
Furthermore, water molecules are in excess relative to the guanidine groups, so it could be defined that [guanidine] = [G–H2O], and based on these conversions, the total kinetics equation can be written as follows:
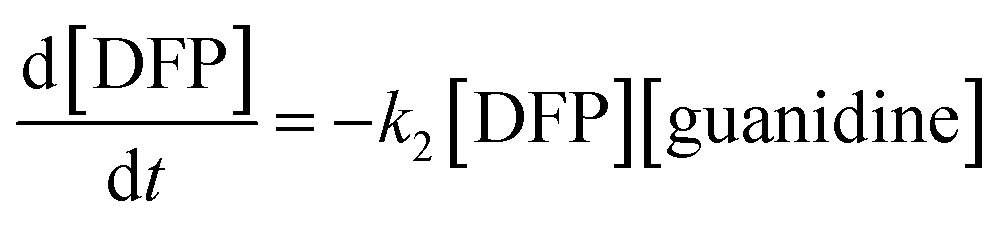 | (2) |
This equation can be further converted to:
| −ln([DFP]/[DFP]0) = k2[guanidine] × t, | (3) |
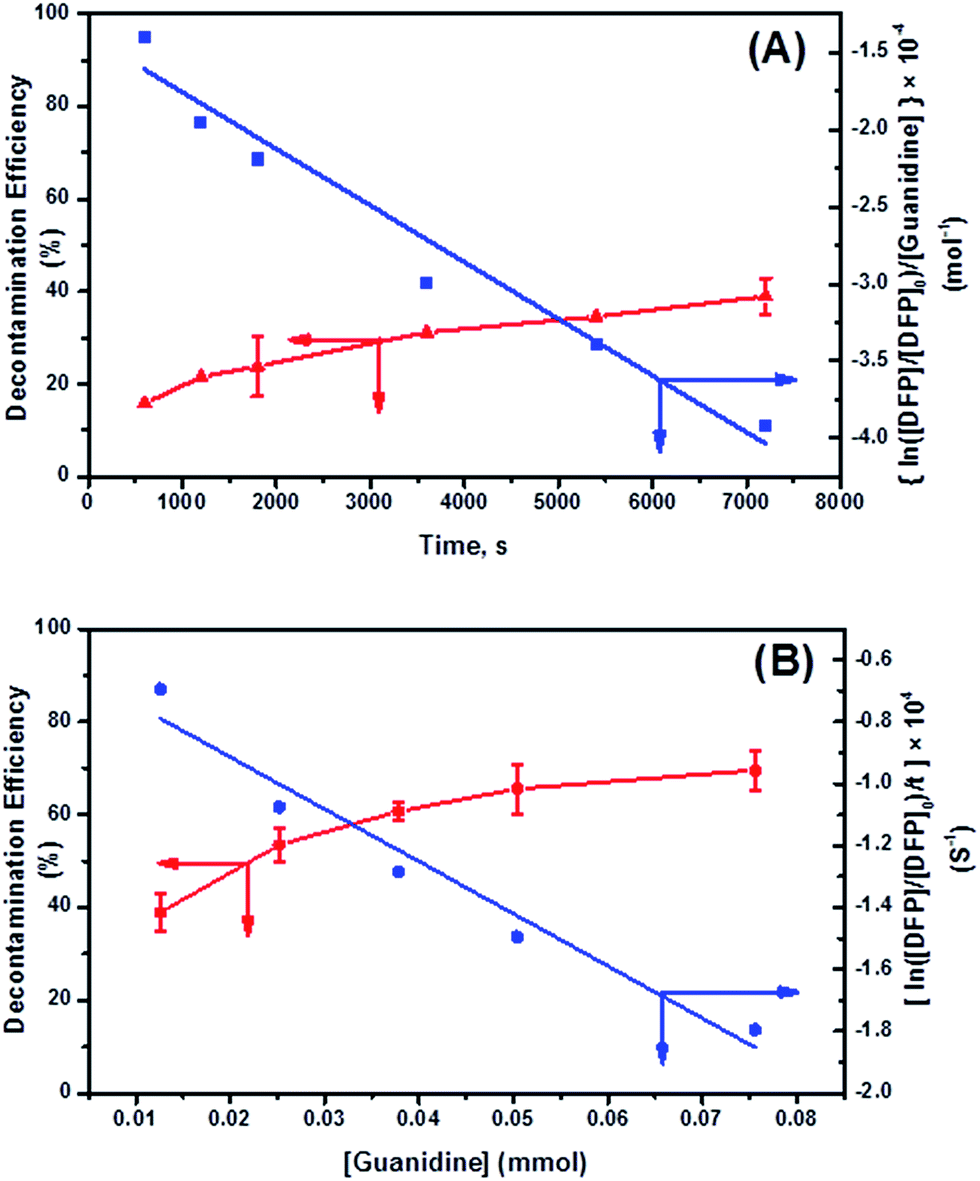 | ||
Fig. 8 Kinetics of DFP conversion with G-PBuOxz/Nylon-6,6 fibers: (A) decontamination kinetics with time and the related observed rate constant (k2); (B) decontamination kinetics with [guanidine] and the related rate constant (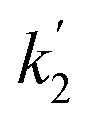 ). ). | ||
Here, the dependency of –ln([DFP]/[DFP]0) on time measured for the G-PBuOxz/Nylon-6,6 fibers exhibited excellent linearity, as shown in Fig. 8(A), and the value of [guanidine] is a constant (1.26 × 10−5 mol), so the value of k2 was calculated from the slope to be 3.6 mol−1 s−1. The decontamination efficiency also increased with an increase in [guanidine]/[DFP]. Fig. 8(B) shows typical DFP conversion vs. [guanidine] kinetics with G-PBuOxz/Nylon-6,6 fibers. The decontamination efficiency increases with [guanidine]/[DFP], and the rate constant for DFP hydrolysis (k2) is obtained from the experimental data using the following equation:
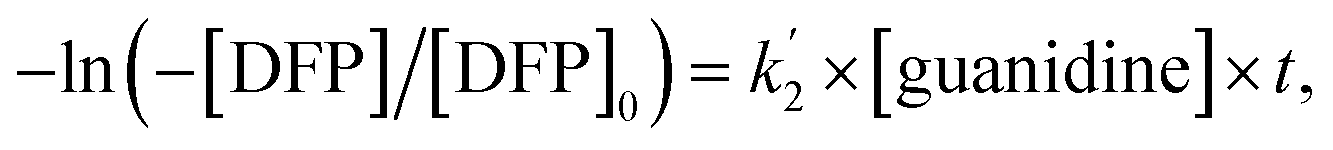
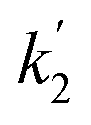 on time measured for the G-PBuOxz/Nylon-6,6 fibers also exhibited excellent linearity, and the time (t) is a constant (7200 s), so the value of
on time measured for the G-PBuOxz/Nylon-6,6 fibers also exhibited excellent linearity, and the time (t) is a constant (7200 s), so the value of 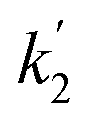 is 1.7 mol−1 s−1, calculated from the slope. The values of k2 and
is 1.7 mol−1 s−1, calculated from the slope. The values of k2 and 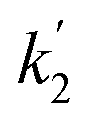 are very close, which can be regarded as good evidence of the accuracy of this kinetics investigation.
are very close, which can be regarded as good evidence of the accuracy of this kinetics investigation.
As a control experiment, when we added just G-PBuOxz/Nylon-6,6 fibers without any water, no decontamination was observed as shown in Fig. S3(A) and (B).† The same result was obtained when there was only water without G-PBuOxz/Nylon-6,6 fibers, as shown in Fig. S3(A) and (C).† These results indicate that the real hydrolysis agents are guanidine–H2O complexes.
Although various synthetic polymers bearing groups with decontamination properties have been reported so far, e.g. hydroxylamine functionalized polyacrylonitrile (PAN), poly(N-vinylguanidine) (PVG) and their combined fiber materials,59–61 the values of the rate constant k2 reported here are much higher than those of previous reports (0.06 mol−1 s−1 to 0.43 mol−1 s−1).
This might be due to (1) the huge amount of functional groups originating from the tailored design of the reactive polymers; (2) the polyamide structure, which might play an important role as a catalyst to increase the decontamination rate constant k2; (3) the porosity of the electrospun fibers, which makes it possible for the guanidine functionality to be used effectively.
4. Conclusions
In conclusion, G-PBuOxz/Nylon-6,6 fibers were conveniently obtained by co-electrospinning Nylon-6,6 and G-PBuOxz, which was synthesized from PBuOxz by amine functionalization and guanidinylation. These fibers appear to be an ardent hydrolyzing species for DFP, which is an organophosphate mimic of CWAs. The detoxification efficiency is relatively high compared with that reported in previous studies. Moreover, the high decontamination efficiency of these fibers against toxic organophosphates indicates their value as a decontamination material that can be used in protective clothing against chemical warfare agents. In addition, the method presented here to prepare functionalized nylon fibers can be extended to prepare nylon-based smart cloths, which can be functionalized with various moieties.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIP) (Sub. No. 2015R1D1A1A01060467 and 2013M3A6A5073175). This work was also supported by the Agency for Defense Development (Sub. No. UD150014ID).References
- L. M. Eubanks, T. J. Dickerson and K. D. Janda, Chem. Soc. Rev., 2007, 36, 458 RSC.
- P. K. Gutch, A. Mazumder and G. Raviraju, RSC Adv., 2016, 6, 2295 RSC.
- B. M. Smith, Chem. Soc. Rev., 2008, 37, 470 RSC.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419 RSC.
- D. R. Goud, A. K. Purohit and V. Tak, et al., Chem. Commun., 2014, 50, 12363 RSC.
- R. K. Totten, P. Ryan and B. Kang, et al., Chem. Cummun., 2012, 48, 4178 RSC.
- Y. Zafrani, L. Yehezkel and M. Goldvaser, et al., Org. Biomol. Chem., 2011, 9, 8445 CAS.
- J. R. Hiscock, M. R. Sambrook and P. B. Cranwell, et al., Chem. Cummun., 2014, 50, 6217 RSC.
- Y. Bai, Y. Dou and L. H. Xie, et al., Chem. Soc. Rev., 2016, 45, 2327 RSC.
- Y. Liu, C. T. Buru and A. J. Howarth, et al., J. Mater. Chem. A, 2016, 4, 13809 CAS.
- H. L. Schreuder-Gibson, Q. Truong and J. E. Walker, et al., MRS Bull., 2003, 28, 574 CrossRef CAS.
- G. H. Dennison, M. R. Sambrook and M. R. Johnston, RSC Adv., 2014, 4, 55524 RSC.
- S. Royo, R. Martínez-Máñez and F. Sancenón, et al., Chem. Cummun., 2007, 4839 RSC.
- K. Desai, K. Kit and J. Li, et al., Polymer, 2009, 50, 3661 CrossRef CAS.
- D. J. Woo and S. K. Obendorf, RSC Adv., 2014, 4, 15727 RSC.
- K. Aoi, H. Suzuki and M. Okada, Macromolecules, 1992, 25, 7073 CrossRef CAS.
- T. Tsunetsugu, J. Furukawa and T. Fueno, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 3529 CrossRef CAS.
- B. Kostova, K. Ivanova-Mileva, D. Rachev and D. Christova, AAPS PharmSciTech, 2013, 14, 352 CrossRef CAS PubMed.
- M. Hartlieb, K. Kempe and U. S. Schubert, J. Mater. Chem. B, 2015, 3, 526 RSC.
- E. Rossegger, V. Schenk and F. Wiesbrock, Polymers, 2013, 5, 956 CrossRef.
- C. Legros, M. C. De Pauw-Gillet and K. C. Tam, et al., Soft Matter, 2015, 11, 3354 RSC.
- K. Lava, B. Verbraeken and R. Hoogenboom, Eur. Polym. J., 2015, 65, 98 CrossRef CAS.
- T. R. Dargaville, K. Lava, B. Verbraeken and R. Hoogenboom, Macromolecules, 2016, 49, 4774 CrossRef CAS.
- U. Mansfeld, S. Hoeppener and K. Kempe, et al., Soft Matter, 2013, 9, 5966 RSC.
- T. Li, H. Tang and P. Wu, Soft Matter, 2015, 11, 1911 RSC.
- M. Hartlieb, D. Pretzel and K. Kempe, et al., Soft Matter, 2013, 9, 4693 RSC.
- J. M. W. Chan, X. Ke and H. Sardon, et al., Chem. Sci., 2014, 5, 3294 RSC.
- M. Hartlieb, D. Pretzel and M. Wagner, et al., J. Mater. Chem. B, 2015, 3, 1748 RSC.
- M. D. Best, S. L. Tobey and E. V. Anslyn, Coord. Chem. Rev., 2003, 240, 3 CrossRef CAS.
- K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67 CrossRef CAS PubMed.
- P. Gilbert and L. E. Moore, J. Appl. Microbiol., 2005, 99, 703 CrossRef CAS PubMed.
- V. Alcázar, J. R. Morán and J. de Mendoza, Tetrahedron Llett., 1995, 36, 3941 CrossRef.
- L. Tauhardt, D. Pretzel and K. Kempe, et al., Polym. Chem., 2014, 5, 5751 RSC.
- T. Hayashi and A. Takasu, Biomacromolecules, 2015, 16, 1259 CrossRef CAS PubMed.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157 CrossRef CAS PubMed.
- C. E. Hoyle, B. A. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- B. A. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC.
- M. A. Gauthier and H. A. Klok, Chem. Cummun., 2008, 2591 RSC.
- I. Tijunelyte, J. Babinot and M. Guerrouache, et al., Polymer, 2012, 53, 29 CrossRef CAS.
- Y. Dzenis, Science, 2004, 304, 1917 CrossRef CAS PubMed.
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59, 1384 CrossRef CAS PubMed.
- L. Buruaga, A. Gonzalez and J. J. Iruin, J. Mater. Sci., 2009, 44, 3186 CrossRef CAS.
- G. Guangming, W. Juntao and Z. Yong, et al., Soft Matter, 2014, 10, 549 RSC.
- P. Samanta, V. Thangapandian and S. Singh, et al., Soft Matter, 2016, 12, 5110 RSC.
- L. Mecozzi, O. Gennari and R. Rega, et al., Soft Matter, 2016, 12, 5542 RSC.
- A. Polini, S. Pagliara and R. Stabile, et al., Soft Matter, 2010, 6, 1668 RSC.
- L. Chen, L. Bromberg, T. A. Hatton and G. C. Rutledge, Polymer, 2007, 48, 4675 CrossRef CAS.
- L. Chen, L. Bromberg, T. A. Hatton and G. C. Rutledge, Polymer, 2008, 49, 1266 CrossRef CAS.
- B. Ding, J. Lin and X. Wang, et al., Soft Matter, 2011, 7, 8376 RSC.
- I. Steyaert, G. Vancoillie and R. Hoogenboom, et al., Polym. Chem., 2015, 6, 2685 RSC.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928 CrossRef CAS.
- L. Bromberg and T. A. Hatton, Ind. Eng. Chem. Res., 2005, 44, 7991 CrossRef CAS.
- G. H. Dennison, M. R. SambrookR and M. R. Johnston, RSC Adv., 2014, 4, 55524 RSC.
- A. M. Costero, S. Gil and M. Parra, et al., Chem. Cummun., 2008, 6002 RSC.
- T. Wagner-Jauregg, J. J. O'Neill and W. H. Summerson, J. Am. Chem. Soc., 1951, 73, 5202 CrossRef CAS.
- G. W. Wagner and P. W. Bartram, J. Mol. Catal. A: Chem., 1999, 144, 419 CrossRef CAS.
- Y. Segall, D. Waysbort and D. Barak, et al., Biochemistry, 1993, 32, 13450 CrossRef.
- E. Bacalum, M. Cheregi and V. David, Rev. Roum. Chim., 2012, 57, 427 CAS.
- L. Chen, L. Bromberg and H. Schreuder-Gibson, et al., J. Mater. Chem., 2009, 19, 2432 RSC.
- L. Bromberg and T. A. Hatton, Polymer, 2007, 48, 7490 CrossRef CAS.
- L. Chen, L. Bromberg and J. A. Lee, et al., Chem. Mater., 2010, 22, 1429 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) Synthesis scheme and structural analysis of monomer; (2) decontamination test of DFP without efficiency. See DOI: 10.1039/c7ra01278k |
| ‡ These authors contribute equally on this work. |
| This journal is © The Royal Society of Chemistry 2017 |