
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing the self-recovery and mechanical property of hydrogels by macromolecular microspheres with thermal and redox initiation systems
Chang Huang†
a,
Yifan Li†b,
Lijie Duana,
Linhui Wanga,
Xiuyan Ren *a and
Guanghui Gaoa
*a and
Guanghui Gaoa
aPolymeric and Soft Materials Laboratory, School of Chemical Engineering, Advanced Institute of Materials Science, Changchun University of Technology, Changchun 130012, China. E-mail: xyren_bio@163.com; Fax: +86-431-85716465; Tel: +86-431-85717352
bDepartment of Anatomy, School of Basic Medical Science, Changchun University of Chinese Medicine, Changchun 130117, China
First published on 14th March 2017
Abstract
In this investigation, a tough hydrogel was reinforced by macromolecular microspheres (MMs) as hydrophobic association centers via free radical polymerization with different initiation systems. The thermal initiator is potassium persulfate and the redox initiators include potassium persulfate and N,N,N′,N′-tetramethylethylenediamine (TMEDA). The mechanical measurements showed that the hydrogel with a redox initiation system possesses a tensile strength of 1.55 MPa, which is much higher than the hydrogel with a thermal initiator. Moreover, the hydrogel with redox initiators exhibited rapid self-recovery to its 90% original dimension in several seconds and low swelling property. The large difference in the morphology of hydrogels before and after swelling was due to the tight entanglement of molecular chains in the internal structure of hydrogels with redox initiators. As a result, the hydrophobic association hydrogels toughened by MMs with excellent mechanical properties would be useful for biomedical applications of tough tissue engineering, such as tendon, muscle, and blood vessel.
Introduction
Hydrogels consisting of a polymer network are soft and wet materials with a large amount of water and have been utilized widely in biomedical, industrial and agricultural fields, such as in drug delivery carriers,1–3 wastewater treatment,4,5 tissue scaffolds,6–8 food chemistry,9,10 biosensors,11,12 and superabsorbent materials.13,14 However, some practical applications of hydrogels were greatly limited by their weak mechanical strength and difficult recovery after deformation. Therefore, some effective toughening mechanisms for hydrogels have been explored, including double network hydrogels,15,16 slide-ring hydrogels,17 nanocomposite hydrogels,18–21 macromolecular microsphere composite hydrogels,22–26 hydrophobic association hydrogels27–29 and other hybrid hydrogels.30,31Wang et al. reported a hydrogel toughened by macromolecular microspheres (MMs). In their system, MMs modified by peroxide groups served as initiators and chemical crosslinking points to toughen the hydrogels by irradiation with 60Co γ-rays. As a result, MMs could not only prevent the further development of cracks, but these hydrogels could also be stretched to deform and absorb a large amount of energy. The resulting MMs hydrogels exhibited excellent compressive properties, but the tensile strength was weak, possibly due to the presence of only chemical crosslinking in the hydrogel network.22 Other inorganic nanoparticles or micelles were also utilized to reinforce the mechanical behaviour of hydrogels. For example, Shi et al. developed a ternarily crosslinked nanocomposite physical hydrogel, which showed a long elongation of 2800% and weak fracture strength of 256 KPa.32 Fu et al. developed a nanomicelle hydrogel crosslinked by vinyl-functionalized thermosensitive pluronic F127 micelles. The obtained hydrogel was super tough, highly resilient and thermo-responsive. However, the tensile strength was reported not to exceed 300 KPa.33,34 In the recent past, Okay et al. reported about hydrophobic association hydrogels, which exhibit excellent self-healing efficiency and ultra-stretchable strain. However, these hydrogels have a low tensile strength of 250 KPa.35,36
In our recent investigation, novel macromolecular microspheres (MMs) were prepared via a conventional emulsion polymerization method and served as a crosslinking centre for hydrophobic segments to enhance the mechanical strength of hydrogels. The resulting MMs-toughened hydrogels were prepared via using a thermal initiation system and exhibited a fracture strength of 0.55 MPa.26 However, the thermal initiation system requires a high reaction temperature, possibly inducing a heterogeneous network structure when the hydrogel is formed. Based on the above conception, we proposed a strategy to improve the mechanical properties of hydrogels, which were formed at low ambient temperatures. It was facile for impart the hydrogels with a homogeneous crosslinking structure. It was envisioned that a redox initiation system of low temperature could be utilized to prepare hydrogels with excellent mechanical behaviours.
Herein, we prepared MMs toughed hydrogels via free radical polymerization with different initiation systems. The thermal initiator was potassium persulfate and the redox initiators consisted of KPS and N,N,N′,N′-tetramethylethylenediamine (TMEDA). It was envisioned that the hydrogels obtained by a redox initiation system could possess tighter entanglements among the polymer molecular chains in its internal structure due to the instantly generated large number of free radicals to trigger monomer polymerization under a low ambient reaction temperature. In this study, we investigated the swelling behavior of hydrogels obtained by different initiation systems to illustrate the degree of the homogeneous distribution of crosslinking centers and physical entanglement of the polymer molecule chains; the morphology of the hydrogels was examined by Scanning Electron Microscopy (SEM). Subsequently, the hydrogels were also measured by tensile, compressive and rheological tests to investigate the effects of different reaction systems on the mechanical properties of the hydrogels. It was envisioned that the tough hydrogels would attract much attention in biomedical applications, such as tendon, muscle, and blood vessel.
Experimental
Materials
Acrylamide (AAm, 99.0%), sodium dodecyl sulfate (SDS, ≥97.0%), potassium persulfate (KPS, 99.5%), N,N,N′,N′-tetramethylethylenediamine (TMEDA, ≥99.5%), sodium chloride (NaCl, 99.5%), dicyclopentyl acrylate (DCPA, 99.5%), sodium carbonate (Na2CO3, 99.0%) were supplied by Aladdin (Shanghai, China). Hexadecyl methacrylate (HMA, 95.0%) and butyl acrylate (BA, 95.0%) were supplied by Zhejiang Kangde New Materials Co., Ltd, China. Deionized water (18.2 Ω cm resistivity at 25 °C) was used in the experiment.Preparation of macromolecular microspheres (MMs)
The macromolecular microspheres (MMs), 349 nm in diameter, were prepared in our previous investigation.26 The preparation was as follows. Appropriate amounts of deionized water, SDS, Na2CO3 were added to a three-necked boiling flask and stirred for dissolution. SDS and Na2CO3 were used as an emulsifier and an electrolyte, respectively. The monomer (BA) and crosslinking agent (DCPA) were added to the reactor and kept at 70 °C, and nitrogen gas was flushed to remove oxygen for 30 min. The initiator KPS (0.02 g) was added to the reactor after dissolving in deionized water. The rate of nitrogen bubbling was decreased to minimize the evaporation. The reaction time was set to 120 min to obtain a high conversion for the experiment. Finally, we obtained MMs latexes with 349 nm.Preparation of hydrogels with a thermal initiator
To illustrate the synthetic procedure, the details were as follows for hydrogels with a thermal initiator of KPS. NaCl (0.4 g) and SDS (0.8 g) were dissolved in 20 mL of deionized water at room temperature by stirring to prepare the SDS–NaCl semi-dilute solution. After the semi-dilute solution became transparent, hexadecyl methacrylate (HMA, 1 mol% to AAm) was added to the SDS–NaCl semi-dilute solution with stirring for 1 h until the solution was clear. The MMs (1 wt% to monomers) as crosslinking centres were then added to the solution by stirring for 5 h for complete dispersion. Acrylamide (5.8 g, AAm) and KPS (0.02 g) were added to the mixture and stirred for 10 min at room temperature to obtain a homogeneous solution. Finally, the solution was carried out at 65 °C for 5 h to obtain MMs hydrogels, which were named as P(AAm/HMA)-MMs-T. For comparison, the hydrophobic association hydrogels without MMs were prepared using the same procedure and called P(AAm/HMA)-T.Preparation of hydrogels with a redox initiation system
As described above, after a semi-dilute solution of NaCl–SDS turned transparent, hexadecyl methacrylate (HMA, 1 mol% to AAm) was added to the SDS–NaCl semi-dilute solution with stirring for 1 h until the solution was clear. The MMs (1 wt% to monomers) as crosslinking centres were then added to the solution by stirring for 5 h for complete dispersion. Acrylamide (5.8 g, AAm), KPS (0.02 g) and TMEDA (40 μL) were added to the mixture and stirred for 10 min at room temperature to obtain a homogeneous solution. Finally, the solution was carried out at 25 °C for 2 h to obtain the P(AAm/HMA)-MMs-R hydrogels. For comparison, the hydrophobic association hydrogels without MMs were prepared using the same procedure and called P(AAm/HMA)-R.Uniaxial elongation measurements
To measure the mechanical property of hydrogels, the tensile measurements (SHIMADZU, model AGS-X, 100N, Japan) were performed on dumbbell-shaped samples (length = 30 mm, width = 4 mm, thickness = 3 mm) at a speed of 40 mm min−1. The tensile strength and elongation at break were recorded. For reproducibility, at least five samples were measured for each sample and the test results were averaged. For the hysteresis measurements, the samples were first stretched to a maximum extension strain of 1000% and then unloaded. The hysteresis energy (U) was estimated by the area based on the loading–unloading curves. The stress (σ) was calculated approximately by σ = F/wt, where F is the loading force, and w and t are the original width and thickness of the specimen, respectively. The strain (ε) under elongation was defined as the change in length (l) relative to the original length (l0) of the freestanding specimen, ε = l/l0 × 100%. The initial elastic modulus (E) was calculated as the slope of the compressive stress–strain curve within the range, ε = 0–20%.
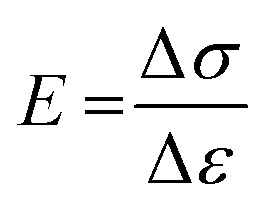 | (1) |
Hysteresis energy (U) was defined as
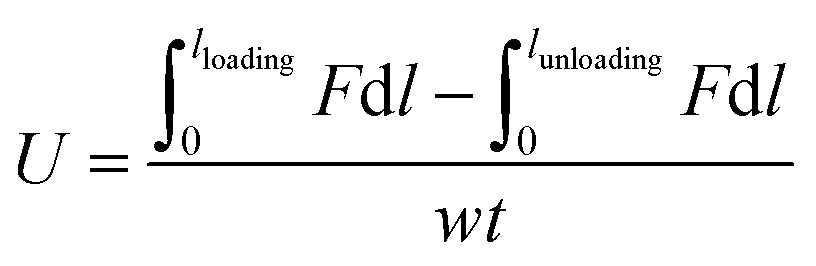 | (2) |
Compression measurements
The compression measurements (SHIMADZU, model AGS-X, 10 KN, Japan) were performed on cylindrical samples, 22 mm in a diameter and 12 mm in height at a strain rate of 2 mm min−1 with the mechanical tester. The hydrogel sample was placed between the plates of the instrument. Before the test, an initial compressive contact to 0.010 ± 0.003 N was applied to ensure complete contact between the hydrogels and the plates. To test the self-recovery rate of the compressed samples. After unloading, we immediately measured the height of the original hydrogel sample at a fixed time interval of 1 min.Self-recovery rate (ω) is defined as
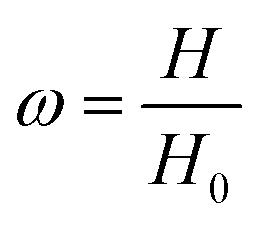 | (3) |
Rheological test
Dynamic rheological measurements were performed on a physical MCR 2000 rheometer (AR 2000ex). The dynamic viscoelastic properties (storage modulus G′ and loss modulus G′′) of the hydrogels were determined at different frequencies of 0.01–100 Hz. Frequency sweep tests for the samples were carried out at 25 °C using a parallel plate geometry (25 mm diameter).Swelling measurements
The hydrogels were cut into samples of about 2 g (diameter 20 mm × 10 mm length). Each sample was placed in an excess of deionized water at room temperature. To reach the swelling equilibrium, the sample was immersed in water for 20 d and water was replaced every 24 h. The gel was wiped with filter paper to remove excess water from the surface, and weighed carefully at a fixed time interval of 1 day. The swelling ratio (SR) was calculated from the following equation:
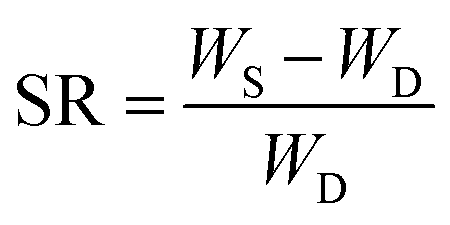 | (4) |
Morphological study
The morphological structure of the hydrogels was examined by Scanning Electron Microscopy (SEM JSM, 6510). The samples were freeze-dried under vacuum by a freeze drier (FDU-2110, Eyela). Before the measurement, all samples were placed in liquid nitrogen for 3 min to break and sputtered with platinum. The magnification factor was 100.Results and discussion
In this approach, the uniform-sized (349 nm) MMs were synthesized using BA as the main component and DCPA as a crosslinker in an aqueous medium emulsion polymerization (as shown in Fig. 1), and corresponding characterization was reported in our previous study.26 The resulting MMs were added to the mixture of acrylamide (AAm) as the hydrogel substrate and hexadecyl methacrylate (HMA) as the hydrophobic component. Subsequently, in different initiation system, AAm and HMA were polymerized to form hydrogels using MMs as the crosslinking centres for hydrophobic association and a large number of PAAm molecular chains would be well distributed around the MMs. Based on this hypothesis, the structure of the MMs-reinforced hydrogels is represented schematically in Fig. 1.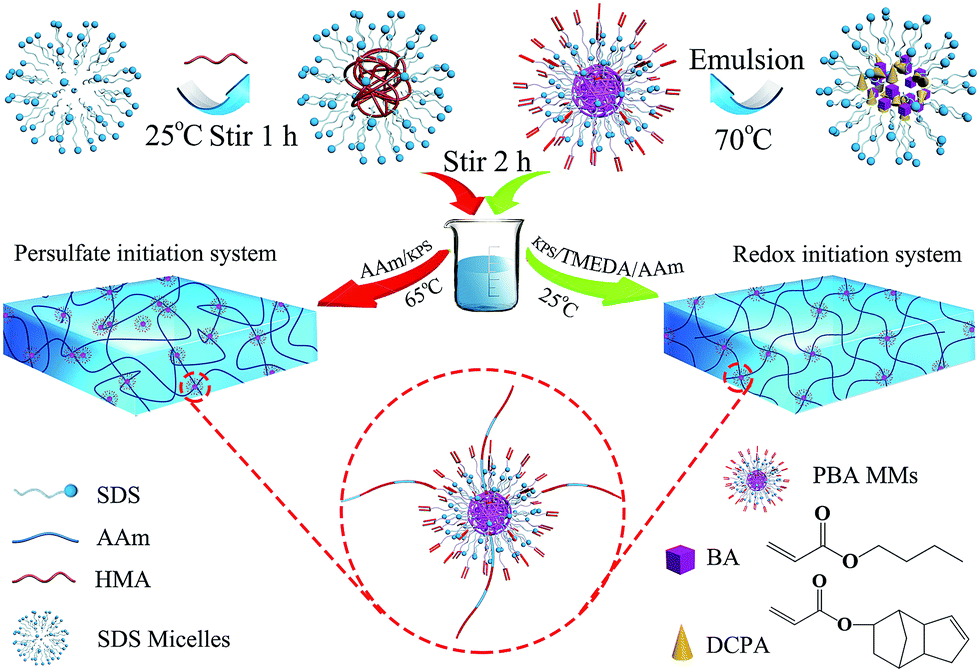 | ||
| Fig. 1 Scheme representing MMs-reinforced hydrogels. | ||
The initiation system played a significant role in the mechanical behaviour of hydrogels. In this investigation, two types of initiation systems including a thermal initiation system and a redox initiation system were used to synthesize MMs-toughening hydrogels. The initial mechanism of the redox initiation system is shown in Fig. 2(a), KPS instantly generated a large number of free radicals under the function of TEMED at 25 °C. The free radical (I) is one of the initial free radicals responsible for the initiation of the vinyl polymerization in addition to the sulfate free radicals (II). However, for a thermal initiator, KPS was decomposed gradually to HO3SO˙ at 65 °C and then the polymerization of the vinyl monomers was initiated, as shown in Fig. 2(b). For the redox initiation system, because the overall activation energy for the decomposition process was lower, a large number of free radicals was instantly produced. These free radicals initiated the polymerization of monomers. At the same time, the resulting polymer molecule chains formed tighter entanglement and the crosslinking centres distributed more evenly due to the instant generation of a large number of free radicals, as shown in Fig. 1. However, for thermal initiation system, the overall activation energy for the decomposition process was higher and the KPS was decomposed gradually to HO3SO˙ at 65 °C, which made the lengths of the resultant polymer chains different.37,38 As a result, the tight degree of physical entanglements among polymer molecule chains declined dramatically, as shown in Fig. 1.
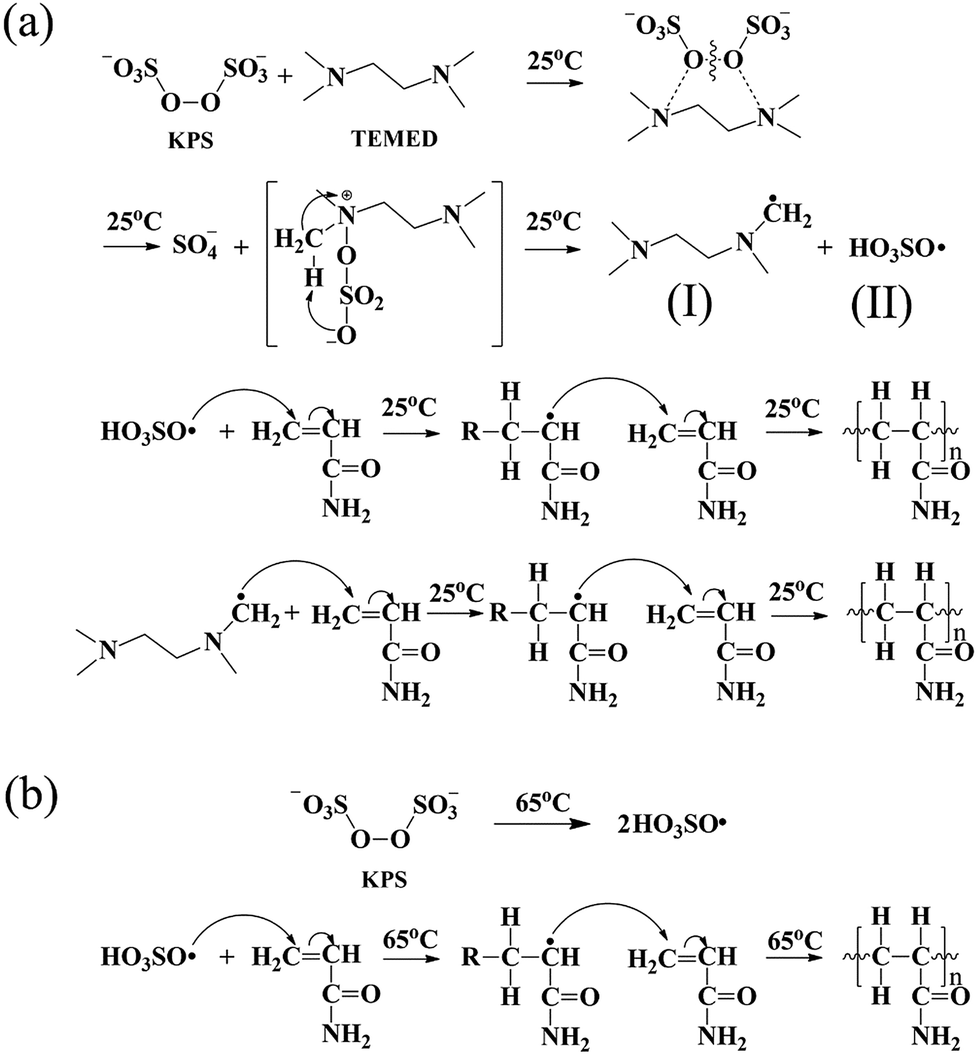 | ||
| Fig. 2 Mechanism of polymerization reaction the synthesis of different initiation systems: (a) redox initial mechanism; (b) thermal initial mechanism. | ||
Swelling characterization is one of the most important methods for analysing the properties of hydrogels. In this study, under different initiation systems, the hydrogels had different network structures and correspondingly exhibited different swelling behaviours. To reach swelling equilibrium, the samples were immersed in abundant water for 20 days, the water was replaced every 24 hours, the gel was wiped with filter paper to remove excess water from the surface, and weighed carefully at fixed time intervals of 1 day. The hydrogel prepared by the thermal initiation system was swollen 45 times (volume ratio) to its original size and the hydrogel with a redox initiation system was swollen only 8 times, as shown in Fig. 3(a). The significantly different swelling behaviours of the two types of hydrogels adequately illustrated that the hydrogel with a redox initiation system possessed tighter internal entanglement structure and homogeneous crosslinking centres distribution compared to the hydrogel with a persulfate initiation system.39 Moreover, the swelling ratio in weight of the hydrogels with and without MMs were measured and is shown in Fig. 3(b). The hydrogels without MMs exhibited a higher swelling ratio than MMs-reinforced ones, indicating that MMs could tightly interact with the hydrophobic segments in hydrogels and result in low swelling behaviour. Scanning electron microscopy (SEM) was utilized to observe the morphology of the hydrogels obtained by different initiation systems. It clearly illustrated that P(AAm/HMA)-MMs-R hydrogels exhibited a more homogeneous structure than P(AAm/HMA)-MMs-T hydrogels before swelling, as shown in Fig. 4(a and c). After swelling for 12 h, a small amount of mesh appeared in P(AAm/HMA)-MMs-R hydrogels, compared with P(AAm/HMA)-MMs-T, due to the tighter internal entanglement structure and homogeneous distribution of crosslinking centres in the hydrogels. However, P(AAm/HMA)-MMs-T hydrogels exhibited a loose network structure, which was consistent with the swelling, as shown in Fig. 4(b and d).
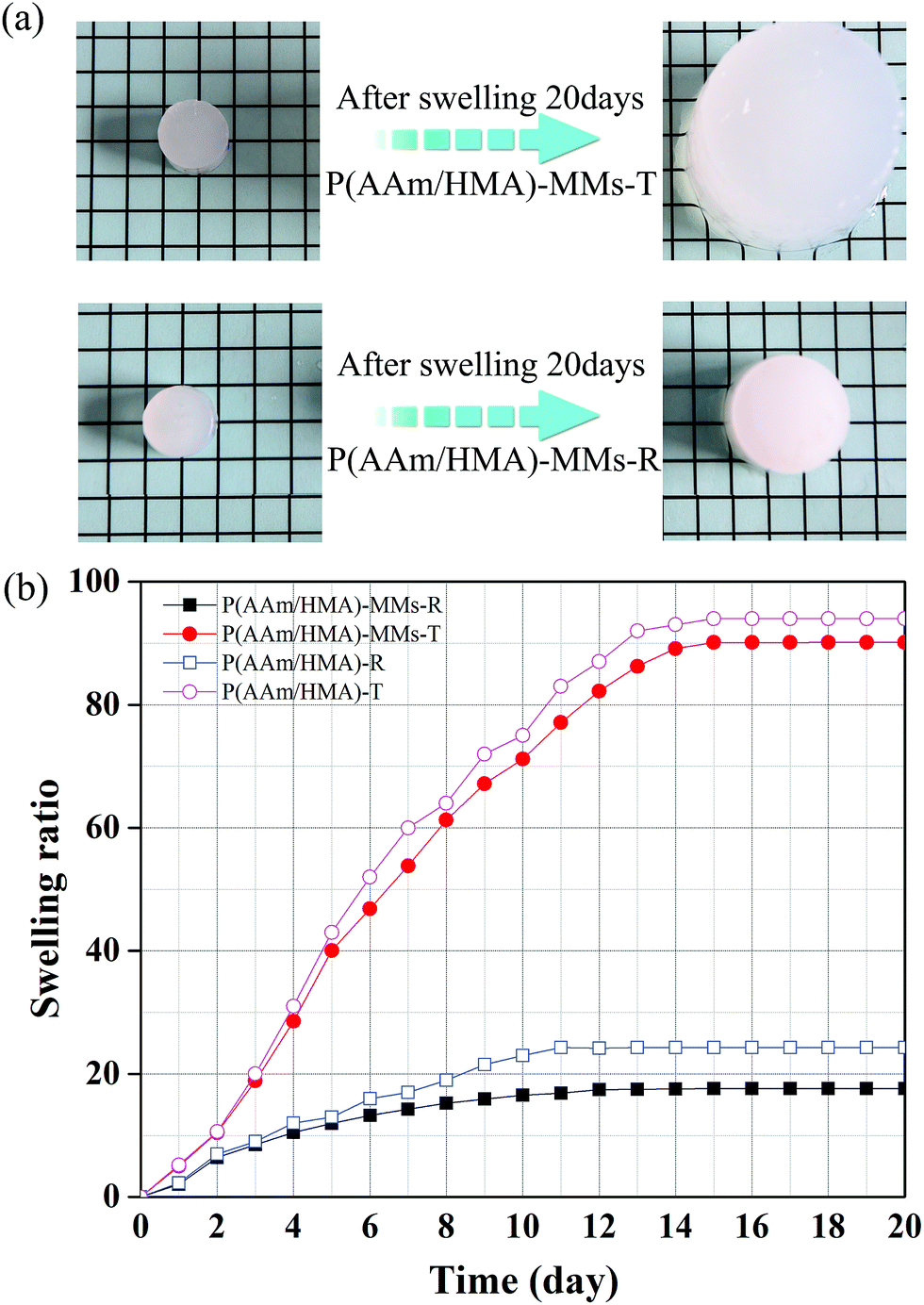 | ||
| Fig. 3 (a) Swelling behaviour and (b) Swelling ratio of P(AAm/HMA)-MMs-R, P(AAm/HMA)-MMs-T, P(AAm/HMA)-R and P(AAm/HMA)-T hydrogels. | ||
 | ||
| Fig. 4 SEM images of before (a, c) and after (b, d) swelling of P(AAm/HMA)-MMs-R (a, b) and P(AAm/HMA)-MMs-T (c, d) hydrogels. | ||
To measure the mechanical behaviour of hydrogels, the tensile test was carried out, and the corresponding tensile curves of hydrogels are shown in Fig. 5(a). The fracture strength of the hydrogels with a redox initiation system was higher than that of hydrogels with a thermal initiation system. For the redox initiation system, the hydrogels possessed a tighter internal entanglement structure and homogeneous crosslinking centres (MMs) distribution due to the instant generation of a large number of free radicals, but the hydrogels with a thermal initiation system possessed a looser internal structure due to the resulting polymer chains with different lengths under the gradual decomposition of the initiator. As a result, the hydrogels exhibited excellent mechanical strength based on the tight internal entanglement structure and homogeneous distribution of crosslinking centres. Moreover, the mechanical behaviour of the hydrogels without MMs was also measured and exhibited slightly lower stress than that of MMs-reinforced hydrogels, indicating that MMs could interact with the hydrophobic segments as crosslinking centres and efficiently enhance the mechanical behaviour of hydrogels. The corresponding elastic moduli of hydrogels are shown in Fig. 5(b), and the elastic modulus of hydrogels with a redox initiation system was higher than that of the hydrogels with a thermal initiation. This was because under the redox initiation system, the entanglement between the resulting polymer molecular chains inside the hydrogels was more compact, and the distribution of crosslinking centres was homogeneous. As a result, when the external force was applied to the hydrogel samples, there would be more polymer chains together to bear the force, which made the elastic modulus higher. However, under the thermal initiation system, the lengths of the resulting polymer molecule chains were different and fewer polymer chains undertook the role of external force, which made its elastic modulus lower. The stress–strain hysteresis loops were also measured at the strain of 1000%, as shown in Fig. 5(c). Compared to the hydrogels with a thermal initiation system, the hydrogels with a redox initiation system exhibited higher dissipation energy because their tight internal entanglement structure and homogeneous crosslinking centres distribution made them store more energy (Fig. 5(d)). Moreover, the corresponding hydrogels without MMs exhibited lower tensile stress and hysteresis energy. These results suggest that the MMs-reinforced hydrogels with a redox initiation system had favourable stiffness and toughness.40,41
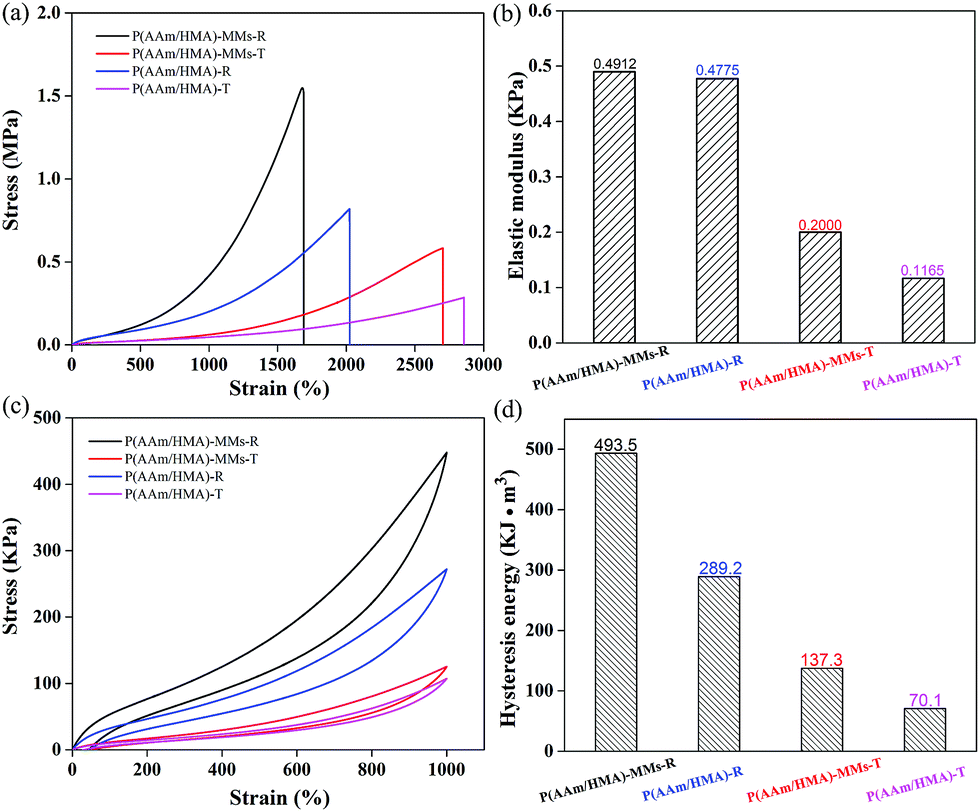 | ||
| Fig. 5 (a) Tensile curves of, and (b) corresponding elastic modulus for 20% strain; (c) tensile cycling tests for 1000% strain and (d) corresponding hysteresis energy of P(AAm/HMA)-MMs-R, P(AAm/HMA)-MMs-T, P(AAm/HMA)-R and P(AAm/HMA)-T hydrogels. | ||
As shown in Fig. 6, the hydrogels with different initiation systems displayed different mechanical properties. Fig. 6(a–d) shows that the P(AAm/HMA)-MMs-R hydrogel and the P(AAm/HMA)-MMs-T hydrogel could be stretched even with a knot. However, the P(AAm/HMA)-MMs-T hydrogel displayed higher elongation than the P(AAm/HMA)-MMs-R hydrogel, indicating that the hydrogels with a thermal initiation system possessed a looser internal structure corresponding to the stretch data. As shown in Fig. 6(e), the P(AAm/HMA)-MMs-T hydrogel could lift the steel block weighing 0.3 kg. However, the P(AAm/HMA)-MMs-R hydrogel exhibited excellent mechanical strength and could lift a steel block weighing 1 kg without breaking.
 | ||
| Fig. 6 Mechanical exhibition of MMs hydrogels with different initiation systems: the P(AAm/HMA)-MMs-R hydrogel with (a and b) knotting and stretching, the P(AAm/HMA)-MMs-T hydrogel with (c and d) knotting and stretching, (e) lifting up the steel block of 0.3 kg and 1 kg in weight by P(AAm/HMA)-MMs-T hydrogel and P(AAm/HMA)-MMs-R hydrogel, respectively. | ||
Subsequently, we investigated the self-recovery behaviour of the hydrogels with different initiation systems using a compression cycle test. When the compression strain was 90%, the stress of the redox-initiated hydrogel achieved 4.45 MPa, which was higher than that of thermal-initiated hydrogel with a stress of 3.1 MPa (Fig. 7(a)). After unloading, as shown in Fig. 7(b), P(AAm/HMA)-MMs-T hydrogel produced a great deformation and reached 82% of its original dimensions. The deformation only recovered 5% after 10 min and the P(AAm/HMA)-MMs-T hydrogel reached 87% of its original dimension. However, P(AAm/HMA)-MMs-R hydrogel quickly restored to 90% of its original dimension in several seconds and reached 96% its original dimension after 10 min. As a result, the P(AAm/HMA)-MMs-R hydrogel obtained by the redox initiation system had favourable and rapid self-recovery properties, which was due to the tight entanglement between the resulting polymer molecular chains, and the homogeneous distribution of crosslinking centers made polymer chains move easily and quickly restore their original state compared to the P(AAm/HMA)-MMs-T hydrogel obtained by the thermal initiation system.42 As shown in Fig. 7(c), the P(AAm/HMA)-MMs-R hydrogel was dyed with methylene blue to better distinguish it. Not surprisingly, the P(AAm/HMA)-MMs-R hydrogel quickly recovered its initial dimensions after the release of the external force, exhibiting prominent self-recovery behaviour. In contrast, the P(AAm/HMA)-MMs-T hydrogel was deformed and it was difficult to maintain its cylindrical shape after unloading.
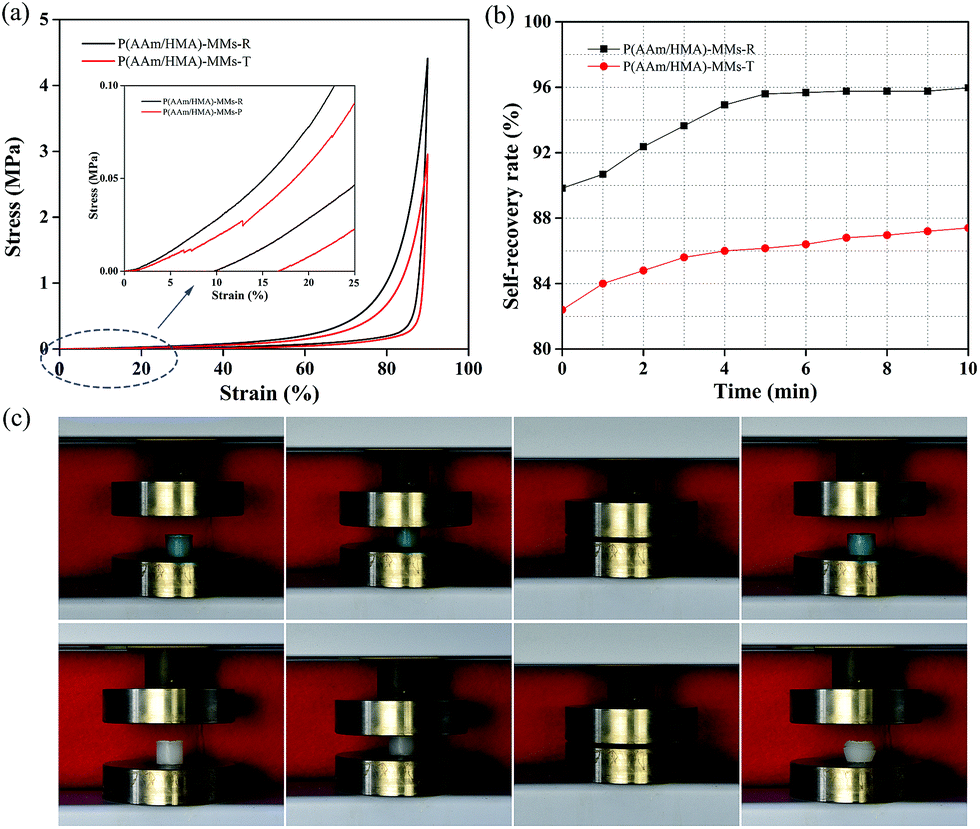 | ||
| Fig. 7 (a) Compression curves of P(AAm/HMA)-MMs-R and P(AAm/HMA)-MMs-T hydrogels; (b) the self-recovery rate of P(AAm/HMA)-MMs-R and P(AAm/HMA)-MMs-T hydrogels in the compression 90%, 0 min mean after unloading and measured height of the original hydrogel sample at fixed time interval of 1 min. (c) The photographs of P(AAm/HMA)-MMs-R hydrogels (blue) and P(AAm/HMA)-MMs-T hydrogels (white) during the compression tests. | ||
To describe the variety of internal structures for the hydrogels, the rheological test was carried out (as shown in Fig. 8). The G′ of the hydrogels was independent of the frequency, which revealed that the network of the hydrogels was stable. The order of magnitude of G′ of the P(AAm/HMA)-MMs-R hydrogels obtained by the redox initiation system was always greater than that of the P(AAm/HMA)-MMs-T hydrogels obtained by the thermal initiation system. Under the thermal initiation system, the lengths of the molecular chains were different and the corresponding internal network structure was loose, which made the network store less energy and the movement of polymer chains consume less energy. As a result, the hydrogel had a lower G′ and G′′. However, for P(AAm/HMA)-MMs-R hydrogel, the tight compact entanglement between the resulting polymer molecular chains and the homogeneous distribution of crosslinking centres made the network store more energy and the movement of polymer chains consume more energy, which resulted in a higher G′ and G′′.43
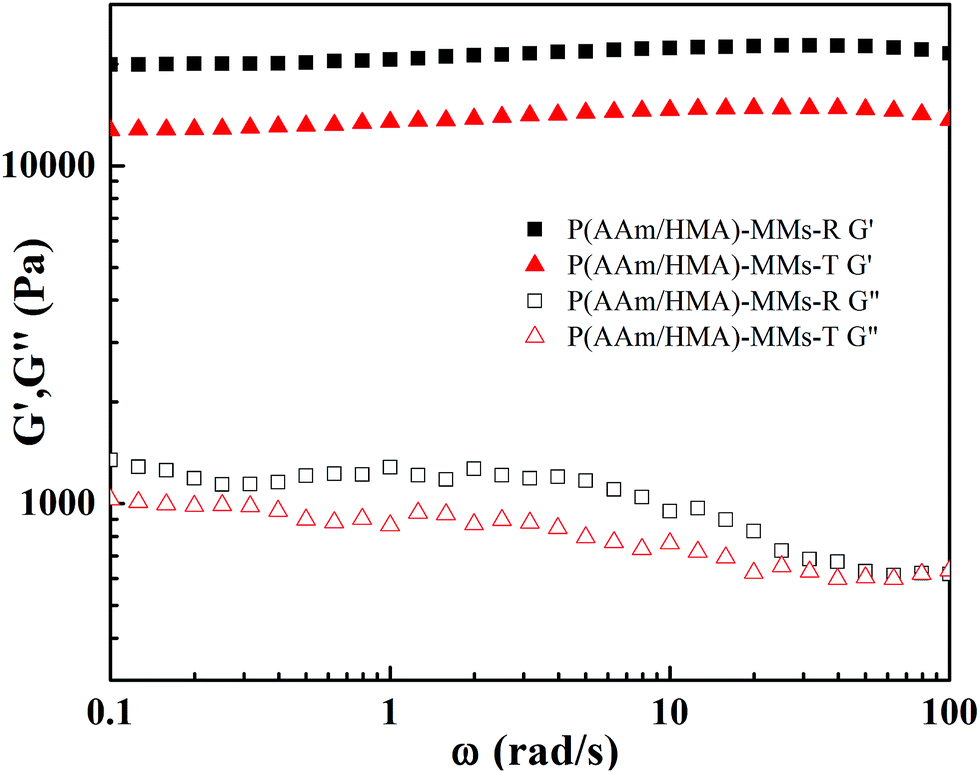 | ||
| Fig. 8 Storage modulus (G′) and loss modulus (G′′) of P(AAm/HMA)-MMs-R and P(AAm/HMA)-MMs-T hydrogels. | ||
Conclusions
MMs-reinforced hydrogels were successfully prepared by two different initiation systems, including thermal initiator KPS and redox initiators KPS and TMEDA. The hydrogels with redox initiators not only possessed high elasticity and strength, but also exhibited quick self-recoverable and lower swelling property. The maximum tensile strength of hydrogels obtained by redox initiators was approximately 1.55 MPa, superior to that of hydrogels obtained by thermal initiation system of 0.6 MPa. The hydrogels could quickly restore to 90% and reached 96% of its original dimension after 10 min. These excellent properties were attributed to the excellent internal structure of the hydrogels. For the redox initiation system, the lower activation energy during the initiating process could instantly produce a large number of free radicals to initiate polymerization of monomers under the mild conditions. All molecular chains would evenly distribute and efficiently interact with MMs as crosslinking centres. As a result, the hydrogels exhibited an excellent mechanical behaviour. However, for the thermally initiated MMs-reinforced hydrogels, the higher activation energy during the initiating process required high temperature to form free radicals, resulting in an increased probability of random chain termination. The molecular chains would bear a non-uniform distribution, adversely influencing the mechanical property of hydrogels. Therefore, the mechanical property of hydrogels was improved effectively using the redox initiation system, which would provide a procedure strategy in designing new generation tough hydrogels. It was anticipated that the hydrogels obtained under the mild conditions would played a significant role in biomedical applications.Acknowledgements
This research was supported by a grant from the National Natural Science Foundation of China (NSFC) (No. 51473023 and 51103014), Science and Technology Department of Jilin Province (No. 20150204064G), Education Department of Jilin Province (No. 201582), Changchun Municipal Science and Technology Bureau (No. 14KG054).References
- C. Silan, A. Akcali, M. T. Otkun, N. Ozbey, S. Butun, O. Ozay and N. Sahiner, Colloids Surf., B, 2012, 89, 248–253 CrossRef CAS PubMed.
- J. K. Oh, S. A. Bencherif and K. Matyjaszewski, Polymer, 2009, 50, 4407–4423 CrossRef CAS.
- N. Sahiner, W. Godbey, G. L. McPherson and V. T. John, Colloid Polym. Sci., 2006, 284, 1121–1129 CAS.
- S. C. N. Tang, D. Y. S. Yan and I. M. C. Lo, Ind. Eng. Chem. Res., 2014, 53, 15718–15724 CrossRef CAS.
- L. Jiang, P. Liu and S. Zhao, Colloids Surf., A, 2015, 470, 31–38 CrossRef CAS.
- S. H. Sontjens, D. L. Nettles, M. A. Carnahan, L. A. Setton and M. W. Grinstaff, Biomacromolecules, 2006, 7, 310–316 CrossRef PubMed.
- T. Gan, Y. Guan and Y. Zhang, J. Mater. Chem., 2010, 20, 5937–5944 RSC.
- E. C. Cho, J.-W. Kim, A. Fernández-Nieves and D. A. Weitz, Nano Lett., 2008, 8, 168–172 CrossRef CAS PubMed.
- H. M. Shewan and J. R. Stokes, J. Food Eng., 2013, 119, 781–792 CrossRef CAS.
- R. Liu, Y. Huang, Y. Ma, S. Jia, M. Gao, J. Li, H. Zhang, D. Xu, M. Wu, Y. Chen, Z. Zhu and C. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 6982–6990 CAS.
- M. R. Islam, A. Ahiabu, X. Li and M. J. Serpe, Sensors, 2014, 14, 8984–8995 CrossRef CAS PubMed.
- Y. J. Seol, J. Y. Park, W. Jeong, T. H. Kim, S. Y. Kim and D. W. Cho, J. Biomed. Mater. Res., 2015, 103, 1404–1413 CrossRef PubMed.
- Y. Lu and M. Ballauff, Prog. Polym. Sci., 2011, 36, 767–792 CrossRef CAS.
- V. Thomas, M. Namdeo, Y. Murali Mohan, S. Bajpai and M. Bajpai, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 45, 107–119 CrossRef.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- C. Li, M. J. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. A. Scherman and D. Liu, Adv. Mater., 2015, 27, 3298–3304 CrossRef CAS PubMed.
- C. Katsuno, A. Konda, K. Urayama, T. Takigawa, M. Kidowaki and K. Ito, Adv. Mater., 2013, 25, 4636–4640 CrossRef CAS PubMed.
- S. H. Jeong, Y. H. Koh, S. W. Kim, J. U. Park, H. E. Kim and J. Song, Biomacromolecules, 2016, 17, 841–851 CrossRef CAS PubMed.
- G. Gao, G. Du, Y. Sun and J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 5029–5037 CAS.
- M. K. Shin, G. M. Spinks, S. R. Shin, S. I. Kim and S. J. Kim, Adv. Mater., 2009, 21, 1712–1715 CrossRef CAS.
- Z. Hu and G. Chen, Adv. Mater., 2014, 26, 5950–5956 CrossRef CAS PubMed.
- T. Huang, H. G. Xu, K. X. Jiao, L. P. Zhu, H. R. Brown and H. L. Wang, Adv. Mater., 2007, 19, 1622–1626 CrossRef CAS.
- C. He, K. Jiao, X. Zhang, M. Xiang, Z. Li and H. Wang, Soft Matter, 2011, 7, 2943 RSC.
- L. W. Xia, R. Xie, X. J. Ju, W. Wang, Q. Chen and L. Y. Chu, Nat. Commun., 2013, 4, 2226 Search PubMed.
- J. Hu, K. Hiwatashi, T. Kurokawa, S. M. Liang, Z. L. Wu and J. P. Gong, Macromolecules, 2011, 44, 7775–7781 CrossRef CAS.
- X. Y. Ren, Z. Yu, B. Liu, X. J. Liu, Y. J. Wang, Q. Su and G. H. Gao, RSC Adv., 2016, 6, 8956–8963 RSC.
- G. Jiang, C. Liu, X. Liu, Q. Chen, G. Zhang, M. Yang and F. Liu, Polymer, 2010, 51, 1507–1515 CrossRef CAS.
- V. Can, Z. Kochovski, V. Reiter, N. Severin, M. Siebenbürger, B. Kent, J. Just, J. P. Rabe, M. Ballauff and O. Okay, Macromolecules, 2016, 49, 2281–2287 CrossRef CAS.
- G. Jiang, C. Liu, X. Liu, G. Zhang, M. Yang and F. Liu, Macromol. Mater. Eng., 2009, 294, 815–820 CrossRef CAS.
- Y. Yang, X. Wang, F. Yang, H. Shen and D. Wu, Adv. Mater., 2016, 28, 7178–7184 CrossRef CAS PubMed.
- H. Yuk, T. Zhang, G. A. Parada, X. Liu and X. Zhao, Nat. Commun., 2016, 7, 12028 CrossRef PubMed.
- F.-K. Shi, M. Zhong, L.-Q. Zhang, X.-Y. Liu and X.-M. Xie, J. Mater. Chem. B, 2016, 4, 6221–6227 RSC.
- Y.-n. Sun, G.-r. Gao, G.-l. Du, Y.-j. Cheng and J. Fu, ACS Macro Lett., 2014, 3, 496–500 CrossRef CAS.
- Y. Sun, S. Liu, G. Du, G. Gao and J. Fu, Chem. Commun., 2015, 51, 8512–8515 RSC.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- D. C. Tuncaboylu, M. Sahin, A. Argun, W. Oppermann and O. Okay, Macromolecules, 2012, 45, 1991–2000 CrossRef CAS.
- X. D. Feng, X. Q. Guo and K. Y. Qiu, Macromol. Chem. Phys., 1988, 189, 77–83 CrossRef CAS.
- M. R. Guilherme, R. da Silva, A. F. Rubira, G. Geuskens and E. C. Muniz, React. Funct. Polym., 2004, 61, 233–243 CrossRef CAS.
- X. J. Liu, H. Q. Li, B. Y. Zhang, Y. J. Wang, X. Y. Ren, S. Guan and G. Hui Gao, RSC Adv., 2016, 6, 4850–4857 RSC.
- C. He, Z. Zheng, D. Zhao, J. Liu, J. Ouyang and H. Wang, Soft Matter, 2013, 9, 2837 RSC.
- Q. Chen, L. Zhu, H. Chen, H. Yan, L. Huang, J. Yang and J. Zheng, Adv. Funct. Mater., 2015, 25, 1598–1607 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583 RSC.
- G. Gao, G. Du, Y. Cheng and J. Fu, J. Mater. Chem. B, 2014, 2, 1539 RSC.
Footnote |
| † These authors contribute equally to this paper. |
| This journal is © The Royal Society of Chemistry 2017 |