
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiwalled carbon nanotube-supported CuCo2S4 as a heterogeneous Fenton-like catalyst with enhanced performance†
Mai Zhang ab,
K. P. Annamalaia,
Lile Liua,
Tianlu Chena,
Jianping Gaoab and
Yousheng Tao*ac
ab,
K. P. Annamalaia,
Lile Liua,
Tianlu Chena,
Jianping Gaoab and
Yousheng Tao*ac
aCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (CAS), Fuzhou 350002, China
bUniversity of Chinese Academy of Sciences, Beijing, 100039, China
cR & D Center of Saline Lake and Epithermal Deposits, Chinese Academy of Geological Sciences, Beijing 100037, China. E-mail: taoys@tom.com
First published on 11th April 2017
Abstract
In this paper, we prepared CuCo2S4/MWCNTs as a heterogeneous iron-free bimetal catalyst for the degradation of methylene blue (MB) dye by uniformly synthesizing CuCo2S4 nanoparticles on the surfaces of multiwalled carbon nanotubes (MWCNTs). The homogeneous nanostructure, tuned by MWCNTs, and the efficient electron transfer from CuCo2S4 to MWCNTs resulted in a quite decent catalytic activity, which was superior to that of pristine CuCo2S4 and monometal sulfides. Moreover, CuCo2S4/MWCNTs maintained a good catalytic performance over a broad pH range of 3 to 12, and exhibited better MB degradation rates under alkaline conditions. Meanwhile, the catalyst was reusable for up to three cycles due to the strong interaction between the nanoparticles and MWCNTs. The results demonstrate that CuCo2S4/MWCNTs is an improved Fenton catalyst and a promising candidate to substitute iron-based catalysts for the treatment of wastewater containing dyes.
1. Introduction
Wastewater containing organic dyes and nitrogen-containing compounds has been an environmental problem in the past decades. In order to address this urgent issue, numerous wastewater treatment techniques have been proposed, such as adsorption, membrane separation, photocatalysis and biodegradation.1–4 Among these physicochemical methods, the Fenton reaction, which uses Fe2+/H2O2 to generate hydroxyl radicals (·OH), attracts extensive attention due to its cost-saving and environmentally friendly nature.5 In addition to commonly used iron salts, copper, cobalt, cerium and manganese salts were also found to be effective in Fenton-like reactions.6–9 However, these homogeneous catalysts cause secondary pollution, because they are difficult to recover. Moreover, a lot of sludge is produced with the use of iron salts during the catalytic process.10 To overcome these negative effects, recyclable heterogeneous catalysts like metal oxides, hydroxides and sulfides have been used to replace homogeneous catalysts.11–16 Since then, enormous progress has been made in heterogeneous catalysis; however, most catalysts still show stability issues at certain values of pH, particularly under strong alkaline conditions. Hence, it is necessary to prepare other types of catalysts with a high catalytic activity and a wide applicable pH range.One method is the preparation of an iron-free bimetal heterogeneous catalyst. The iron-based bimetal catalysts such as CoFe2O4 and CuFe2O4 showed high catalytic efficiencies with an increased activity of Fe2O3 but were limited by the narrow applicable pH range of Fe2+.17,18 Moreover, copper and cobalt catalysts have been studied separately, but the combined effect of copper and cobalt in the Fenton reaction has not been studied.19,20 Thus, we drew our attention on their bimetal sulfides. CuCo2S4 has been demonstrated to be effective in providing multiple redox states, which is beneficial for reaction activity.21 Furthermore, the higher electrical conductivity of CuCo2S4 compared to that of CuCo2O4 can accelerate the electron transfer. In addition, the respective workable pH ranges for the Cu+/H2O2 and Co2+/H2O2 systems are broader than that for Fe2+/H2O2, and the synergistic effect between copper and cobalt may further broaden the applicable pH range for the reaction. The other method involves the use as substrates of carbon nanomaterials, such as multiwalled carbon nanotubes (MWCNTs) and graphene. Owing to their large surface area and good conductivity, MWCNTs effectively enhance the activity of catalysts and introduce additional functional properties as matrix materials.22 For the Fenton reaction, an optimum addition of MWCNTs can enhance the degradation performance of the catalysts.23 The surface functionalities and structural defects of MWCNTs can act as active sites to catalyze H2O2 involved reactions.24 Moreover, the robust matrix composed of MWCNTs can improve the reusability of the catalysts and broaden the practicable pH range.25 Fe3O4–MWCNT nanocomposite can be used in the pH range from 1 to 10, which indicates the better stability of the catalyst after the incorporation of MWCNTs.26
In this study, we prepared CuCo2S4/MWCNTs by a mild two-step hydrothermal method, in which CuCo2S4 nanoparticles were deposited on MWCNTs. Compared to monometal catalysts (CuS and CoS) and pristine CuCo2S4, the CuCo2S4/MWCNT composite exhibited a higher Fenton-like catalytic efficiency. Furthermore, CuCo2S4/MWCNTs tolerated a broad range of pH and exhibited a good reusability. The achieved high catalytic performance demonstrated that the synthesized CuCo2S4/MWCNT composite was an efficient Fenton-like catalyst for potential future applications. The mechanism for catalytic enhancement in CuCo2S4/MWCNTs is also discussed at the end of the paper.
2. Experimental
2.1. Materials
Copper acetate monohydrate (Cu(CH3COO)2·H2O, 99.0%), cobalt acetate tetrahydrate (Co(CH3COO)2·4H2O, 99.5%) and L-cysteine (C3H7NO2S, 99%) were purchased from Aladdin, China. Sodium carbonate (Na2CO3, ≥99%) and methylene blue (MB, ≥98%) were obtained from Adamas Reagent Co., Ltd. Triethylamine (C6H15N, 99.0%), hydrogen peroxide (H2O2, 30% w/w), nitric acid (HNO3, 65.0–68.0%) and sulfuric acid (H2SO4, 95–98%) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All the abovementioned chemicals were used directly without any further purification. MWCNT powder (purity > 95%, diameter: 30–50 nm) was provided by Chengdu Organic Chemicals Co., Ltd and the Chinese Academy of Sciences.2.2. Preparation of CuCo2S4/MWCNTs
MWCNTs were first acidified with a HNO3/H2SO4 mixture at 60 °C, as reported in the literature.27 The functionalization process was effective in removing impurities.28 Then, 20.0 mg of functionalized MWCNTs were added to 10.0 ml of distilled water and sonicated for 3 hours. The copper and cobalt solution was prepared by adding 64.5 mg of Cu(CH3COO)2·H2O and 161.0 mg of Co(CH3COO)2·4H2O into 10 ml of distilled water and stirred for 30 minutes. Subsequently, the functionalized MWCNT suspension was added to the abovementioned solution and further stirred for 30 minutes to form a uniform mixture. Then an aqueous solution of 32.0 mg of Na2CO3 was added to the mixture, followed by the addition of 0.5 ml of triethylamine. The mixture was then transferred to a 50.0 ml Teflon-lined stainless steel autoclave, heated at 180 °C for 12 h and allowed to cool to room temperature. The precipitate was washed with distilled water and ethanol several times and dried at 70 °C to obtain copper and cobalt hydroxide/MWCNTs.CuCo2S4/MWCNTs was subsequently prepared. First, 50.0 mg of hydroxide/MWCNTs was dispersed in 10.0 ml of distilled water. An L-cysteine solution was prepared by dissolving 75.0 mg of L-cysteine in 10.0 ml of distilled water. Then, this solution was added to the hydroxide/MWCNT suspension, followed by stirring and heating at 180 °C for 6 h in a Teflon-lined stainless steel autoclave. Finally, the sample was washed and dried at 70 °C. CuCo2S4, CuS and CoS were prepared without MWCNTs by the same experimental conditions.
2.3. Characterization
The structural phase of the samples was investigated by X-ray diffraction (XRD) with a Miniflex II (Rigaku) using a monochromatized X-ray beam from Cu Kα (λ = 0.154050 nm) radiation. The morphology of the samples was studied via scanning electron microscopy (SEM, JSM-6700, JEOL) and transmission electron microscopy (TEM, TecnaiF2, Philips) with energy dispersive spectrometry (EDS). To study the surface area and pore structure of the samples, the nitrogen adsorption–desorption isotherms were recorded using ASAP2020 (Micromeritics) at 77 K. Raman spectra were recorded on a Labram Hr800 Evolution (Horiba). Furthermore, the surface electronic structure was analyzed by X-ray photoelectron spectroscopy (XPS) using an ESCALAB 250Xi (Thermo Fisher).For the catalytic activity tests, 10.0 mg of catalyst was added to an MB aqueous solution and stirred for 3 hours to reach adsorption equilibrium. Subsequently, 1.0 ml of H2O2 was added and the concentration of MB solution was determined using Spectrometer UV-1100 (Mapada). To study the stability of the catalysts under different pH values, the pH in the mixtures of MB solution and catalysts was adjusted with HCl or NaOH solution before the catalytic reaction. The degradation experiments were then conducted and the MB concentration in solution was determined after 20 minutes. To test the reusability of the catalyst, 10.0 mg of catalyst was used to degrade MB during 20 minutes for three cycles. The catalyst was collected after each reaction by filtration and washed with ethanol and water several times. Afterwards, the catalyst was used again in the adsorption and degradation experiments under the same conditions as described above.
3. Results and discussion
XRD experiments were taken to investigate the chemical structures of CuCo2S4 and CuCo2S4/MWCNTs. As shown in Fig. S1,† the diffraction peaks at 2θ = 26.6°, 31.2°, 37.9°, 46.9°, 49.9° and 54.8° can be indexed to the diffraction of the (022), (113), (004), (224), (115) and (044) planes of the CuCo2S4 standard structure (JCPDS file no. 42-1450), respectively. Compared to pristine CuCo2S4, the peaks of CuCo2S4 in CuCo2S4/MWCNTs are sharper and no apparent peaks of secondary phases are observed. The higher purity and crystallinity of CuCo2S4 in CuCo2S4/MWCNTs demonstrate that MWCNTs can assist the nucleation of CuCo2S4. Typical diffraction peaks of MWCNTs were not detected, which indicates the uniform dispersion of MWCNTs in the CuCo2S4/MWCNT composite.The SEM and TEM images of samples are shown in Fig. 1 and S2–S5.† As the scanning electron microscopy (SEM) image shows in Fig. 1a, the MWCNTs were decorated and separated by CuCo2S4 nanoparticles without apparent agglomeration, exactly in agreement with the XRD results. The transmission electron microscopy (TEM) image was also recorded as shown in Fig. 1b, which demonstrates that CuCo2S4 nanoparticles had a diameter of approximately 20 nm and were dispersed uniformly on the surfaces of MWCNTs or their interlayers. Such uniform dispersion of the CuCo2S4 nanoparticles in an interlaced structure inhibited their aggregation, which prevented the efficiency loss of the catalyst after use. A typical HRTEM image, shown in Fig. 1c, demonstrates that the CuCo2S4 nanoparticles were attached to the outer walls of MWCNTs, and exhibited clear lattice fringes. The interplanar space was calculated from the inverse FFT image (Fig. 1c, inset), and it was determined to be 0.33 nm, which belongs to the (022) plane of CuCo2S4 and is in good agreement with the XRD results.
 | ||
| Fig. 1 SEM image (a), TEM image (b) and HRTEM image (c) of CuCo2S4/MWCNTs. The image inset in (c) is the inverse Fast Fourier Transform (FFT) image of the square marked region in (c). | ||
An excellent dye and H2O2 adsorption is an important factor for the efficiency of the Fenton reaction in the degradation of dyes. Nitrogen adsorption was measured to evaluate the adsorptive properties and verify the surmise of surface area increase based on the CuCo2S4/MWCNT morphology. Fig. 2 shows the nitrogen adsorption/desorption isotherms of CuCo2S4 and CuCo2S4/MWCNTs, and the nitrogen adsorption/desorption isotherms of MWCNTs are shown in Fig. S6.† The BET surface area of CuCo2S4 is 29 m2 g−1, while that of CuCo2S4/MWCNTs is 38 m2 g−1. The pore volume of CuCo2S4/MWCNTs is 0.21 cm3 g−1, which is higher than that of CuCo2S4 (0.14 cm3 g−1). As a result, CuCo2S4/MWCNTs provides more active sites and can adsorb a larger amount of dye and H2O2 molecules.
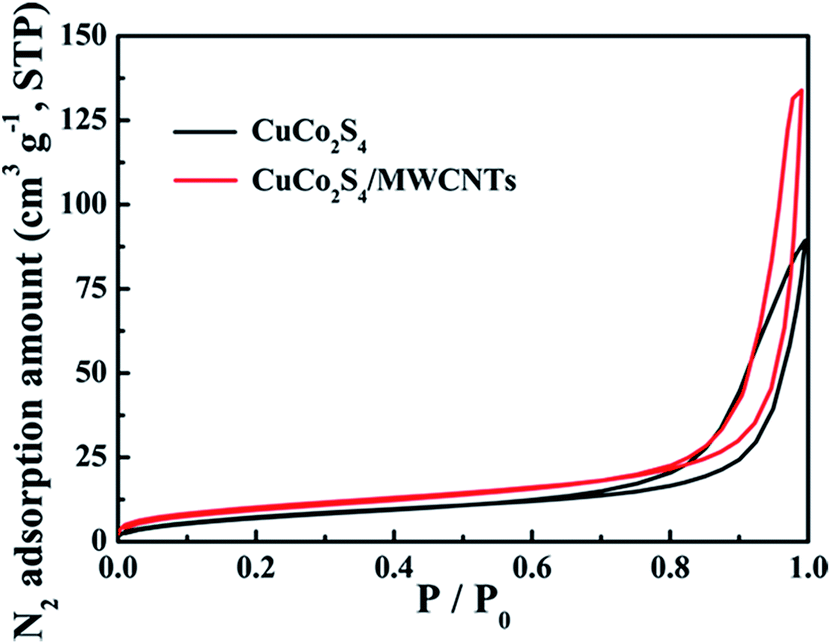 | ||
| Fig. 2 Nitrogen adsorption/desorption isotherms of CuCo2S4 and CuCo2S4/MWCNTs at 77 K. | ||
Fig. S7† shows the Raman spectra. The peaks of the MWCNTs and CuCo2S4/MWCNTs spectra can be grouped into seven bands, depending on the model proposed by Rebelo et al.29 The relative areas of satellite bands (S, Dl, Dr, Gl and Gr bands) and those of graphitic-type domain bands (D and G bands) are summed respectively and their ratio is used to reflect the chemical functionalization degree. The ratio for MWCNTs was 0.5, while for CuCo2S4/MWCNTs it was 0.7. The results show an increased structural complexity in MWCNTs which can be ascribed to the aromaticity disruption and vacancies formation.30
X-ray photoelectron spectroscopy (XPS) spectra, shown in Fig. 3, were recorded to identify the elemental chemical states and to help elucidate the mechanism for the Fenton reaction of CuCo2S4 and CuCo2S4/MWCNTs. All results were calibrated according to the peak position of C 1s, which was assumed to be at 284.8 eV. Fig. 3a is the full scan survey XPS spectrum of CuCo2S4/MWCNTs. Fig. 3b shows the C 1s spectrum of CuCo2S4/MWCNTs, which can be divided into five types of peaks, attributed to different types of carbon in MWCNTs: C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O, CO, COO and O–COO.31–33 The presence of oxygen-containing functional groups confirms the functionalization of MWCNTs, which facilitated the deposition of CuCo2S4 nanoparticles on MWCNTs. CuCo2S4 was considered to have a valence distribution of Cu1.2+(Co2.4+)2(S1.5−)4, with coexistence of Cu+, Co2+ and Co3+.34 Fig. 3c shows the Cu 2p spectra of CuCo2S4 and CuCo2S4/MWCNTs. In CuCo2S4 the doublet peaks at 932.2 eV and 952.1 eV are attributed to Cu+ 2p3/2 and 2p1/2, respectively, and in CuCo2S4/MWCNTs, the doublet peaks at 932.6 eV and 952.6 eV are attributed to Cu+ 2p3/2 and 2p1/2. Cu2+ is generally considered as nonexistent in pure CuCo2S4. However, the doublets at 934.5 eV and 954.4 eV in CuCo2S4 and the doublets at 935.1 eV and 955.1 eV in CuCo2S4/MWCNTs demonstrate the presence of Cu2+, which was caused by exposure to air.35 The peak at 944.2 eV in CuCo2S4 and the peak at 944.9 eV in CuCo2S4/MWCNTs are shake-up satellites. The Co 2p spectra of CuCo2S4 and CuCo2S4/MWCNTs are presented in Fig. 3d and can be fitted into two spin–orbit doublets and two shake-up satellites. In the Co 2p3/2 spectrum of CuCo2S4, the peaks at 778.6 eV and 780.7 eV were attributed to Co2+ and Co3+, while in the Co 2p3/2 spectrum of CuCo2S4/MWCNTs the peaks attributed to Co2+ and Co3+ appeared at 779.1 eV and 781.2 eV, respectively. Thus, all results suggest the coexistence of Co2+ and Co3+. We can see that upon incorporation of MWCNTs, both Cu 2p and Co 2p peaks shifted to higher binding energies. The shift may be attributed to the excellent dispersion of the CuCo2S4 nanoparticles on the MWCNTs and the great conductivity of MWCNTs. Therefore, the CuCo2S4 nanoparticles serve as electron donors and the electrons can be transferred to the MWCNTs.36 These cause a decrease in the density of the outer electron cloud of Cu and Co, and the increase in the binding energies of Cu 2p and Co 2p. Moreover, the surface ionic ratios of Co2+/Co3+ were 1.1
C, C–O, CO, COO and O–COO.31–33 The presence of oxygen-containing functional groups confirms the functionalization of MWCNTs, which facilitated the deposition of CuCo2S4 nanoparticles on MWCNTs. CuCo2S4 was considered to have a valence distribution of Cu1.2+(Co2.4+)2(S1.5−)4, with coexistence of Cu+, Co2+ and Co3+.34 Fig. 3c shows the Cu 2p spectra of CuCo2S4 and CuCo2S4/MWCNTs. In CuCo2S4 the doublet peaks at 932.2 eV and 952.1 eV are attributed to Cu+ 2p3/2 and 2p1/2, respectively, and in CuCo2S4/MWCNTs, the doublet peaks at 932.6 eV and 952.6 eV are attributed to Cu+ 2p3/2 and 2p1/2. Cu2+ is generally considered as nonexistent in pure CuCo2S4. However, the doublets at 934.5 eV and 954.4 eV in CuCo2S4 and the doublets at 935.1 eV and 955.1 eV in CuCo2S4/MWCNTs demonstrate the presence of Cu2+, which was caused by exposure to air.35 The peak at 944.2 eV in CuCo2S4 and the peak at 944.9 eV in CuCo2S4/MWCNTs are shake-up satellites. The Co 2p spectra of CuCo2S4 and CuCo2S4/MWCNTs are presented in Fig. 3d and can be fitted into two spin–orbit doublets and two shake-up satellites. In the Co 2p3/2 spectrum of CuCo2S4, the peaks at 778.6 eV and 780.7 eV were attributed to Co2+ and Co3+, while in the Co 2p3/2 spectrum of CuCo2S4/MWCNTs the peaks attributed to Co2+ and Co3+ appeared at 779.1 eV and 781.2 eV, respectively. Thus, all results suggest the coexistence of Co2+ and Co3+. We can see that upon incorporation of MWCNTs, both Cu 2p and Co 2p peaks shifted to higher binding energies. The shift may be attributed to the excellent dispersion of the CuCo2S4 nanoparticles on the MWCNTs and the great conductivity of MWCNTs. Therefore, the CuCo2S4 nanoparticles serve as electron donors and the electrons can be transferred to the MWCNTs.36 These cause a decrease in the density of the outer electron cloud of Cu and Co, and the increase in the binding energies of Cu 2p and Co 2p. Moreover, the surface ionic ratios of Co2+/Co3+ were 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2.5:1 in CuCo2S4 and CuCo2S4/MWCNTs, respectively. The relative increase in Co2+ content may be ascribed to electron transfer during the synthesis.37 Finally, the S 2p spectrum of CuCo2S4/MWCNTs can be divided into four peaks, indicating the complex valence state of S, as shown in Fig. S8.†
:1 and 2.5:1 in CuCo2S4 and CuCo2S4/MWCNTs, respectively. The relative increase in Co2+ content may be ascribed to electron transfer during the synthesis.37 Finally, the S 2p spectrum of CuCo2S4/MWCNTs can be divided into four peaks, indicating the complex valence state of S, as shown in Fig. S8.†
 | ||
| Fig. 3 Full survey scan XPS spectrum (a) and XPS spectrum of C (b) of CuCo2S4/MWCNTs. XPS spectra of Cu (c) and Co (d) of CuCo2S4 and CuCo2S4/MWCNTs. | ||
The Fenton-like catalytic rates of CuS, CoS, CuCo2S4 and CuCo2S4/MWCNTs are shown in Fig. 4a. CuCo2S4 and CuCo2S4/MWCNTs showed a competitive catalytic efficiency when used in small amounts. Among these catalysts, CuCo2S4/MWCNTs showed the highest catalytic performance and completed the degradation within 6 minutes. However, when pristine CuCo2S4 was used as catalyst, the degradation reaction took up to 12 min, while their corresponding monometal sulfides exhibited a poor efficiency at the specified time. Thus, it was evident that bimetal sulfide CuCo2S4 exhibits a higher Fenton-like catalytic activity than their corresponding monometal sulfides. Futhermore, the addition of MWCNTs further enhanced the catalytic rate. The high catalytic rate can be mainly attributed to the increased surface area and improved charge transfer in the composites.
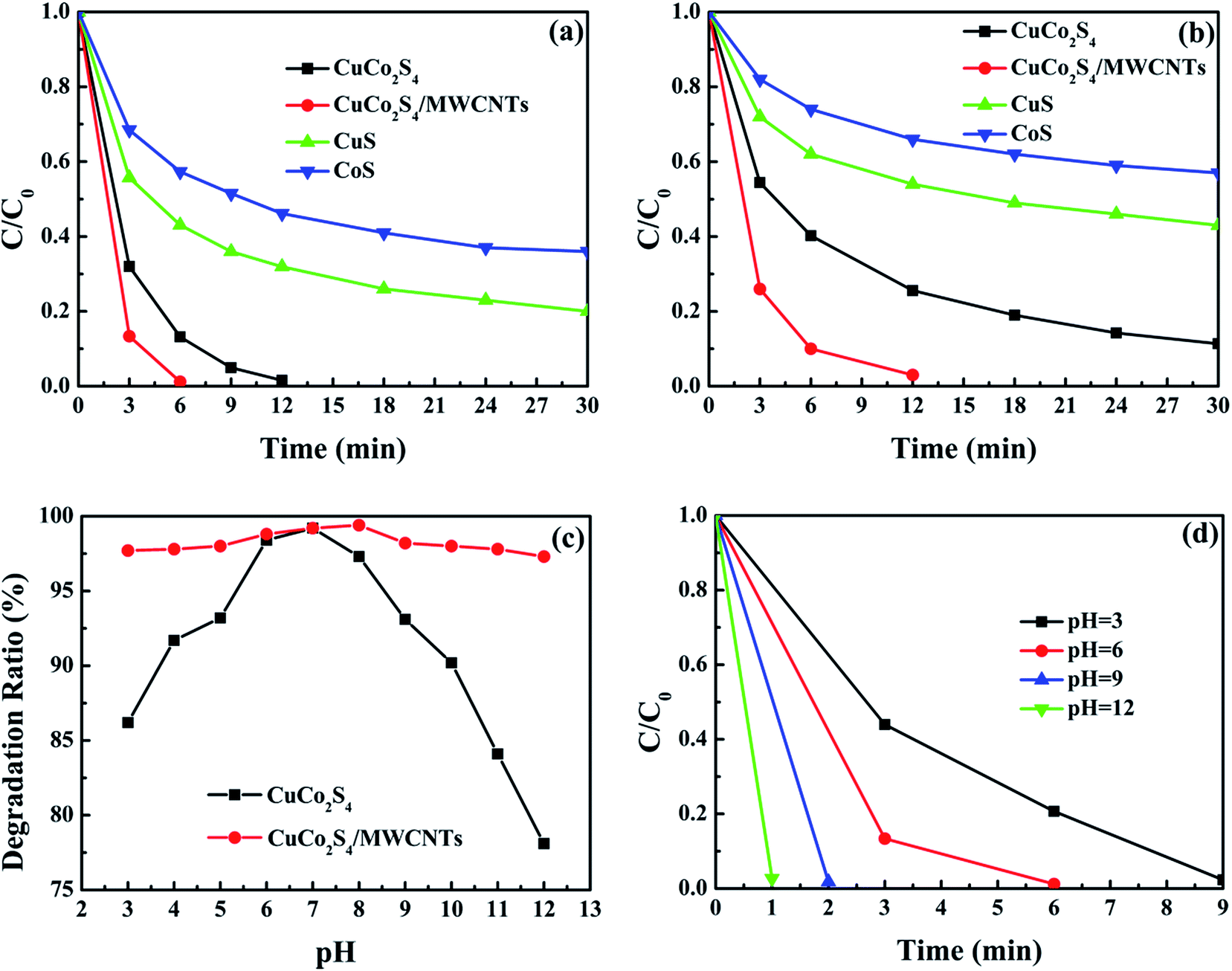 | ||
| Fig. 4 (a) MB degradation rate using CuS, CoS, CuCo2S4 and CuCo2S4/MWCNTs as catalysts. Conditions: original concentration of MB = 20.0 mg l−1, volume of MB solution = 30.0 ml, additional H2O2 = 1.0 ml, catalyst amount = 10.0 mg, room temperature, initial pH = 6. (b) MB degradation rate at high concentration using CuS, CoS, CuCo2S4 and CuCo2S4/MWCNTs as catalysts. Conditions: original concentration of MB = 50.0 mg l−1, volume of MB solution = 30.0 ml, additional H2O2 = 1.0 ml, catalyst amount = 10.0 mg, room temperature, initial pH = 6. (c) MB degradation efficiency using CuCo2S4 and CuCo2S4/MWCNTs at different initial pHs during 20 minutes and (d) MB degradation rate using CuCo2S4/MWCNTs at different initial pHs. Conditions: original concentration of MB = 20.0 mg l−1, volume of MB solution = 30.0 ml, additional H2O2 = 1.0 ml, catalyst amount = 10.0 mg, room temperature. | ||
Catalytic performance was also tested under a different initial dye concentration (50 mg l−1) with the same amount of catalyst. As shown in Fig. 4b, CuCo2S4/MWCNTs degraded 97.0% of MB within 12 min, whereas after the same time, CuCo2S4 degraded only 74.4%. Photographs of the MB solution are shown in Fig. S9.† The results demonstrate that the addition of MWCNTs improved the degradation rate to a larger extent. The catalytic rates of our samples (no. 1–4) and relevant Fenton-like catalysts reported in the literature, namely, CuS (no. 5–7), Co3O4 (no. 6), iron-based bimetal composites (no. 9–16) and CNTs-based composites (no. 17–19) are listed in Table S1.† It is noted that the as-prepared CuCo2S4/MWCNT composite stands out as an efficient catalyst owing to the combination of low catalytic dosage, convenient application method, high catalytic rate and highly efficient degradation of MB. The catalytic performance of CuCo2S4/MWCNTs is superior to that of monometal sulfides CuS and Co3O4, and is comparable to that of iron-based bimetal composites. Additionally, compared with CNT-based composites, our samples can be prepared by a simple synthetic procedure, and exhibit an uniform morphology and high catalytic activity.
Furthermore, a series of degradation experiments were conducted to investigate the catalytic performance of CuCo2S4 and CuCo2S4/MWCNTs at different initial pHs. Before adding H2O2, the mixtures of MB solution and catalysts were adjusted to a pH value of 3–12. By contrast to iron-based catalysts, which are restricted to a certain pH range, the iron-free CuCo2S4 and CuCo2S4/MWCNTs catalysts worked over a broad pH range and their MB degradation efficiency exceeded 75.0% within 20 minutes, as shown in Fig. 4c. For CuCo2S4, Cu+/H2O2 system is stable in acidic and neutral conditions, whereas the Fe2+/H2O2 system is stable only at pH ≤ 4; moreover, the Co2+/H2O2 system exhibits the similar or even higher catalytic activity under alkaline conditions.38 Therefore, the combination of copper and cobalt results in a high tolerance of CuCo2S4 towards a broad pH range. In addition, CuCo2S4 showed better performance in a neutral pH range. The declining catalytic activity under acidic/alkaline conditions was ascribed to the relatively inferior stability of CuCo2S4 at extreme pHs. The improved performance of CuCo2S4/MWCNTs compared to pristine CuCo2S4 at extreme pHs was attributed to the additional active sites of the functionalized MWCNTs and their stable tailored structure. Fig. S10† shows the catalytic rate of CuCo2S4 and Fig. 4d exhibits the MB degradation rate using CuCo2S4/MWCNTs at different pHs. It can be seen that CuCo2S4/MWCNTs performed better at alkaline pHs than at acidic pHs. The pH-dependent activity may be due to the cationic character of the MB molecules and the catalytic properties of CuCo2S4.39 Firstly, the pH of the solution can change the surface charge in CuCo2S4. When the pH value exceeds pHpzc (the point of zero charge), the surface charge of CuCo2S4 becomes negative and larger amounts of cationic MB molecules can be adsorbed on the catalysts. Besides, in acidic and near-neutral conditions a portion of Cu+ ions are oxidized to Cu2+ by molecular oxygen and cannot generate ·OH radicals. It is worth mentioning that the degradation efficiency of CuS/graphene under acidic conditions is much lower than under neutral conditions, as described in the literature.40 Hence, the high efficiency of CuCo2S4 and CuCo2S4/MWCNTs under acidic conditions can be attributed to the synergistic effect of copper and cobalt. In conclusion, the results demonstrate that, unlike iron-based catalysts, the iron-free bimetal CuCo2S4 and CuCo2S4/MWCNTs catalysts can be directly used in wastewater treatment without any pH limitations.
The reusability of CuCo2S4 and CuCo2S4/MWCNTs was evaluated and the results are shown in Fig. 5. After 3 cycles of reactions, the catalytic activity of CuCo2S4 decreased and 60.5% of MB was degraded after 20 minutes. An improvement in the reusability could be observed when MWCNTs were introduced. The results show that CuCo2S4/MWCNTs maintained a high removal efficiency better than CuCo2S4, and 98.0% of MB could be degraded by CuCo2S4/MWCNTs in the second cycle, and 84.8% in the third cycle. The results further verify the strong anchoring of CuCo2S4 in CuCo2S4/MWCNTs, which avoids the aggregation of CuCo2S4 upon reuse, hence strengthening the stability of the CuCo2S4/MWCNTs catalyst in the Fenton reaction. XRD experiments of CuCo2S4/MWCNTs after 1 and 2 cycles were conducted to study the predominant effects of the catalytic reaction after different cycles, and the results are shown in Fig. S11.† We can see that the XRD pattern of CuCo2S4/MWCNTs after 1 cycle displays both the peaks of CuCo2S4 and weak peaks of MWCNTs. Thus, the XRD spectrum reveals that the CuCo2S4/MWCNTs catalyst is stable, and only a small part of CuCo2S4 is lost after the first cycle. After the second cycle, some peaks attributed to impurities appeared; this was due to the dissolution of cobalt in the reaction media. The losing of cobalt ions leads to the decrease in catalytic activity in the third cycle. The results are confirmed by energy dispersive spectrometry (EDS, shown in Fig. S12 and Table S2†).
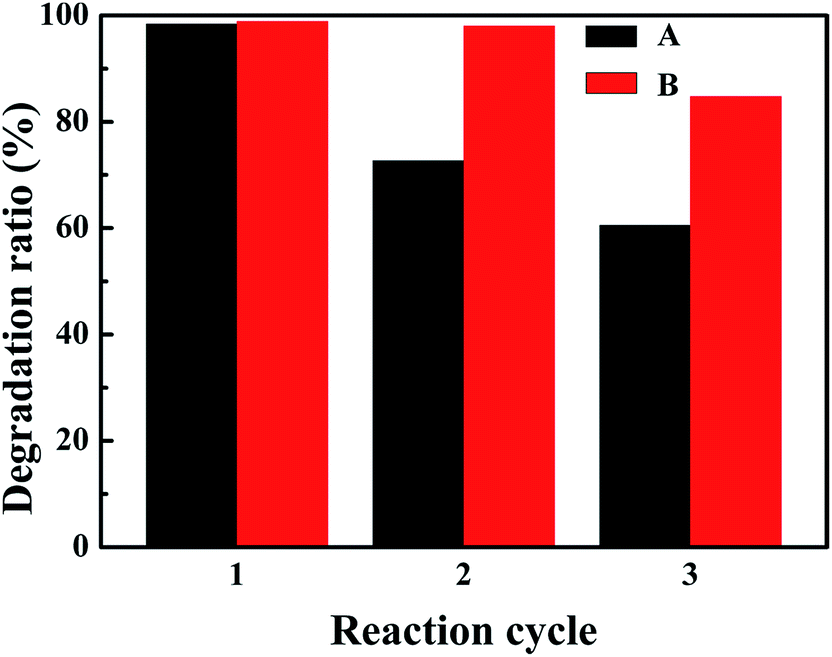 | ||
| Fig. 5 Reusability study of CuCo2S4 (A) and CuCo2S4/MWCNTs (B) for the MB degradation. Conditions: initial concentration of MB = 20.0 mg l−1, volume of MB solution = 30.0 ml, added H2O2 = 1.0 ml, catalyst amount = 10.0 mg, room temperature, initial pH = 6, degradation time = 20 min. | ||
The catalytic process of CuCo2S4/MWCNTs can be described as follows: before the Fenton reaction begins, large amounts of MB molecules are adsorbed by CuCo2S4/MWCNTs; upon addition of H2O2, H2O2 molecules are readily adsorbed, and the catalyzed reaction occurs on the CuCo2S4 nanoparticle surface and MWCNTs to generate ·OH, which will attack MB molecules, either adsorbed or in solution; as the adsorbed MB molecules are degraded, the free MB molecules in solution are adsorbed and then decomposed until the degradation process is completed.
To further understand the reasons for the enhanced catalytic activity, we first considered the catalytic mechanism of CuCo2S4 individually. The CuCo2S4/H2O2 system, formed on the surface of CuCo2S4, is a combination of the Cu+/H2O2, Co2+/H2O2 and Cu+/Co3+ redox systems. The Cu+/H2O2 reaction systems are similar to Fe2+/H2O2 (eqn (1) and (2)):40
| Cu+ + H2O2 → Cu2+ + OH− + ·OH | (1) |
| Cu2+ + H2O2 → H+ + ·O2H + Cu+ | (2) |
The proposed mechanisms for Co2+/H2O2 are shown below (eqn (3) and (4)).41–43
| Co2+ + H2O2 → Co3+ + OH− + ·OH | (3) |
| Co3+ + H2O2 → Co2+ + ·O2H + H+ | (4) |
In combination with the experimental results, the better performance of CuCo2S4 compared to monometal sulfides can be ascribed to the following reasons:
(i) Co3+ ions are thermodynamically susceptible to be reduced by Cu+ ions (eqn (5)). The redox reaction of Cu+/Co3+ will accelerate the redox reactions of both Cu+/Cu2+ and Co2+/Co3+, thus promoting the overall Fenton reaction cycle, and consequently, improving the catalytic rate.
| Cu+ + Co3+ → Cu2+ + Co2+ | (5) |
(ii) The operating pH range of the iron-free Fenton-like Cu+/H2O2 and Co2+/H2O2 systems are both broader than that of Fe2+/H2O2. The stability of CuCo2S4 at extreme pHs and the combination of the Cu+/H2O2 and Co2+/H2O2 systems result in a broader workable pH range.
(iii) CuCo2S4 has good electrical conductivity and hence electrons can be readily transfer to the MWCNTs.44
Thus, the function of MWCNTs in CuCo2S4/MWCNTs is concluded as follows:
(i) MWCNTs significantly tuned the nanostructure of CuCo2S4/MWCNTs. Firstly, as the XRD results showed, the MWCNTs provide nucleation sites such that the CuCo2S4 nanoparticles on MWCNTs exhibit a higher purity and crystallinity than pristine CuCo2S4. Secondly, the CuCo2S4 nanoparticles are loaded on the MWCNTs uniformly so aggregation of CuCo2S4 nanoparticles can be inhibited. Furthermore, the size of the CuCo2S4 nanoparticles was controlled by MWCNTs. Thirdly, the larger surface area of CuCo2S4/MWCNTs compared to pristine CuCo2S4 helps to adsorb a larger amount of MB and H2O2 molecules. The adsorbed H2O2 can be catalyzed quickly by the active sites on the surface of CuCo2S4/MWCNTs, and the generated ·OH radicals can immediately attack the nearby adsorbed MB molecules.45 Fourthly, CuCo2S4/MWCNTs forms a more stable structure than CuCo2S4. MWCNTs act as a matrix such that CuCo2S4 nanoparticles are not easily leaked. The stable structure helps CuCo2S4/MWCNTs to operate well at extreme pHs or after several reaction cycles.46
(ii) Electrons from the CuCo2S4 nanoparticles can move to MWCNTs, due to the good conductivity of MWCNTs, which will accelerate electron transfer, promote the redox rate of the Fenton reaction cycle and further enhance the catalytic rate compared to CuCo2S4.47 It is notable that a larger ratio of Co2+/Co3+ in CuCo2S4/MWCNTs than in pure CuCo2S4 is conducive to the initiation of the catalytic reaction, and meanwhile, it helps to economize H2O2 for fewer Co3+ are needed to be reduced at the beginning.
(iii) The defects in MWCNTs are deemed as active sites to produce ·OH, which further enhance catalytic efficiency. The diffused MB and H2O2 molecules can be trapped by the incomplete dangling bonds and the strongly distorted π-electron system around the defects.48 Additionally, defects with an increased electron density can serve as reducing active sites to reduce H2O2 and produce ·OH.24
A scheme with the mechanism of the enhanced catalytic process is shown in Fig. 6.
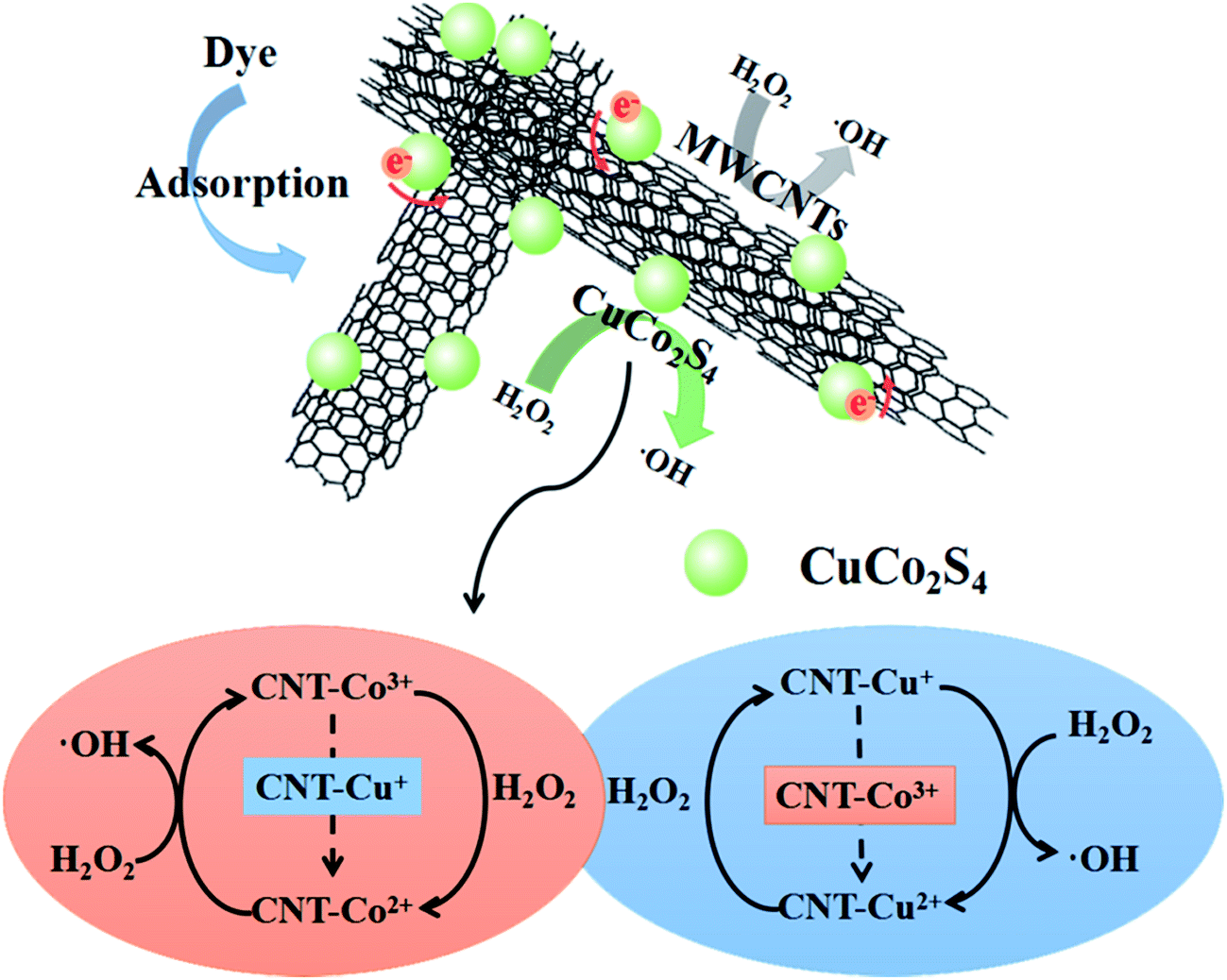 | ||
| Fig. 6 Scheme illustrating the mechanism of the enhanced catalytic process of CuCo2S4/MWCNTs. | ||
4. Conclusions
In summary, a CuCo2S4/MWCNT composite was synthesized as a heterogeneous Fenton-like catalyst by a mild two-step hydrothermal method. The catalyst CuCo2S4 exhibited a higher catalytic rate than CuS and CoS. Moreover, the iron-free catalysts CuCo2S4 exhibited good performance over a broader pH range of 3–12. MWCNTs provide stable frameworks where CuCo2S4 nanoparticles could be anchored and strongly bonded, favoring the uniform growth of the nanoparticles. As a result, the addition of MWCNTs enhanced the catalytic rate and the catalytic performance at extreme pHs, and improved the reusability. Furthermore, the mechanism for the enhanced catalytic process with CuCo2S4/MWCNTs was discussed. In conclusion, the CuCo2S4/MWCNT composite is a promising Fenton-like catalyst for wastewater containing dyes.Acknowledgements
This study was supported by the National Natural Science Foundation of China (21273236) and the Science and Technology Planning Projects of the Fujian Province, China (2014H2008 and 2015I0008) and STS Project of Fujian-CAS (2016T3036).Notes and references
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- K. Lutchmiah, A. R. D. Verliefde, K. Roest, L. C. Rietveld and E. R. Cornelissen, Water Res., 2014, 58, 179–197 CrossRef CAS PubMed.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed.
- B. Guieysse and Z. N. Norvill, J. Hazard. Mater., 2014, 267, 142–152 CrossRef CAS PubMed.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- V. Nicolini, E. Gambuzzi, G. Malavasi, L. Menabue, M. C. Menziani, G. Lusvardi, A. Pedone, F. Benedetti, P. Luches, S. D'Addato and S. Valeri, J. Phys. Chem. B, 2015, 119, 4009–4019 CrossRef CAS PubMed.
- E. Contreras, J. Urra, C. Vásquez and C. Palma, Biotechnol. Prog., 2012, 28, 114–120 CrossRef CAS PubMed.
- F. Chen, X. Zhao, H. Liu and J. Qu, Appl. Catal., B, 2014, 158–159, 85–90 CrossRef CAS.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, RSC Adv., 2015, 5, 7628–7636 RSC.
- F. M. Duarte, F. J. Maldonado-Hódar and L. M. Madeira, Appl. Catal., A, 2013, 458, 39–47 CrossRef CAS.
- J. Y. T. Chan, S. Y. Ang, E. Y. Ye, M. Sullivan, J. Zhang and M. Lin, Phys. Chem. Chem. Phys., 2015, 17, 25333–25341 RSC.
- F. Xiao, W. Li, L. Fang and D. Wang, J. Hazard. Mater., 2016, 308, 11–20 CrossRef CAS PubMed.
- V. Kavitha and K. Palanivelu, Int. J. Environ. Sci. Technol., 2016, 13, 927–936 CrossRef CAS.
- H. Chen, Z. Zhang, Z. Yang, Q. Yang, B. Li and Z. Bai, Chem. Eng. J., 2015, 273, 481–489 CrossRef CAS.
- D. Wu, Y. Chen, Y. Zhang, Y. Feng and K. Shih, Sep. Purif. Technol., 2015, 154, 60–67 CrossRef CAS.
- Y. Wang, L. Zhang, H. Jiu, N. Li and Y. Sun, Appl. Surf. Sci., 2014, 303, 54–60 CrossRef CAS.
- Y. Wang, H. Zhao, M. Li, J. Fan and G. Zhao, Appl. Catal., B, 2014, 147, 534–545 CrossRef CAS.
- Y. Du, W. Ma, P. Liu, B. Zou and J. Ma, J. Hazard. Mater., 2016, 308, 58–66 CrossRef CAS PubMed.
- G. Nie, Z. Li, X. Lu, J. Lei, C. Zhang and C. Wang, Appl. Surf. Sci., 2013, 284, 595–600 CrossRef CAS.
- Y.-P. Zhu, T.-Z. Ren and Z.-Y. Yuan, Nanoscale, 2014, 6, 11395–11402 RSC.
- J. Tang, Y. Ge, J. Shen and M. Ye, Chem. Commun., 2016, 52, 1509–1512 RSC.
- V. Cleveland, J.-P. Bingham and E. Kan, Sep. Purif. Technol., 2014, 133, 388–395 CrossRef CAS.
- W. Li, T. Sun and F. Li, Ind. Eng. Chem. Res., 2014, 53, 18095–18103 CrossRef CAS.
- R. S. Ribeiro, A. M. T. Silva, J. L. Figueiredo, J. L. Faria and H. T. Gomes, Appl. Catal., B, 2016, 187, 428–460 CrossRef CAS.
- J. Ma, M. Yang, F. Yu and J. Chen, J. Colloid Interface Sci., 2015, 444, 24–32 CrossRef CAS PubMed.
- H. Wang, H. Jiang, S. Wang, W. Shi, J. He, H. Liu and Y. Huang, RSC Adv., 2014, 4, 45809–45815 RSC.
- J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, Y.-S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253–1256 CrossRef CAS PubMed.
- P.-X. Hou, C. Liu and H.-M. Cheng, Carbon, 2008, 46, 2003–2025 CrossRef CAS.
- S. L. H. Rebelo, A. Guedes, M. E. Szefczyk, A. M. Pereira, J. P. Araujo and C. Freire, Phys. Chem. Chem. Phys., 2016, 18, 12784–12796 RSC.
- D. Hines, M. H. Rummeli, D. Adebimpe and D. L. Akins, Chem. Commun., 2014, 50, 11568–11571 RSC.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- Y. Wei, X. Ling, L. Zou, D. Lai, H. Lu and Y. Xu, Colloids Surf., A, 2015, 482, 507–513 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS.
- R. A. D. Pattrick, V. S. Coker, C. I. Pearce, N. D. Telling and G. van der Laan, Can. Mineral., 2008, 46, 1317–1322 CrossRef CAS.
- A. N. Buckley, W. M. Skinner, S. L. Harmer, A. Pring and L.-J. Fan, Geochim. Cosmochim. Acta, 2009, 73, 4452–4467 CrossRef CAS.
- Y. Fan, D. Han, B. Cai, W. Ma, M. Javed, S. Gan, T. Wu, M. Siddiq, X. Dong and L. Niu, J. Mater. Chem. A, 2014, 2, 13565–13570 CAS.
- B. Cai, X. Lv, S. Gan, M. Zhou, W. Ma, T. Wu, F. Li, D. Han and L. Niu, Nanoscale, 2013, 5, 1910–1916 RSC.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- Y.-R. Zhang, S.-Q. Wang, S.-L. Shen and B.-X. Zhao, Chem. Eng. J., 2013, 233, 258–264 CrossRef CAS.
- J. Qian, K. Wang, Q. Guan, H. Li, H. Xu, Q. Liu, W. Liu and B. Qiu, Appl. Surf. Sci., 2014, 288, 633–640 CrossRef CAS.
- T. Warang, N. Patel, R. Fernandes, N. Bazzanella and A. Miotello, Appl. Catal., B, 2013, 132–133, 204–211 CrossRef CAS.
- C. Deng, X. Ge, H. Hu, L. Yao, C. Han and D. Zhao, CrystEngComm, 2014, 16, 2738–2745 RSC.
- Y. Yavuz, A. S. Koparal, A. Artık and Ü. B. Öğütveren, Desalination, 2009, 249, 828–831 CrossRef CAS.
- L.-l. Liu, K. P. Annamalai and Y.-s. Tao, New Carbon Mater., 2016, 31, 336–342 CrossRef.
- J. R. Kim and E. Kan, J. Ind. Eng. Chem., 2015, 21, 644–652 CrossRef CAS.
- J. Xu, X. Liu, G. V. Lowry, Z. Cao, H. Zhao, J. L. Zhou and X. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 7333–7342 CAS.
- J. Wang, Y. Lin, M. Pinault, A. Filoramo, M. Fabert, B. Ratier, J. Bouclé and N. Herlin-Boime, ACS Appl. Mater. Interfaces, 2015, 7, 51–56 Search PubMed.
- J. A. Rodríguez-Manzo, O. Cretu and F. Banhart, ACS Nano, 2010, 4, 3422–3428 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01269a |
| This journal is © The Royal Society of Chemistry 2017 |