
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of 1,3-diyne compounds as novel and potent antidepressant agents: synthesis, cell-based assay and behavioral studies†
Kai-Qing Ma *a,
Yan-Hong Miaoab,
Xiao Liab,
Yu-Zhi Zhoua,
Xiao-Xia Gaoa,
Xiang Zhangd,
Jian-Bin Chaoc and
Xue-Mei Qin*a
*a,
Yan-Hong Miaoab,
Xiao Liab,
Yu-Zhi Zhoua,
Xiao-Xia Gaoa,
Xiang Zhangd,
Jian-Bin Chaoc and
Xue-Mei Qin*a
aModern Research Center for Traditional Chinese Medicine, Shanxi University, Taiyuan 030006, P. R. China. E-mail: makaiqing@sxu.edu.cn
bCollege of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, P. R. China
cScientific Instrument Center, Shanxi University, Taiyuan 030006, P. R. China
dDepartment of Chemistry, University of Louisville, Louisville, KY 40208, USA
First published on 13th March 2017
Abstract
Sixteen 1,3-diynes compounds were designed and synthesized. Their protective effects on corticosterone-injured PC12 cells were evaluated. Among them, five compounds (7a, 7c, 7d, 11, 12) displayed significant protective activity at 10 μmol. Moreover, the protective activity of compound 7a was proved to be associated with the regulation of the apoptosis related proteins. The mice forced swim test showed that 7a had a concentration-dependent antidepressant-like effect. Overall, our findings support that compound 7a is a promising lead candidate that deserves further evaluation in the design of antidepressants.
1. Introduction
Natural product structures remain a rich source of inspiration in the discovery and development of novel small molecule modulators of bioactivity for chemical biology and medicinal chemistry research.1,2 1,3-Diynes are common functionalities in the skeletons of polyacetylenes natural products, such as panaxytriol (1) and falcarinol (2) (Fig. 1).3 The diverse structures of these compounds, along with their interesting biological activities, which include anti-tumor,4 anti-obesity,5 anti-diabetic,6 immunosuppressive7 and neurotoxicity effects8 provide compelling reasons for the considerable interest of researchers in this class of natural products.9 In our previous study, bupleurynol (3), RB-2 (4) and their isomers were isolated from Radix Bupleuri (RB). Furthermore, the polyacetylenes natural products appeared to be the key components responsible for antidepressant activity, which was determined primarily through pharmacokinetic studies.10 However, to the best of our knowledge, structure–activity relationships (SARs) related to their antidepressant effects have not been studied due to the low abundance of these natural products.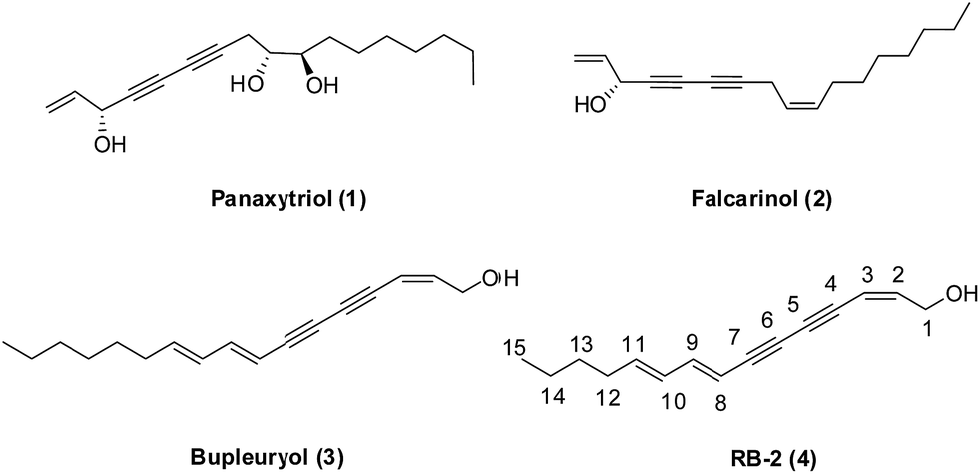 | ||
| Fig. 1 The structures of several polyacetylenes natural products. | ||
Depression is a common psychiatric disorder, although the pathogenesis of depression is not well understood.11 Up until now, antidepressants have functioned by increasing the levels of monoamine neurotransmitters (serotonin (5-HT), norepinephrine (NE) and dopamine (DA)) in the brain.12
However, treatment with these antidepressants shows modest efficacy and the majority of patients fail to achieve full remission.13 Therefore, the discovery of new scaffolds with novel mechanisms of action as antidepressants has recently garnered significant interest.14
The PC12 cell line is derived from a pheochromocytoma of the rat adrenal medulla and is widely employed as a model system to investigate a variety of neuronal functions.15 It has been demonstrated that different antidepressants drugs are capable of exhibiting protective effects against cytotoxicity in PC12 cells.16,17 Hence, corticosterone-induced cytotoxicity in PC12 cells has been widely employed as a rapid in vitro screening tool for a primary assessment of neuroprotective activity.18,19
In a continuation of our efforts to explore the antidepressant activity of polyacetylenes natural products,10,20 a novel series of compounds bearing the 1,3-diyne scaffold were designed and synthesized by using a convenient procedure. The anti depression activity of these compounds were subsequently evaluated using corticosterone-injured PC12 cells, and we found that five compounds possess high neuroprotective activity. To elucidate the mechanism of this neuroprotective activity, the effect of compound 7a on the expressions of apoptosis-related proteins (Bax, Bcl-2, Cyt-C and caspase-3) was investigated. Finally, we also assessed the effects of compound 7a on mice through the forced swim tests (FST) to verify its antidepressant properties.
2. Results and discussion
2.1 Chemistry
The key intermediate, compound 5, was prepared as previously reported.20 The final products (7a–7j) were prepared from the coupling of compound 5 and several different terminal alkynes using the Cadiot–Chodkiewicz reaction and subsequent removal of the TBDPS group (Scheme 1).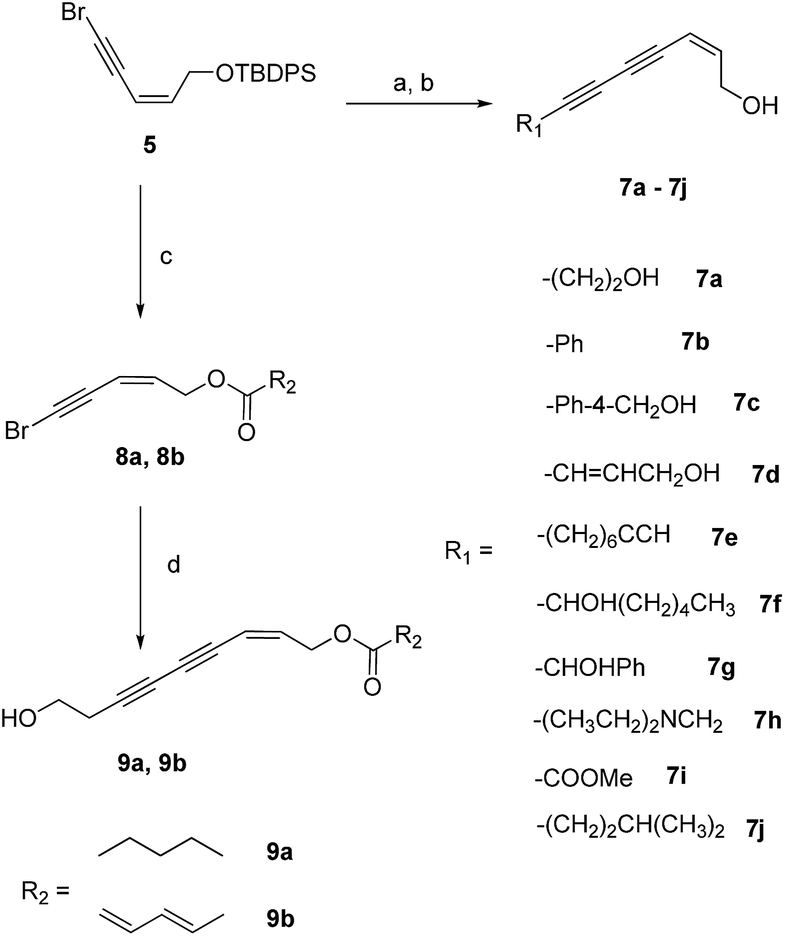 | ||
Scheme 1 Synthetic route of compounds 7a–7j and 9a, 9b. Reagent and conditions: (a) CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR, CuI, piperidine, rt, 3 h, 40–71% or CHCR, CuI, Pd(PPh3)4, i-Pr2NH, THF, rt, 3 h, 44–52%; (b) Et3N·3HF, THF, rt, overnight, 49–56% or TBAF, THF, 1.5 h, 55%; (c) Et3N·3HF, THF, rt, overnight; RCOOH, DMAP, DCC, DCM, 5 h, rt, 40–41% (two steps); (d) but-3-yn-1-ol (10), CuI, Pd(PPh3)4, i-Pr2NH, THF, rt, 3 h, 58–59%. CR, CuI, piperidine, rt, 3 h, 40–71% or CHCR, CuI, Pd(PPh3)4, i-Pr2NH, THF, rt, 3 h, 44–52%; (b) Et3N·3HF, THF, rt, overnight, 49–56% or TBAF, THF, 1.5 h, 55%; (c) Et3N·3HF, THF, rt, overnight; RCOOH, DMAP, DCC, DCM, 5 h, rt, 40–41% (two steps); (d) but-3-yn-1-ol (10), CuI, Pd(PPh3)4, i-Pr2NH, THF, rt, 3 h, 58–59%. | ||
The structure–activity relationships (SARs) of compound 7a were explored by decoration of the 1-OH group with different ester groups. As illustrated in Scheme 1, compound 5 was deprotected to give the free alcohol. The crude product reacted with hexanoic acid or (2E,4E)-hexa-2,4-dienoic acid to afford compounds 8a or 8b, respectively. The final compounds (9a and 9b) were obtained from 8a or 8b via the Cadiot–Chodkiewicz coupling reaction with 3-butyn-1-ol (10).
Additionally, the SARs of 7a were explored by the decoration of the 9-OH group with different ester groups and alkyl groups. Compound 6 reacted with either dimethoxymethane, acetic anhydride or methyl iodide to give the corresponding ether or ester, which was deprotected to give target molecules 11, 12 and 13, respectively (Scheme 2). Meanwhile, an enyne group was chosen to replace the 1,3-diyne group to furnish derivative 16 (Scheme 3). Intermediate 15 was prepared from 3-butyn-1-ol (10) through hydrozirconation–iodination followed by Sonogashira cross coupling.21 The TBDPS group was removed to give compound 16.
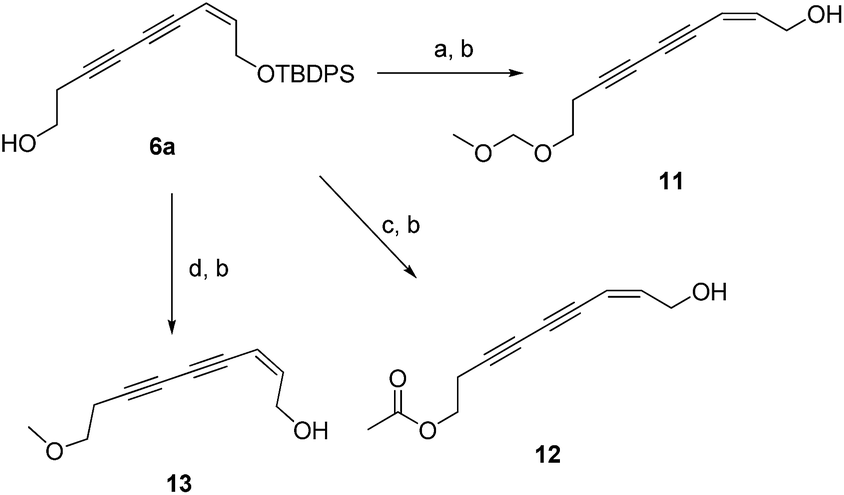 | ||
| Scheme 2 Synthetic route of compounds 11–13. Reagent and conditions: (a) dimethoxymethane, DCM, P2O5, 16 h, 54%; (b) Et3N·3HF, THF, rt, overnight, 43–59%; (c) acetic anhydride, Et3N, DMAP, DCM, 43%; (d) CH3I, NaH, THF, rt, 3 h, 46%. | ||
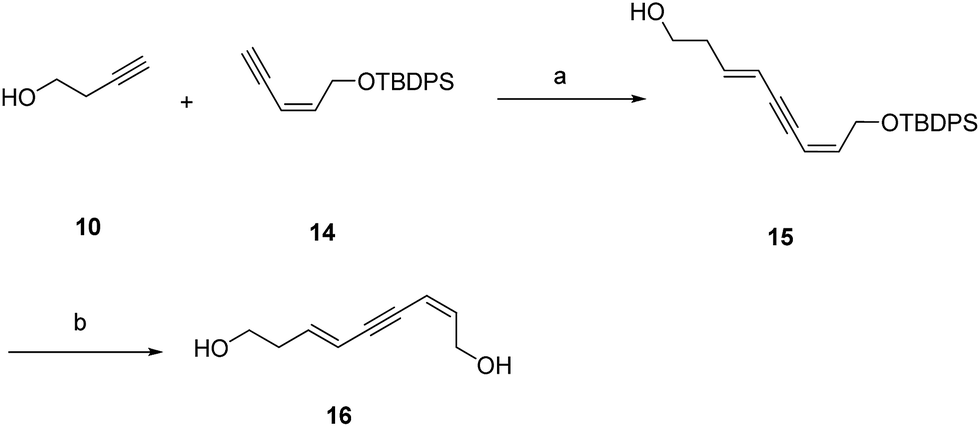 | ||
| Scheme 3 Synthetic route of compound 16. Reagent and conditions: (a) ZrCp2Cl2, AlH(i-Bu)2, I2, THF, overnight, PdCl2(PPh3)2, CuI, i-Pr2NH, THF, rt, 12 h, two steps 41%; (b) Et3N·3HF, THF, rt, 44%. | ||
2.2 Protective activities on the corticosterone-injured PC12 cells
Preliminary screening of the synthesized compounds and natural product RB-2 was performed to evaluate their protective activities in corticosterone-injured PC12 cells. The protection rates of the tested compound were calculated17 (Table 1).| Compound | PRa (%) | ||
|---|---|---|---|
| 10 μM | 20 μM | 50 μM | |
| a PR is the protection rate of the tested compound obtained from five independent experiments measured at 48 h after treatment with the test compound at three different concentrations (10 μM, 20 μM and 50 μM). PR = (Ad − Ac)/Ac × 100% ± S.D., where Ac represents the mean absorbance value of five independent experiments of the control group treated only with corticosterone, Ad indicate the mean absorbance value of five independent experiments of the test group treated with corticosterone and the test drug. S.D. represents the standard deviation. Data are presented as mean ± S.D. ‘—’ represents compounds did not exhibit protection effects on the corticosterone-injured PC12 cells. | |||
| 2 | — | — | — |
| 7a | 13.1 ± 2.9 | 17.8 ± 3.6 | 9.7 ± 3.4 |
| 7b | — | — | — |
| 7c | 13.9 ± 2.0 | — | — |
| 7d | 14.5 ± 2.4 | — | — |
| 7e | — | — | — |
| 7f | — | — | — |
| 7g | 3.9 ± 2.7 | — | — |
| 7h | — | 4.6 ± 3.9 | — |
| 10a | — | — | — |
| 10b | — | — | — |
| 11 | 13.4 ± 1.9 | 19.6 ± 2.3 | — |
| 12 | 11.3 ± 1.6 | 16.7 ± 2.0 | — |
| 13 | — | — | — |
| 7i | — | — | — |
| 7j | — | — | — |
| 16 | 6.2 ± 2.1 | 6.4 ± 2.6 | 3.8 ± 2.1 |
| 17 | — | — | — |
| Ago. | 17.4 ± 3.6 | 22.5 ± 5.5 | — |
Unfortunately, RB-2 did not show any protective effects on the corticosterone-injured PC12 cells. However, five analogues (7a, 7c, 7d, 11, 12) demonstrated potent protective effects on the PC12 cells at a concentration of 10 μM, which was equivalent to the protective effects of the positive control (agomelatine: 17.4 ± 3.6). Replacement of the diene moiety of RB-2 with an alkyl primary alcohol (7a) or allyl alcohol (7d) moiety showed promising and significant protective effects at 10 μM, while compounds with the secondary alcohol functional group displayed either no or weak protective effects (7f and 7g). Replacement of the diene unit with phenyl and alkyl groups did not show any protective effects (7b, 7j and 7e), while compound 7c, which featured a benzyl alcohol, showed protective effects at 10 μM. Incorporation of the di-substituted amino group into the structure afforded compound 6h, which displayed weak protective activity at 20 μM. Conversion of the 1-OH group of 7a to the corresponding ester removed the protective effects (9a and 9b), demonstrating that the 1-OH group was necessary for the protective activity. When the 9-OH group was converted to either corresponding ethyl ester or methoxymethyl ether, the protective effect was retained. However, the methyl ether showed no protective effect. When the 1,3-diyne units were replaced with an enyne group (16), the protective effects were decreased, which indicated that the 1,3-diyne moiety was important. Hexa-2,4-diyne-1,6-diol (17), which was commercial available, was also selected to evaluate its protective effects. The results suggested that it displayed no protective effect. Comparing its structure to that of compound 7a suggested that the vinyl alcohol was crucial to the activity.
2.3 The effects of compound 7a on the expressions of apoptosis-related proteins
To determine whether these 1,3-diyne compounds exerted their protective effects on inhibiting the apoptosis of the PC12 cells, the expression of apoptosis-related proteins in PC12 cells in response to corticosterone and treatment with compound 7a were assessed. When PC12 cells were treated with corticosterone alone, the results showed that the expression of the pro-apoptotic proteins Bax, caspase-3 and cytochrome c were up-regulated, while the expression of the anti-apoptotic Bcl-2 protein was simultaneously down-regulated (Fig. 2).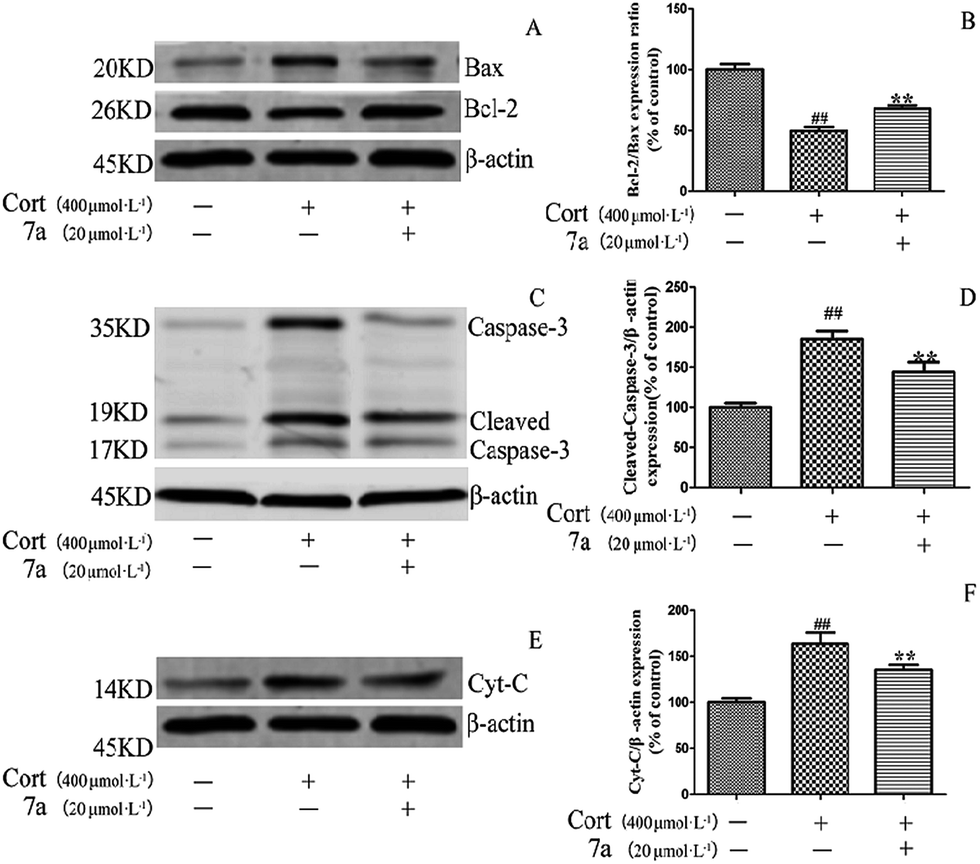 | ||
| Fig. 2 Effect of compound 7a on the expression levels of Bcl-2, Bax, Caspase-3 and Cyt-C in PC12 cells. PC12 cells were preincubated with 7a before treatment with 400 μmol L−1 corticosterone for 24 h. Data are expressed as percentage of the control, and the results are expressed as the means ± SD (n = 5). **P < 0.01 vs. corticosterone group; ##P < 0.01 vs. control group. Cort: corticosterone. (A/B) The expression of Bcl-2 and Bax. (C/D) The expression of caspase-3. (E/F) The expression of Cyt-C. | ||
These results indicated that corticosterone had induced apoptosis in the PC12 cells. However, when compound 7a was added to the corticosterone-treated group, expression of the Bcl-2 protein increased remarkably, while the expression of Bax, caspase-3 and cytochrome c proteins decreased significantly compared to the group treated only with corticosterone. These results suggested that the alteration of the expression of the apoptosis-related proteins contributed to the protective effect of compound 7a on corticosterone-induced apoptosis in PC12 cells.
2.4 The behavioral studies
We then investigated whether the excellent potency of the 1,3-diyne analogues in PC12 cells translate into antidepressant-like efficacy in a behavioral model. The effects of compound 7a was investigated in a mouse forced swim test, an assay in which mice are placed into a beaker of water and the time mouse spends passively floating in the water (immobility) is recorded. Most traditional antidepressants decrease the amount of time the mice spend immobile. Ten mice were administered intragastrically with venlafaxine as a positive control (50 mg kg−1). Compound 7a demonstrated a antidepressant-like effect in a concentration-dependent manner when administered intraperitoneally, with a significant reduction in immobility at the minimal dose of 0.0125 mg kg−1 (Fig. 3).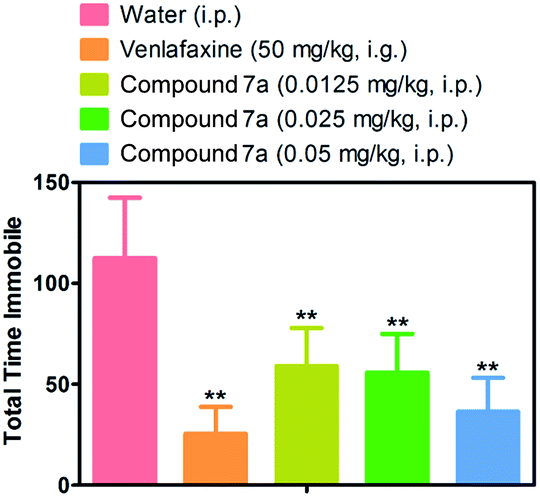 | ||
| Fig. 3 Mouse forced swim data for compound 7a. Compound 7a, which shows the protective effects on corticosterone-injured PC12 cells, also produced the expected decrease in immobility. Data represent the mean ± S.D. of 10 mice per group. **p < 0.001. | ||
3. Experimental
3.1 Chemistry
Unless otherwise noted, all reactions were carried out under an argon atmosphere with dry solvents. Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on Bruker AV-600 spectrometers at ambient temperature with CDCl3 as the solvent unless otherwise stated. High-resolution mass spectra (HRMS) were recorded at Shanxi University Mass Spectrometry Laboratory using a Thermo Scientific Q Exactive.3.1.1.1 (Z)-9-((tert-Butyldiphenylsilyl)oxy)-7-nonlyene-3,5-diyn-1-ol (6a). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the 3-butyne-1-ol (12.3 μL, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 6a (29.0 mg, 60%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.68 (m, 4H), 7.44–7.39 (m, 6H), 6.22–6.21 (m, 1H), 5.49 (d, J = 10.8 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 3.75 (t, J = 6.0 Hz, 2H), 2.59 (t, J = 6.0 Hz, 2H), 1.06 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 146.3, 135.6, 133.4, 129.7, 127.7, 107.9, 81.8, 79.2, 71.4, 66.8, 62.5, 60.7, 26.8, 24.0, 19.1; HRMS (APCI) [M + H]+ calculated for C25H29O2Si: 389.1937, found: 389.1930.
3.1.1.2 (Z)-tert-Butyldiphenyl(7-phenyl-2-heptene-4,6-diyn-1-oxy)silane (6b). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the phenylacetylene (17.8 μL, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 1/0) to afford 6b (26.6 mg, 51%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.71–7.70 (m, 4H), 7.49–7.47 (m, 2H), 7.45–7.40 (m, 6H), 7.38–7.31 (m, 3H), 6.28–6.24 (m, 1H), 5.60 (d, J = 10.8 Hz, 1H), 4.53 (d, J = 6.0 Hz, 2H); 1.08 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 146.3, 135.6, 133.4, 132.4, 129.7, 129.2, 128.4, 127.7, 121.8, 108.1, 82.4, 79.1, 77.5, 73.8, 62.6, 26.8, 19.2; HRMS (APCI) [M + H]+ calculated for C29H28OSi: 421.1988, found: 421.1980.
3.1.1.3 (2E,8Z)-10-((tert-Butyldiphenylsilyl)oxy)-2,8-decadien-4,6-diyn-1-ol (6c). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the (E)-2-penten-4-yn-1-ol (34.4 mg, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 6c (20.0 mg, 40%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.68 (m, 4H), 7.44–7.39 (m, 6H), 6.41–6.37 (m, 1H), 6.24–6.21 (m, 1H), 5.84 (d, J = 15.6 Hz, 1H), 5.55 (d, J = 11.4 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 4.25 (d, J = 4.2 Hz, 2H), 1.26 (s, 1H), 1.06 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 146.3, 145.3, 135.6, 133.4, 129.7, 127.8, 109.0, 108.0, 80.7, 79.1, 77.3, 74.4, 62.8, 62.6, 26.8, 19.2; HRMS (APCI) [M + H]+ calculated for C26H29O2Si: 401.1937, found: 401.1927.
3.1.1.4 (Z)-(4-(7′-(tert-Butyldiphenylsilyl)oxy)-5-heptene-1,3-diyn)benzyl alcohol (6d). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the 4-ethynylbenzyl alcohol (21.5 mg, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 6/1) to afford 6d (39.5 mg, 71%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.69 (d, J = 7.8 Hz, 4H), 7.47 (d, J = 7.8 Hz, 2H), 7.44–7.39 (m, 6H), 7.33 (d, J = 7.8 Hz, 2H), 6.27–6.23 (m, 1H), 5.59 (d, J = 10.8 Hz, 1H), 4.71 (s, 2H), 4.51 (d, J = 6.0 Hz, 2H), 1.55 (s, 1H), 1.07 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 146.4, 142.0, 135.6, 133.4, 132.6, 129.7, 127.7, 126.8, 120.9, 108.0, 82.2, 79.1, 77.6, 73.8, 64.8, 62.6, 26.8, 19.2; HRMS (APCI) [M + H]+ calculated for C30H31O2Si: 451.2093, found: 451.2086.
3.1.1.5 (Z)-(-2-Pentadecene-4,6,14-triyn-1-oxy)tert-butyldiphenylsilane (6e). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the 4-ethynylbenzyl alcohol (21.5 mg, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was dried over anhydrous Na2SO4 and concentrated under reduced pressure to give a crude oil, which was used directly in the next step.
3.1.1.6 (Z)-13-((tert-Butyldiphenylsilyl)oxy)-11-tridecene-7,9-diyn-6-ol (6f). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the 1-octyne-3-ol (23.7 μL, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 6/1) to afford 6f (25.0 mg, 45%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.67 (m, 4H), 7.45–7.38 (m, 6H), 6.23 (dt, J = 11.4, 6.0 Hz, 1H), 5.51 (d, J = 10.8 Hz, 1H), 4.47 (d, 6.0 Hz, 3H), 1.75–1.68 (m, 2H), 1.56 (s, 1H), 1.47–1.42 (m, 2H), 1.31 (d, J = 1.8 Hz, 4H), 1.06 (s, 9H), 0.90 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 146.7, 135.6, 133.4, 129.7, 127.7, 107.7, 84.3, 78.5, 74.6, 69.4, 63.0, 62.5, 37.5, 31.4, 26.8, 24.7, 22.5, 19.2, 14.0; HRMS (APCI) [M + H]+ calculated for C29H37O2Si: 445.2563, found: 445.2552.
3.1.1.7 (Z)-8-((tert-Butyldiphenylsilyl)oxy)-N,N-diethyl-6-octene-2,4-diyn-1-amine (6h). Following the general procedure using the bromoalkyne (50 mg, 0.13 mmol), CuI (6.0 mg, 0.03 mmol), the N,N-diethylprop-2-yn-1-amine (22.5 μL, 0.17 mmol) and piperidine (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 2/1) to afford 6h (26.1 mg, 48%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.68 (d, J = 7.2 Hz, 4H), 7.44–7.38 (m, 6H), 6.23–6.19 (m, 1H), 5.50 (d, J = 10.8 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 3.54 (s, 2H), 2.54 (q, J = 7.2 Hz, 4H), 1.08–1.06 (m, 15H); 13C NMR (150 MHz, CDCl3) δ: 146.2, 135.6, 133.5, 129.7, 127.7, 108.0, 80.0, 79.2, 71.9, 69.3, 62.6, 47.4, 41.4, 26.8, 19.2, 12.7; HRMS: (APCI) [M + H]+ calculated for C28H36NOSi: 430.2566, found: 430.2563.
3.1.2.1 (Z)-8-((tert-Butyldiphenylsilyl)oxy)-1-phenyl-6-octene-2,4-diyn-1-ol (6g). Following the general procedure using the bromoalkyne (50.0 mg, 0.125 mmol), the 1-phenyl-2-propyne-1-ol (18.3 μL, 0.15 mmol), Pd(PPh3)4 (4.3 mg, 0.004 mmol), CuI (0.7 mg, 0.004 mmol), diisopropylamine (35.1 μL, 0.25 mmol) and THF (0.3 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 6g (25.0 mg, 44%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.70–7.68 (m, 4H), 7.51 (d, J = 7.2 Hz, 2H), 7.44–7.34 (m, 9H), 6.26 (dt, J = 11.4, 6.0 Hz, 1H), 5.55–5.52 (m, 2H), 4.48 (d, J = 6.0 Hz, 2H), 2.26 (d, J = 6.0 Hz, 1H), 1.07 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 147.0, 139.6, 135.5, 133.3, 129.7, 128.7, 128.7, 127.7, 126.6, 107.6, 82.4, 78.3, 75.4, 71.1, 65.1, 62.5, 26.8, 19.1; HRMS: (APCI) [M + H]+ calculated for C30H31O2Si: 451.2093, found: 451.2089.
3.1.2.2 (Z)-Methyl-8-((tert-butyldiphenylsilyl)oxy)-6-octene-2,4-diynoate (6i). Following the general procedure using the bromoalkyne (50.0 mg, 0.125 mmol), the methyl propiolate (13.4 μL, 0.15 mmol), Pd(PPh3)4 (4.3 mg, 0.004 mmol), CuI (0.7 mg, 0.004 mmol), diisopropylamine (35.1 μL, 0.25 mmol) and THF (0.3 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 50/1) to afford 6i (24.9 mg, 50%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.67 (d, J = 7.2 Hz, 4H), 7.46–7.39 (m, 6H), 6.40–6.36 (m, 1H), 5.53 (d, J = 11.4 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H), 3.80 (s, 3H), 1.06 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 153.3, 149.9, 135.5, 133.1, 129.8, 127.8, 127.8, 106.7, 79.5, 72.3, 71.0, 62.6, 53.0, 26.8, 19.2; HRMS (APCI) [M + H]+ calculated for C25H27O3Si: 403.1729, found: 403.1721.
3.1.2.3 (Z)-((10-Methyl-2-undecene-4,6-diyn)-1-oxyl)tert-butyldiphenylsilane (6j). Following the general procedure using the bromoalkyne (50.0 mg, 0.125 mmol), the 5-methyl-hex-1-yne (19.7 μL, 0.15 mmol), Pd(PPh3)4 (4.3 mg, 0.004 mmol), CuI (0.7 mg, 0.004 mmol), diisopropylamine (35.1 μL, 0.25 mmol) and THF (0.3 mL), the resulting residue was purified by silica gel column chromatography (PE) to afford 6j (27.0 mg, 52%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.68 (m, 4H), 7.45–7.38 (m, 6H), 6.18 (dt, J = 11.4, 6.0 Hz, 1H), 5.49 (d, J = 11.4 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.72–1.65 (m, 1H), 1.43 (q, J = 7.2 Hz, 2H), 1.06 (s, 9H), 0.9 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ: 145.6, 135.6, 133.5, 129.6, 127.7, 108.2, 85.8, 79.7, 70.7, 64.9, 62.5, 37.0, 27.1, 26.8, 22.1, 19.2, 17.6; HRMS: (APCI) [M + H]+ calculated for C28H35OSi: 415.2457, found: 415.2451.
3.1.3.1 (Z)-2-Nonene-4,6-diyne-1,9-diol (7a). Following the general procedure using 6a (17 mg, 0.044 mmol), Et3N·3HF (87.7 μL, 0.54 mmol) and THF (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 2/1) to afford 7a (3.5 mg, 54%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.24 (dt, J = 10.8 Hz, 1H), 5.61 (d, J = 11.4 Hz, 1H), 4.43 (t, J = 5.4 Hz, 2H), 3.78 (q, J = 6.0 Hz, 2H), 2.62 (t, J = 6.6 Hz, 2H), 1.56 (s, 2H); 13C NMR (150 MHz, CDCl3) δ: 145.3, 109.4, 82.3, 79.6, 71.1, 66.6, 61.2, 60.7, 24.0; HRMS (APCI) [M + H]+ calculated for C9H11O2: 151.0759, found: 151.0752.
3.1.3.2 (Z)-7-Phenyl-2-hepten-4,6-diyn-1-ol (7b). Following the general procedure using 6b (17.0 mg, 0.044 mmol), Et3N·3HF (87.7 μL, 0.54 mmol) and THF (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 2/1) to afford 7b (3.5 mg, 54%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.51–7.50 (m, 2H), 7.38–7.32 (m, 3H), 6.30–6.26 (m, 1H), 5.71 (d, J = 11.4 Hz, 1H) 4.48 (d, J = 5.4 Hz, 2H), 1.26 (s, 1H); 13C NMR (150 MHz, CDCl3) δ: 145.4, 132.5, 129.3, 128.4, 121.6, 109.5, 82.8, 79.5, 73.5, 61.2, 29.7; HRMS (APCI) [M + H]+ calculated for C13H11O: 183.0810, found: 183.0803.
3.1.3.3 (2Z,8E)-2,8-Decadiene-4,6-diyne-1,10-diol (7c). Following the general procedure using 6c (16.0 mg, 0.040 mmol), Et3N·3HF (80.0 μL, 0.492 mmol) and THF (0.6 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 2/1) to afford 7c (3.6 mg, 56%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 6.44–6.42 (d, J = 15.0 Hz, 1H), 6.27–6.23 (m, 1H), 5.88 (d, J = 16.8 Hz, 1H), 5.76 (d, J = 10.8 Hz, 1H), 4.44 (s, 2H), 4.27 (s, 2H); 13C NMR (150 MHz, CDCl3) δ: 145.6, 145.3, 109.5, 108.8, 81.0, 79.5, 62.7, 61.2, 29.7; HRMS (APCI) [M − H]− calculated for C10H9O2: 161.0603, found: 161.0602.
3.1.3.4 (Z)-7-(4-(Hydroxymethyl)phenyl)-2-heptene-4,6-diyn-1-ol (7d). Following the general procedure using 6d (34.0 mg, 0.075 mmol), Et3N·3HF (151.3 μL, 0.928 mmol) and THF (1.1 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 3/1) to afford 7d (9.2 mg, 58%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.50 (d, J = 7.8 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 6.29–6.25 (m, 1H), 5.71 (d, J = 10.8 Hz, 1H), 4.71 (s, 2H), 4.47 (d, J = 6.0 Hz, 2H), 1.25 (2, 1H); 13C NMR (150 MHz, CDCl3) δ: 145.4, 142.2, 132.7, 126.8, 120.8, 109.5, 82.7, 79.5, 73.6, 64.8, 61.2, 29.7; HRMS (APCI) [M − H]− calculated for C14H11O2: 211.0759, found: 211.0762.
3.1.3.5 (Z)-2-Pentadecene-4,6,14-triyn-1-ol (7e). Following the general procedure using crude compound 6e (16.0 mg, 0.035 mmol), Et3N·3HF (70.8 μL, 0.435 mmol) and THF (0.5 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 10/1) to afford 7e (4.0 mg, 53%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.23–6.19 (m, 1H), 5.61 (d, J = 10.8 Hz, 1H), 4.43 (d, J = 6.0 Hz, 2H), 2.35 (t, J = 6.6 Hz, 2H), 2.20 (t, J = 7.2 Hz, 2H), 1.95 (s, 1H), 1.42 (s, 4H), 1.25 (s, 4H); 13C NMR (150 MHz, CDCl3) δ: 144.7, 109.8, 86.1, 86.0, 84.4, 68.2, 63.0, 61.2, 29.7, 28.3, 28.2, 28.1, 28.0, 19.5, 18.3; HRMS (APCI) [M − H]− calculated for C15H17O: 213.1279, found: 213.1282.
3.1.3.6 (Z)-2-Tridecene-4,6-diyne-1,8-diol (7f). Following the general procedure using 6f (15.0 mg, 0.034 mmol), Et3N·3HF (67.6 μL, 0.415 mmol) and THF (0.5 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 3/1) to afford 7f (3.5 mg, 51%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.68 (m, 4H), 7.45–7.39 (m, 6H), 6.23 (dt, J = 11.4, 6.0 Hz, 1H), 5.51 (d, J = 10.8 Hz, 1H), 4.47 (d, J = 6.0 Hz, 3H), 1.75–1.68 (m, 2H), 1.56 (s, 1H), 1.47–1.42 (m, 2H), 1.31 (d, J = 1.8 Hz, 4H), 1.06 (s, 9H), 0.9 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 145.8, 109.2, 84.7, 78.8, 74.3, 69.2, 63.1, 61.2, 37.5, 31.4, 24.7, 22.5, 14.0; HRMS (APCI) [M − H]− calculated for C13H17O2: 205.1229, found: 205.1231.
3.1.3.7 (Z)-8-Phenylocta-2-octene-4,6-diyne-1,8-diol (7g). Following the general procedure using 6g (25.0 mg, 0.055 mmol), Et3N·3HF (111.2 μL, 0.677 mmol) and THF (0.8 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 1/1) to afford 7g (6.0 mg, 51%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.53–7.52 (m, 2H), 7.41–7.36 (m, 3H), 6.27 (dt, J = 11.4, 6.6 Hz, 1H), 5.63 (d, J = 10.8 Hz, 1H), 5.58 (d, J = 6.0 Hz, 2H), 4.43 (s, 2H), 2.31 (d, J = 6.0 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ: 146.1, 139.5, 128.8, 128.8, 126.6, 109.1, 82.8, 78.7, 75.1, 70.9, 65.2, 61.2; HRMS: (APCI) [M − H]− calculated for C14H11O2: 211.0759, found: 211.0760.
3.1.3.8 (Z)-8-(Diethylamino)-2-octene-4,6-diyn-1-ol (7h). Following the general procedure using 6h (25.0 mg, 0.058 mmol), TBAF (75.6 μL, 0.075 mmol) and THF (0.5 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 1/2) to afford 7h (5.9 mg, 53%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.26–6.22 (m, 1H), 5.62 (d, J = 11.4 Hz, 1H), 4.43 (d, J = 6.6 Hz, 2H), 3.59 (s, 2H), 2.59 (q, J = 7.2 Hz, 4H), 1.25 (s, 1H), 1.09 (t, J = 7.2 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ: 145.4, 132.1, 128.5, 109.4, 79.4, 71.8, 61.2, 47.5, 41.4, 29.7; HRMS (APCI) [M + H]+ calculated for C12H18NO: 192.1388, found: 192.1381.
3.1.3.9 (Z)-Methyl-8-hydroxy-6-octene-2,4-diynoate (7i). Following the general procedure using 6i (12.0 mg, 0.030 mmol), Et3N·3HF (59.1 μL, 0.369 mmol) and THF (0.5 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 7i (2.4 mg, 49%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.44–6.40 (m, 1H), 5.66 (d, J = 10.8 Hz, 1H), 4.45 (t, J = 5.4 Hz, 2H), 3.81 (s, 3H), 1.51 (s, 1H); 13C NMR (150 MHz, CDCl3) δ: 151.1, 149.1, 107.9, 79.2, 72.6, 70.7, 61.3, 53.1, 29.7; HRMS: (APCI) [M + H]+ calculated for C9H9O3: 165.0552, found: 165.0546.
3.1.3.10 (Z)-10-Methyl-2-undecaen-4,6-diyn-1-ol (7j). Following the general procedure using 6j (24.0 mg, 0.058 mmol), Et3N·3HF (116 μL, 0.713 mmol) and THF (0.8 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 7j (5.6 mg, 55%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 6.22–6.18 (m, 1H), 5.60 (d, J = 10.8 Hz, 1H), 4.42 (d, J = 6.0 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.73–1.66 (m, 1H), 1.44 (q, J = 7.2 Hz, 2H), 1.25 (s, 1H), 0.90 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ: 144.6, 109.8, 86.3, 80.1, 70.4, 64.6, 61.1, 37.0, 27.2, 22.1, 17.6; HRMS: (APCI) [M - H]− calculated for C12H15O: 175.1123, found: 175.1125.
3.1.4.1 (Z)-5-Bromo-2-pentene-4-yn-1-ol. To a solution of 5 (0.3 g, 0.75 mmol) in THF (11.0 mL) was added triethylamine trihydrofluride (1.5 mL, 9.23 mmol) at room temperature. The resulting solution was stirred at room temperature for 8 h and quenched with H2O (3 mL). The reaction mixture was extracted with EtOAc (6 mL × 3), and the combined extracts were washed with saturated NaCl solution, dried over anhydrous Na2SO4 and the filtrate was concentrated under reduced pressure to give a crude oil, which was used directly in the next step.
3.1.4.2 General procedure for the DCC coupling reaction between the alcohols and acids (8a, 8b). Dicyclohexylcarbodiimide (1.1 equiv.) and N,N-dimethylaminopyridine (0.2 equiv.) were added to a stirred solution of acid (1.1 equiv.) in dichloromethane (0.15 M) at 0 °C. The reaction mixture was stirred for 30 min and then a solution of the crude compound alcohol (1 equiv.) in dichloromethane (0.25 M) was added dropwise. The mixture was allowed to warm to room temperature over 12 h, diluted with diethyl ether, and then sequentially washed with saturated aqueous solution of NH4Cl, a saturated aqueous solution of NaHCO3 and brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure.
3.1.4.2.1 (2E,4E)-(Z)-5-Bromo-2-pentene-4-yn-1-oxylhexanoate (8a). Following the general procedure using dicyclohexylcarbodiimide (142.0 mg, 0.688 mmol), N,N-dimethylaminopyridine (15.3 mg, 0.125 mmol), hexanoic acid (69.4 mg, 0.616 mmol), the crude compound (60 mg, 0.63 mmol), and dichloromethane (8 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 20/1) to afford 8a (40.0 mg, 41%, 2 steps) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.07–6.03 (m, 1H), 5.64 (d, J = 10.8 Hz, 1H), 4.81 (d, J = 6.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.66–1.61 (m, 2H), 1.34–1.29 (m, 4H), 0.90 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 173.6, 138.3, 112.1, 75.6, 62.0, 56.1, 34.1, 31.3, 24.6, 22.3, 13.9; HRMS: (APCI) [M + H]+ calculated for C11H16BrO2: 259.0334, found: 259.0323.
3.1.4.2.2 (2E,4E)-(Z)-5-Bromo-2-pentene-4-yn-1-oxyl-2,4-hexadienoate (8b). Following the general procedure using dicyclohexylcarbodiimide (127.7 mg, 0.618 mmol), N,N-dimethylaminopyridine (13.7 mg, 0.112 mmol), sorbic acid (69.4 mg, 0.616 mmol), the crude compound (60 mg, 0.63 mmol), and dichloromethane (8 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 10/1) to afford 8b (3.8 mg, 40%, 2 steps) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.30–7.27 (m, 1H), 6.22–6.12 (m, 2H), 6.09 (dt, J = 10.8, 6.0 Hz, 1H), 5.78 (d, J = 15.0 Hz, 1H), 5.65 (d, J = 10.8 Hz, 1H), 4.88 (d, J = 6.6 Hz, 2H), 1.86 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 166.9, 145.6, 139.8, 138.5, 129.7, 118.3, 112.0, 75.6, 62.0, 56.1, 18.7; HRMS: (APCI) [M + H]+ calculated for C11H12BrO2: 255.0021, found: 255.0013.
3.1.4.3 General procedure for the coupling reaction between terminal alkynes and bromoalkynes (9a, 9b). A solution of the bromoalkyne (1 equiv.) in THF (0.4 M) was sparged with argon over 20 min and then PdCl2(PPh3)2 (0.03 equiv.), CuI (0.03 equiv.) and diisopropylamine (2 equiv.) were added sequentially at room temperature, followed immediately by the terminal alkyne (1.2 equiv.). The reaction mixture was stirred at room temperature for 12 h and then concentrated under reduced pressure.
3.1.4.3.1 (Z)-9-Hydroxy-2-nonene-4,6-diyn-1-oxyl hexanoate (9a). Following the general procedure using the bromoalkyne ester 8a (20 mg, 0.077 mmol), the 3-butyn-1-ol (7.1 μL, 0.093 mmol), PdCl2(PPh3)2 (1.6 mg, 0.002 mmol), CuI (0.4 mg, 0.002 mmol), diisopropylamine (21.6 μL, 0.154 mmol) and THF (0.7 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 9a (11.2 mg, 59%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.16–6.12 (m, 1H), 5.68 (d, J = 10.8 Hz, 1H), 4.83 (d, J = 6.6 Hz, 2H), 3.78 (t, J = 6.0 Hz, 2H), 2.62 (t, J = 6.0 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.64 (dt, J = 15.6, 7.8 Hz, 2H), 1.34–1.27 (m, 4H), 1.25 (s, 1H), 0.90 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 173.5, 140.2, 111.5, 82.6, 80.3, 70.6, 66.6, 62.2, 60.7, 34.1, 31.3, 24.6, 24.0, 22.3, 13.9; HRMS: (APCI) [M − H]− calculated for C15H19O3: 247.1334, found: 247.1339.
3.1.4.3.2 (2E,4E)-(Z)-9-Hydroxy-2-nonene-4,6-diyn-1-oxyl-2,4-hexadienoate (9b). Following the general procedure using the bromoalkyne ester 8b (21.0 mg, 0.082 mmol), the 3-butyn-1-ol (7.5 μL, 0.099 mmol), PdCl2(PPh3)2 (1.7 mg, 0.003 mmol), CuI (0.5 mg, 0.003 mmol), diisopropylamine (23.2 μL, 0.164 mmol) and THF (0.8 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 4/1) to afford 9b (11.0 mg, 58%) as a pale colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.30–7.27 (m, 1H), 6.22–6.13 (m, 3H), 5.78 (d, J = 15.0 Hz, 1H), 5.68 (d, J = 10.8 Hz, 1H), 4.90 (d, J = 6.0 Hz, 2H), 3.78 (d, J = 6.0 Hz, 2H), 2.62 (d, J = 6.0 Hz, 2H), 1.86 (d, J = 6.0 Hz, 3H), 1.83 (s, 1H); 13C NMR (150 MHz, CDCl3) δ: 166.9, 145.7, 140.3, 139.8, 129.7, 118.3, 111.4, 82.6, 80.3, 70.7, 66.6, 62.2, 60.7, 24.0, 18.7; HRMS: (APCI) [M + H]+ calculated for C15H17O3: 245.1178, found: 245.1171.
3.1.5.1 (Z)-16,16-Dimethyl-15,15-diphenyl-2,4,14-trioxa-15-silanyl-11-heptadecene-7,9-diyne (11-1). To a stirred solution of 6a (25 mg, 0.064 mmol) in dry DCM (2 mL) was added dimethoxymethane (57.0 μL, 0.643 mmol), then the reaction mixture was cooled to 0 °C, phosphorous pentoxide (45.7 mg, 0.322 mmol) was added to the reaction mixture. The mixture was warmed to room temperature and stirred for 16 h. The resulting mixture was diluted with DCM, washed with saturated NaHCO3, dried with anhydrous K2CO3, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EtOAc = 20/1) to afford 11-1 (15.0 mg, 54%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.68–7.67 (m, 4H), 7.44–7.38 (m, 6H), 6.21–6.17 (m, 1H), 5.48 (d, J = 10.8 Hz, 1H), 4.64 (s, 2H), 4.47 (d, J = 5.4 Hz, 2H), 3.65 (t, J = 7.2 Hz, 2H), 3.36 (s, 3H), 2.62 (t, J = 6.6 Hz, 2H), 1.05 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 146.1, 135.6, 133.5, 129.7, 127.7, 108.0, 96.4, 82.0, 79.5, 71.2, 66.0, 65.4, 62.5, 55.3, 26.8, 21.2, 19.2; HRMS: (APCI) [M + H]+ calculated for C27H33O3Si: 433.2199, found: 433.2192.
3.1.5.2 (Z)-9-(Methoxymethoxy)nona-2-en-4,6-diyn-1-ol (11). Following the general procedure using 11-1 (15.0 mg, 0.035 mmol), triethylamine trihydrofluoride (69.6 μL, 0.431 mmol) and THF (0.5 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 4/1) to afford 11 (2.9 mg, 43%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.24–6.20 (m, 1H), 5.60 (d, J = 11.4 Hz, 1H), 4.66 (s, 2H), 4.41 (d, J = 4.2 Hz, 2H), 3.68 (t, J = 6.6 Hz, 2H), 3.39 (s, 3H), 2.65 (t, J = 6.6 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ: 145.1, 109.6, 96.5, 82.6, 79.8, 70.9, 65.8, 65.3, 61.2, 55.4, 29.7; HRMS: (APCI) [M − H]− calculated for C11H13O3: 193.0865, found: 193.0866.
3.1.6.1 (Z)-9-((tert-Butyldiphenylsilyl)oxy)-7-nonene-3,5-diyn-1-oxyl acetate (12-1). Acetic anhydride (0.32 mL, 3.42 mmol) was added dropwise to a solution of 5a (0.2 g, 2.85 mmol), DMAP (34.9 mg, 0.28 mmol) and triethylamine (0.8 mL, 5.71 mmol) in 30 mL of dry DCM. After stirring for 16 h at room temperature, the reaction mixture was poured into a separatory funnel containing 30 mL of 10% NH4Cl. The organic phase was isolated, and the aqueous phase was extracted with CH2Cl2 (30 mL × 3). All the organic phases were combined, washed with brine, dried over anhydrous Na2SO4, and the filtrate was concentrated under reduced pressure to give a crude oil. The resulting residue was purified by silica gel column chromatography (PE/EtOAc = 30/1) to afford 12-1 (95.3 mg, 43%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.67 (m, 4H), 7.45–7.38 (m, 6H), 6.21 (dt, J = 11.4, 6.0 Hz, 1H), 5.49 (d, J = 10.8 Hz, 1H), 4.47 (d, J = 6.0 Hz, 2H), 4.16 (t, J = 6.6 Hz, 2H), 2.66 (t, J = 6.6 Hz, 2H), 2.07 (s, 3H), 1.05 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 170.7, 146.4, 135.6, 133.4, 129.7, 127.7, 107.9, 80.7, 79.2, 71.5, 66.4, 62.5, 61.7, 26.8, 20.8, 20.1, 19.1; HRMS: (APCI) [M + H]+ calculated for C27H31O3Si: 431.2042, found: 431.2036.
3.1.6.2 (Z)-9-Hydroxy-7-nonene-3,5-diyn-1-oxyl acetate (12). Following the general procedure using 12-1 (22.0 mg, 0.051 mmol), triethylamine trihydrofluoride (0.1 mL, 0.627 mmol) and THF (0.7 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 12 (4.8 mg, 49%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.26–6.22 (m, 1H), 5.60 (d, J = 10.8 Hz, 1H), 4.43 (d, J = 6.0 Hz, 2H), 4.19 (t, J = 6.6 Hz, 2H), 2.69 (t, J = 6.6 Hz, 2H), 2.09 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 170.8, 145.4, 109.4, 81.2, 79.6, 71.2, 66.2, 61.6, 61.2, 29.7, 20.8; HRMS: (APCI) [M + H]+ calculated for C11H13O3: 193.0865, found: 193.0858.
3.1.7.1 (Z)-((9-Methoxy-2-nonene-4,6-diyn)-1-oxyl)tert-butyldiphenylsilane (13-1). A solution of 6a (25 mg, 0.064 mmol) and methyl iodide (80.1 μL, 1.287 mmmol) in dry THF (0.5 mL) was cooled to 0 °C, then sodium hydride (60% dispersion in mineral oil, 3 mg, 0.077 mmol) was added portionwise over 10 min. After 5 min more, the cooling was removed and the reaction mixture was stirred at room temperature for 3 h, H2O (1 mL) was added to the reaction flask and the resulting solution was extracted with EtOAc (1 mL × 3), the combined extracts were washed with saturated NaCl solution (1 mL × 2), dried over anhydrous Na2SO4 and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EtOAc = 20/1) to afford 13-1 (12.0 mg, 46%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 7.69–7.68 (m, 4H), 7.45–7.39 (m, 6H), 6.19 (dt, J = 11.4, 6.0 Hz, 1H), 5.48 (d, J = 10.8 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 3.51 (t, J = 7.2 Hz, 2H), 3.37 (s, 3H), 2.59 (t, J = 6.6 Hz, 2H), 1.06 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 146.1, 135.6, 133.5, 129.7, 127.7, 108.0, 82.0, 79.5, 71.2, 70.1, 65.9, 62.5, 58.7, 266.8, 20.9, 19.1; HRMS: (APCI) [M − H]− calculated for C26H29O2Si: 401.1937, found: 401.1929.
3.1.7.2 (Z)-9-Methoxy-2-nonene-4,6-diyn-1-ol (13). Following the general procedure using 13-1 (20.0 mg, 0.050 mmol), triethylamine trihydrofluoride (0.1 mL, 0.615 mmol) and THF (0.7 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 4/1) to afford 13 (4.0 mg, 49%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 6.24–6.20 (dt, J = 11.4, 6.6 Hz, 1H), 5.60 (d, 10.8 Hz, 1H), 4.42 (d, J = 6.0 Hz, 2H), 3.53 (t, J = 6.6 Hz, 2H), 3.38 (s, 3H), 2.62 (t, J = 6.6 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ: 145.1, 109.6, 82.5, 79.8, 70.9, 70.1, 65.7, 61.2, 58.8, 29.7; HRMS: (APCI) [M + H]+ calculated for C10H13O2: 165.0916, found: 165.0909.
3.1.8.1 (3E,7Z)-9-((tert-Butyldiphenylsilyl)oxy)-3,7-nonadien-5-yn-1-ol (15). In a two-neck flask filled with argon, Cp2ZrCl2 (1.25 g, 4.276 mmol) was placed in of dry THF (7 mL). The mixture was cooled in an ice bath, and DIBAL-H (1 M, 3.4 mL, 3.4 mmol) was added. The reaction mixture turned a pale pink and it was allowed to stir for 1 h. In a second flask filled with argon, a solution of 3-butyn-1-ol (0.2 g, 2.85 mmol) in dry THF (2.3 mL) was chilled in an ice bath, and DIBAL-H (1 M, 2.9 mL, 2.9 mmol) was added to this solution. The resulting yellow solution was allowed to stirred under argon for 1 h. Then the 3-butyn-1-ol solution was added via syringe into the prepared zirconium hydride reagent. The reaction mixture was allowed to warm to room temperature, and then to stirred for 5 hours. The mixture was cooled to −78 °C, and to that, crystalline iodine (869 mg, 3.424 mmol) was added. The reaction mixture stirred overnight and then water (4 mL) was added. After stirring for 2.5 hours, the mixture was poured into an aqueous solution prepared from sat. Na2S2O3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sat. NaHCO3 (2:1). The aqueous layer was extracted with EtOAc (2 mL × 3), and the combined extracts were washed with saturated NaCl solution (3 mL × 2), dried over anhydrous Na2SO4 and the filtrate was concentrated under reduced pressure to give a crude oil, which was used directly in the next step.
:sat. NaHCO3 (2:1). The aqueous layer was extracted with EtOAc (2 mL × 3), and the combined extracts were washed with saturated NaCl solution (3 mL × 2), dried over anhydrous Na2SO4 and the filtrate was concentrated under reduced pressure to give a crude oil, which was used directly in the next step.A solution of the crude compound vinyl iodide (43 mg, 0.217 mmol) in THF (1 mL) was sparged with argon over 10 min and then PdCl2(PPh3)2 (1.5 mg, 0.002 mmol), CuI (0.4 mg, 0.002 mmol) and triethylamine (60.2 μL, 0.434 mmol) were sequentially added at 0 °C. Followed immediately by (Z)-(2-pentene-4-yn-1-oxyl)tert-butyldiphenylsilane (76.7 mg, 0.239 mmol). The stirred reaction mixture was allowed to warm to room temperature over 12 h and then filtered through silica and washed with diethyl ether. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EtOAc = 5/1) to afford 15 (35.0 mg, 41%, 2 steps) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 7.70–7.66 (m, 4H), 7.44–7.36 (m, 6H), 6.09–6.05 (m, 1H), 5.97–5.92 (m, 1H), 5.63 (d, J = 10.8 Hz, 1H), 5.59 (d, J = 12.6 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H), 3.67 (t, J = 6.0 Hz, 2H), 2.36 (q, J = 6.6 Hz, 2H), 1.06 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 141.8, 140.1, 135.6, 135.4, 133.6, 129.6, 127.7, 127.6, 112.4, 109.2, 62.5, 61.5, 36.4, 26.8, 19.2; HRMS: (APCI) [M + H]+ calculated for C25H31O2Si: 391.2093, found: 391.2083.
3.1.8.2 (2Z,6E)-2,6-Nonadien-4-yne-1,9-diol (16). Following the general procedure using 15 (30.0 mg, 0.077 mmol), triethylamine trihydrofluoride (0.15 mL, 0.947 mmol) and THF (1.1 mL), the resulting residue was purified by silica gel column chromatography (PE/EtOAc = 2/1) to afford 16 (5.1 mg, 44%) as a pale yellow oil. 1H NMR (600 MHz, CDCl3) δ: 6.15 (dt, J = 15.6, 7.2 Hz, 1H), 6.07 (dt, 10.8, 6.0 Hz, 1H), 5.72 (dd, J = 16.2, 10.8 Hz, 2H), 4.41 (d, J = 5.4 Hz, 2H), 3.71 (t, J = 6.0 Hz, 2H), 2.41 (q, J = 6.0 Hz, 2H), 1.56 (s, 2H); 13C NMR (150 MHz, CDCl3) δ: 140.8, 140.8, 112.2, 110.8, 93.7, 84.3, 61.5, 61.1, 36.4; HRMS (APCI) [M − H]− calculated for C9H11O2: 151.0759, found: 151.0752.
3.2 Biology assay
000 rpm min−1 for 15 min at 4 °C, and the supernatants were collected. Protein concentrations were determined by bicinchoninic acid assay. Equal amounts of protein (50 μg) were separated by electrophoresis on 12% sodium dodecyl sulphate polyacrylamide gels and transferred onto PVDF membranes. These membranes were incubated with 5% (w/v) non-fat milk powder in tris-buffered saline containing 0.1% (v/v) Tween-20 (TBST) for 2 h to block nonspecific binding sites. The membranes were then incubated overnight at 4 °C with the primary antibodies. After washing with TBST, the membranes were incubated for 2 h at room temperature with the fluorescent secondary antibodies. After rewashing with TBST, the membranes were scanning by fluorescent scanner (Odclyssey CLX, Gene Company limited, USA).3.3 General procedures for behavioral studies
Kunming mice were obtained from the experimental animal center of Military Medical Science Academy of the PLA, China. All animal experiments were done in accordance with the National Guidelines for Experimental Animal Welfare (MOST, PR China, 2006), which had full accreditation from the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC Intl.). This study was approved by the Committee on the Ethics of Animal Experiments of Shanxi University. Maximal effort was made to minimize animal suffering and the number of animals necessary for the acquisition of reliable data. All mice in this study were treated humanely throughout the experimental period. Compound 7a was synthesized as described above, and Venlafaxine hydrochloride capsules were purchased from Chengdu Kanghong Pharmaceutical Group Co., Ltd. The Venlafaxine hydrochloride was administered intragastrically (IG). Compound 7a was dissolved with a small amount of DMSO and diluted with sterile water for injection before use. The final concentration of DMSO is less than 0.1%. Compound 7a was administered by intraperitoneal (IP) injection. The purified water was administered by intraperitoneal (IP) injection. The mice were assigned into 5 groups (n = 10); venlafaxine hydrochloride group (50 mg kg−1), group, 7a group (0.0125 mg kg−1), 7a group (0.025 mg kg−1), 7a group (0.05 mg kg−1). Mice were individually placed into clear glass cylinders (diameter 20 cm, height 50 cm) containing 25 ± 1 °C water 10 cm deep. Mice were placed in the water and forced to swim for 6 min. The time the animal spent immobile was recorded over a 4 min trial. Immobility was defined as the postural position of floating in the water.3.4 Statistical analysis
All values were expressed as mean ± S.D. A two-tailed unpaired t-test by SPSS 16.0 (Chicago, IL, USA) was applied to analyze those significant differences between two groups, and the significance threshold was considered at p < 0.001.4. Conclusions
In summary, based on the structure of natural product RB-2, sixteen 1,3-diynes compounds were designed and synthesized. Subsequently, their protective effects on corticosterone-injured PC12 cells were evaluated. Five compounds showed significant protective activity in this assay. The SAR studies suggested that the 1,3-diyne and the allyl alcohol motifs were crucial to the protective activity. Moreover, the protective activity of compound 7a was shown to be associated with the regulation of apoptosis-related proteins. Finally, the mice forced swim test showed that 7a had a concentration-dependent antidepressant-like effect. Together, our findings supported that compound 7a is a promising lead candidate that deserves further evaluation in the design of antidepressants with unique scaffolds.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21402111, 81473415), Basic Research Program of Shanxi Province (No. 2015021039). The research project was supported by Shanxi Scholarship Council of China (No. 2015-020) and science and technology innovation project of Shanxi Province (2016115).Notes and references
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- A. L. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed. Engl., 2006, 45, 1034–1057 CrossRef PubMed.
- W. Heydenreuter, E. Kunold and S. A. Sieber, Chem. Commun., 2015, 51, 15784–15787 RSC.
- L. F. Liang, T. Wang, Y. S. Cai, W. F. He, P. Sun, Y. F. Li, Q. Huang, O. Taglialatela-Scafati, H. Y. Wang and Y. W. Guo, Eur. J. Med. Chem., 2014, 79, 290–297 CrossRef CAS PubMed.
- H. Kim, J. Chin, H. Choi, K. Baek, T. G. Lee, S. E. Park, W. Wang, D. Hahn, I. Yang, J. Lee, B. Mun, M. Ekins, S. J. Nam and H. Kang, Org. Lett., 2012, 15, 100–103 CrossRef PubMed.
- B. Zhang, Y. Wang, S. P. Yang, Y. Zhou, W. B. Wu, W. Tang, J.-P. Zuo, Y. Li and J. M. Yue, J. Am. Chem. Soc., 2012, 134, 20605–20608 CrossRef CAS PubMed.
- Z. Zhang, C. Lu, X. Liu, J. Su, W. Dai, S. Yan, A. Lu and W. Zhang, J. Proteome Res., 2013, 13, 925–933 CrossRef PubMed.
- Z. F. Zhou, M. Menna, Y. S. Cai and Y. W. Guo, Chem. Rev., 2015, 115, 1543–1596 CrossRef CAS PubMed.
- J. Liu, Y. Fang, L. Yang, X. Qin, G. Du and X. Gao, J. Pharm. Biomed. Anal., 2015, 111, 257–265 CrossRef CAS PubMed.
- O. Berton and E. J. Nestler, Nat. Rev. Neurosci., 2006, 7, 137–151 CrossRef CAS PubMed.
- B. Haenisch and H. Bönisch, Pharmacol. Ther., 2011, 129, 352–368 CrossRef CAS PubMed.
- F. Artigas, ACS Chem. Neurosci., 2013, 4, 5–8 CrossRef CAS PubMed.
- D. M. Gerhard, E. S. Wohleb and R. S. Duman, Drug Discovery Today, 2016, 21, 454–464 CrossRef CAS PubMed.
- D. Vaudry, P. Stork, P. Lazarovici and L. Eiden, Science, 2002, 296, 1648–1649 CrossRef CAS PubMed.
- Y. F. Li, Y. Q. Liu, W. C. Huang and Z. P. Luo, Acta Pharmacol. Sin., 2003, 24, 996–1000 CAS.
- B. Zeng, Y. Li, B. Niu, X. Wang, Y. Cheng, Z. Zhou, T. You, Y. Liu, H. Wang and J. Xu, J. Mol. Neurosci., 2016, 59, 567–578 CrossRef CAS PubMed.
- W. Ang, G. Chen, L. Xiong, Y. Chang, W. Pi, Y. Liu, C. Li, J. Zheng, L. Zhou, B. Yang, Y. Deng, S. Yang, Y. Luo and Y. Wei, Eur. J. Med. Chem., 2014, 82, 263–273 CrossRef PubMed.
- Y. Yang, W. Ang, H. Long, Y. Chang, Z. Li, L. Zhou, T. Yang, Y. Deng and Y. Luo, Sci. Rep., 2016, 6, 34711 CrossRef CAS PubMed.
- K. Q. Ma, Y. H. Miao, X. X. Gao, J. B. Chao, X. Zhang and X. M. Qin, Chin. Chem. Lett., 2017 DOI:10.1016/j.cclet.2016.11.032.
- D. J. Galler and K. A. Parker, Org. Lett., 2015, 17, 5544–5546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01268c |
| This journal is © The Royal Society of Chemistry 2017 |