DOI:
10.1039/C7RA00954B
(Paper)
RSC Adv., 2017,
7, 15997-16004
Semisynthesis of some matrine ether derivatives as insecticidal agents†
Received
22nd January 2017
, Accepted 27th February 2017
First published on 13th March 2017
Abstract
In continuation of our program to discover new potential natural-product-based crop protection agents, we synthesized a series of 14-formyl-15-aryloxy/methoxymatrine and 14-aryloxymethylidenylmatrine derivatives as pesticidal agents by structural modification of matrine, a biorenewable quinolizidine alkaloid isolated from Sophora flavescens. The structural assignment was based on spectroscopic and X-ray analysis data. Their pesticidal activities were carried out against two typically crop-threatening agricultural insect pests, Mythimna separata Walker and Plutella xylostella Linnaeus. Compounds 4i and 4k exhibited more potent oral toxicity than matrine against 3rd-instar larvae of P. xylostella. As compared with matrine, all derivatives displayed a growth inhibitory property against early 3rd-instar larvae of M. separata, and in particular compounds 4i–k displayed more promising insecticidal activity than toosendanin. Some interesting results of structure–activity relationships were also observed.
Introduction
Mythimna separata Walker (oriental armyworm) and Plutella xylostella Linnaeus (diamondback moth) are two widely distributed and serious typical lepidopteran insect pests. Their infestations are very troublesome and hard to control.1,2 In 2012, intermittent outbreaks of M. separata occurred widely in China, and about 4 million hectares of crops were completely lost.3 Although lots of chemical pesticides have been extensively applied to deal with insect pest outbreaks, insect pest resistance and negative impacts on human health and environmental safety have emerged.4–7 Therefore, the development of new potential alternatives to effectively and selectively control insect pests is highly urgent.
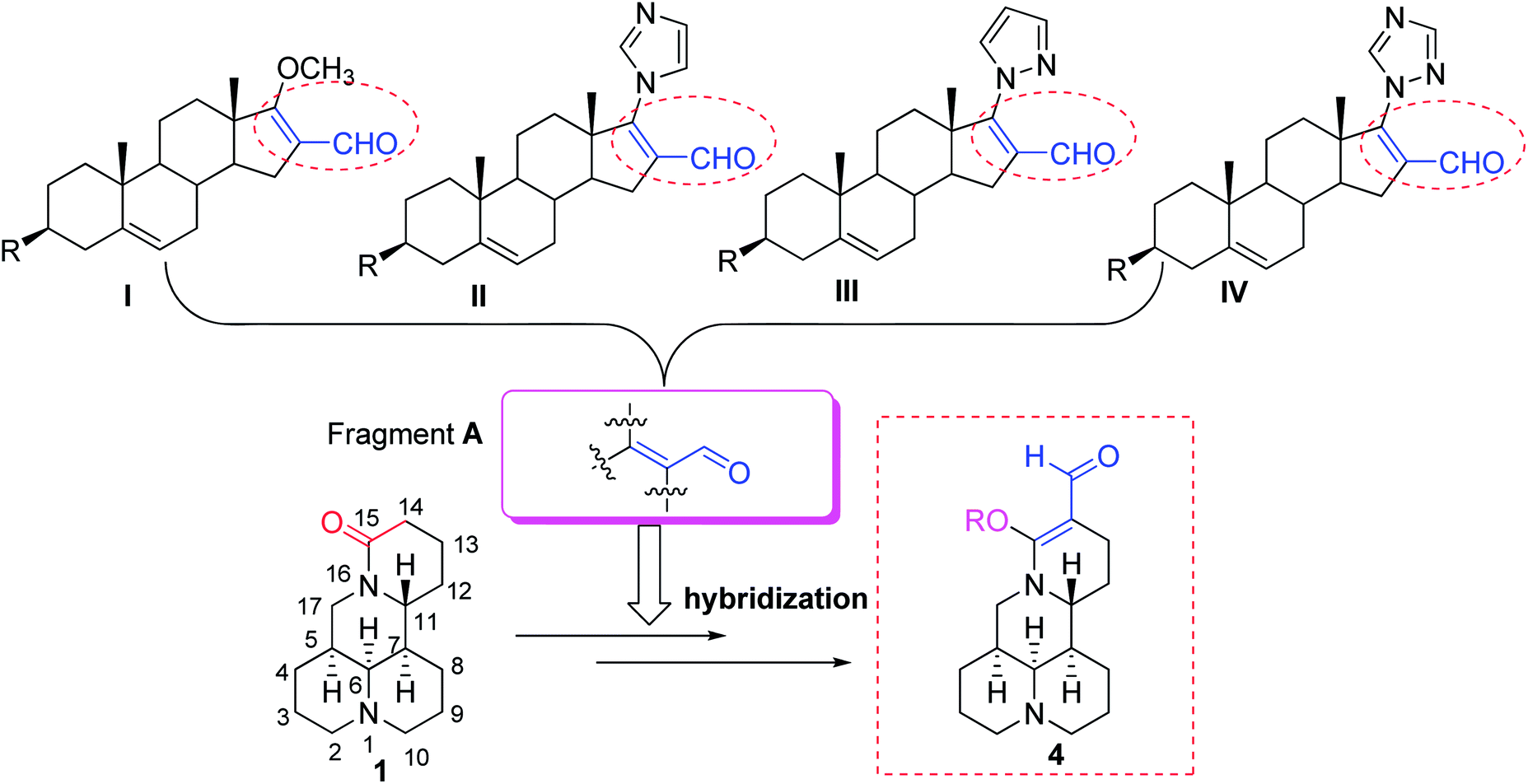
Matrine (1, Fig. 1) is a biorenewable quinolizidine alkaloid isolated from the roots of Sophora flavescens (Kushen), which is widely distributed in Asia, Oceanica, and the Pacific islands.8,9 Matrine and its derivatives exhibited a broad scope of biological properties such as anticancer, anti-inflammatory, and antiviral activities.10–16 Meanwhile, compound 1 also exhibited potent insecticidal activity in the agricultural field.17–19 On the other hand, it was found that the introduction of an acrylic aldehyde scaffold (fragment A, Fig. 1) into dehydroepiandrosterone could produce derivatives I–IV20–22 (Fig. 1), which showed potent antiproliferative effects. Moreover, their lipophilicity, which improves their ability to cross through cell membranes, was increased, whereas their side effects and toxicity were decreased.21 Meanwhile, to the best of our knowledge, little attention has been paid to the structural modifications of compound 1 as pesticidal agents. Based upon the above interesting results, to discover biorenewable matrine-based pesticides, herein an acrylic aldehyde scaffold was introduced into compound 1, followed by transformation to 14-formyl-15-aryloxy/methoxymatrine (4, Fig. 1). Their pesticidal activities were evaluated against two typically crop-threatening insect pests, M. separata and P. xylostella.
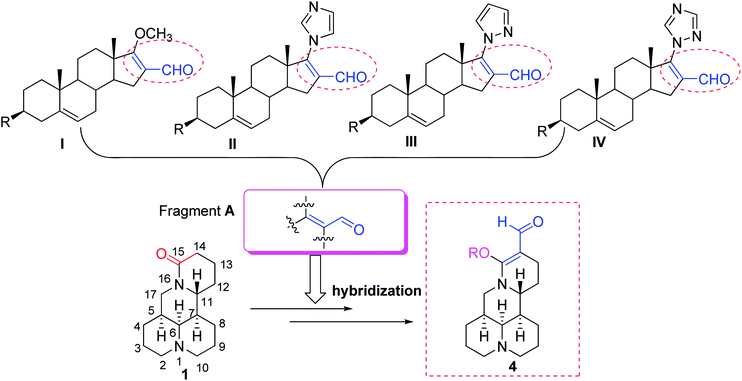 |
| | Fig. 1 Chemical structures of matrine (1), its derivatives (4), and other dehydroepiandrosterone derivatives (I–IV). | |
Materials and methods
General
All chemical reagents were purchased and utilized without further purification. Compound 1 was purchased from Baoji Haoxiang Bio-technology Co. Ltd. Melting point (mp) was determined using the XT-4 digital mp apparatus. Optical rotation was measured using an Autopol III automatic polarimeter. Infrared (IR) spectra were measured by a TENSOR 27 spectrometer. Proton nuclear magnetic resonance spectra (1H NMR) were measured with Avance III 500 MHz equipment. High-resolution mass spectra (HRMS) were carried out with an LTQ FT Ultra instrument. Microwave irradiation was performed in a CEM Discover Synthesis Unit.
Synthesis of 14-formyl-15-chloromatrine (2)
POCl3 (15 mmol) was added slowly to a solution of DMF (1.2 mL) in dry CH2Cl2 (5 mL) at 0 °C. After addition, the mixture was stirred for 1 h at 0 °C. Then, a solution of 1 (5 mmol) in dry CH2Cl2 (15 mL) was added dropwise to the above mixture. After addition, the reaction temperature was slowly raised to room temperature, and the reaction process was checked by TLC analysis. After 9 h, the mixture was concentrated in vacuo, and 5 mL of ice water was added. The pH value of the mixture was adjusted with 40% aq. NaOH to 8–9. Subsequently, hydrolysis of the mixture was conducted at 60 °C for 2 h. After cooling, the pale yellow precipitate was filtered and washed with water. Finally, the solid was purified by silica gel column chromatography eluted with dichloromethane/methanol to give 2 (92% yield) as a pale yellow solid.
Data for 2
Mp 106–108 °C; [α]20D = −83 (c 2.6 mg mL−1, CHCl3); IR cm−1 (KBr): 2776, 1635, 1564; 1H NMR (500 MHz, CDCl3) δ: 9.67 (s, 1H), 4.06–4.10 (m, 1H), 4.01 (dd, J = 13.0, 4.0 Hz, 1H), 3.59 (t, J = 13.0 Hz, 1H), 2.79–2.86 (m, 2H), 2.39–2.44 (m, 1H), 2.27–2.33 (m, 1H), 2.18 (s, 1H), 1.96–2.03 (m, 2H), 1.80–1.92 (m, 4H), 1.55–1.71 (m, 5H), 1.43–1.49 (m, 2H), 1.32–1.40 (m, 1H); HRMS (ESI): calcd for C16H24ON2Cl ([M + H]+), 295.1572; found, 295.1571.
General procedure for synthesis of 4a–e, k, l
A mixture of 2 (0.3 mmol), KOH (0.6 mmol), and different phenols (3a–d, k, l, 0.9 mmol) in DMF (5 mL) were stirred at 120 °C for 2–9 h; or a mixture of 2 (0.3 mmol) and KOH (1.5 mmol) in MeOH (3e, 5 mL) was refluxed for 14 h. Then the reaction mixture was diluted with water (10 mL), and extracted with ethyl acetate (15 mL × 3). Subsequently, the combined organic phase was washed by saturated aq. Na2CO3 (20 mL × 2), dried over anhydrous Na2SO4, concentrated in vacuo, and purified by PTLC eluted with dichloromethane/methanol to afford target compounds 4a–e, k, l.
General procedure for synthesis of 4f–j
Compound 2 (0.3 mmol) reacted with different phenols (3f–j, 0.9 mmol) in the presence of K2CO3 (0.66 mmol) and DMF (0.5 mL) under microwave irradiation at 120 °C for 20 min. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2 (10 mL), and washed by saturated aq. Na2CO3 (5 mL × 2), dried over anhydrous Na2SO4, concentrated in vacuo, and purified by PTLC eluted with dichloromethane/methanol to afford target compounds 4f–j.
Data for 4a
Yield: 65%, pale yellow solid, mp 97–99 °C; [α]20D = −16 (c 3.3 mg mL−1, CHCl3); IR cm−1 (KBr): 3051, 2759, 1635, 1576; 1H NMR (500 MHz, CDCl3) δ: 9.39 (s, 1H), 7.29–7.32 (m, 2H), 7.04 (t, J = 7.5 Hz, 1H), 7.01 (d, J = 8.0 Hz, 2H), 3.95–4.00 (m, 1H), 3.47 (dd, J = 13.0, 3.5 Hz, 1H), 3.32 (t, J = 13.0 Hz, 1H), 2.74–2.84 (m, 2H), 2.44–2.50 (m, 1H), 2.35–2.41 (m, 1H), 2.11 (s, 1H), 1.89–2.05 (m, 4H), 1.73–1.79 (m, 1H), 1.55–1.67 (m, 4H), 1.36–1.46 (m, 5H); HRMS (ESI): calcd for C22H29O2N2 ([M + H]+), 353.2224; found, 353.2221.
Data for 4b
Yield: 77%, pale yellow solid, mp 134–136 °C; [α]20D = −18 (c 3.2 mg mL−1, CHCl3); IR cm−1 (KBr): 3052, 2757, 1637, 1564, 1577, 1505; 1H NMR (500 MHz, CDCl3) δ: 9.33 (s, 1H), 7.18 (d, J = 7.0 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 3.97–3.99 (m, 1H), 3.34 (d, J = 8.0 Hz, 2H), 2.74–2.84 (m, 2H), 2.41–2.45 (m, 2H), 2.34 (s, 3H), 2.12 (s, 1H), 1.90–2.03 (m, 4H), 1.78 (s, 1H), 1.56–1.70 (m, 4H), 1.37–1.46 (m, 5H); HRMS (ESI): calcd for C23H31O2N2 ([M + H]+), 367.2380; found, 367.2380.
Data for 4c
Yield: 64%, pale yellow solid, mp 159–161 °C; [α]20D = −24 (c 3.0 mg mL−1, CHCl3); IR cm−1 (KBr): 3052, 2762, 1635, 1564, 1505; 1H NMR (500 MHz, CDCl3) δ: 9.38 (s, 1H), 7.29 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 3.95–4.00 (m, 1H), 3.51 (dd, J = 13.0, 4.5 Hz, 1H), 3.32 (t, J = 13.0 Hz, 1H), 2.74–2.84 (m, 2H), 2.35–2.47 (m, 2H), 2.12 (s, 1H), 1.90–2.03 (m, 4H), 1.75–1.80 (m, 1H), 1.59–1.69 (m, 4H), 1.37–1.46 (m, 5H), 1.29 (s, 9H); HRMS (ESI): calcd for C26H37O2N2 ([M + H]+), 409.2850; found, 409.2849.
Data for 4d
Yield: 52%, pale yellow solid, mp 101–103 °C; [α]20D = −19 (c 2.0 mg mL−1, CHCl3); IR cm−1 (KBr): 3059, 2764, 1635, 1571, 1485; 1H NMR (500 MHz, CDCl3) δ: 9.29 (s, 1H), 7.19 (d, J = 11.0 Hz, 2H), 6.89 (d, J = 11.0 Hz, ArH, 2H), 3.87–3.93 (m, 1H), 3.24–3.37 (m, 2H), 2.67–2.78 (m, 2H), 2.27–2.42 (m, 2H), 2.05 (s, 1H), 1.83–1.99 (m, 4H), 1.64–1.72 (m, 2H), 1.49–1.60 (m, 4H), 1.33–1.40 (m, 4H); HRMS (ESI): calcd for C26H28O2N2Cl ([M + H]+), 387.1834; found, 387.1833.
Data for 4e
Yield: 51%, pale yellow solid, mp 84–86 °C; [α]20D = −40 (c 2.8 mg mL−1, CHCl3); IR cm−1 (KBr): 2757, 1623, 1555;1H NMR (500 MHz, CDCl3) δ: 9.50 (s, CHO, 1H), 3.86–3.90 (m, 1H), 3.83 (s, 3H), 3.57 (dd, J = 12.5, 4.5 Hz, 1H), 3.38 (t, J = 12.5 Hz, 1H), 2.78–2.85 (m, 2H), 2.25–2.37 (m, 2H), 2.15 (s, 1H), 1.85–2.01 (m, 4H), 1.72–1.75 (m, 2H), 1.56–1.67 (m, 5H), 1.41–1.48 (m, 3H); HRMS (ESI): calcd for C17H27O2N2 ([M + H]+), 291.2067; found, 291.2067.
Data for 4f
Yield: 74%, pale yellow solid, mp 100–102 °C; [α]20D = −30 (c 2.5 mg mL−1, CHCl3); IR cm−1 (KBr): 3043, 2761, 1632, 1574; 1H NMR (500 MHz, CDCl3) δ: 9.34 (s, 1H), 7.16 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 6.79–6.82 (m, 2H), 3.97–4.01 (m, 1H), 3.49 (dd, J = 13.0, 4.0 Hz, 1H), 3.32 (t, J = 12.5 Hz, 1H), 2.75–2.84 (m, 2H), 2.38–2.50 (m, 2H), 2.33 (s, 3H), 2.12 (s, 1H), 1.92–2.05 (m, 4H), 1.74–1.80 (m, 1H), 1.58–1.68 (m, 4H), 1.37–1.45 (m, 5H); HRMS (ESI): calcd for C23H31O2N2 ([M + H]+), 367.2380; found, 367.2380.
Data for 4g
Yield: 72%, pale yellow solid, mp 90–92 °C; [α]20D = −15 (c 2.7 mg mL−1, CHCl3); IR cm−1 (KBr): 3002, 2743, 1628, 1562, 1507; 1H NMR (500 MHz, CDCl3) δ: 9.35 (s, 1H), 7.08 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 3.95–3.99 (m, 1H), 3.48 (dd, J = 13.0, 4.5 Hz, 1H), 3.31 (t, J = 13.0 Hz, 1H), 2.84–2.74 (m, 2H), 2.35–2.50 (m, 2H), 2.30 (s, 3H), 2.11 (s, 1H), 1.89–2.04 (m, 4H), 1.72–1.78 (m, 1H), 1.55–1.67 (m, 4H), 1.39–1.45 (m, 5H); HRMS (ESI): calcd for C23H31O2N2 ([M + H]+), 367.2380; found, 367.2379.
Data for 4h
Yield: 70%, pale yellow solid, mp 87–89 °C; [α]20D = −18 (c 2.6 mg mL−1, CHCl3); IR cm−1 (KBr): 3057, 2762, 1635, 1579, 1501; 1H NMR (500 MHz, CDCl3) δ: 9.40 (s, 1H), 6.94 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 3.93–3.97 (m, 1H), 3.77 (s, 3H), 3.48 (dd, J = 14.0, 5.0 Hz, 1H), 3.31 (t, J = 12.5 Hz, 1H), 2.74–2.84 (m, 2H), 2.33–2.50 (m, 2H), 2.10 (s, 1H), 1.91–2.03 (m, 4H), 1.71–1.76 (m, 1H), 1.52–1.64 (m, 4H), 1.37–1.45 (m, 5H); HRMS (ESI): calcd for C23H31O3N2 ([M + H]+), 383.2329; found, 383.2326.
Data for 4i
Yield: 65%, white solid, mp 100–102 °C; [α]20D = −30 (c 2.6 mg mL−1, CHCl3); IR cm−1 (KBr): 3055, 2774, 1628, 1562, 1505; 1H NMR (500 MHz, CDCl3) δ: 9.45 (s, 1H), 7.79 (t, J = 8.0 Hz, 2H), 7.71 (d, J = 8.5 Hz, 1H), 7.46 (t, J = 8.0 Hz, 1H), 7.39 (d, J = 7.5 Hz, 1H), 7.33 (s, 1H), 7.26–7.28 (m, 1H), 4.01–4.03 (m, 1H), 3.51 (dd, J = 13.0, 4.0 Hz, 1H), 3.34 (t, J = 12.5 Hz, 1H), 2.73–2.84 (m, 2H), 2.41–2.54 (m, 2H), 2.09–2.11 (m, 2H), 1.88–2.00 (m, 3H), 1.80–1.84 (m, 1H), 1.58–1.71 (m, 4H), 1.34–1.47 (m, 5H); HRMS (ESI): calcd for C26H31O2N2 ([M + H]+), 403.2380; found, 403.2377.
Data for 4j
Yield: 58%, white solid, mp 103–105 °C; [α]20D = −11 (c 2.8 mg mL−1, CHCl3); IR cm−1 (KBr): 3057, 2757, 1635, 1595, 1576, 1507; 1H NMR (500 MHz, CDCl3) δ: 9.41 (s, 1H), 8.28–8.30 (m, 1H), 7.85–7.87 (m, 1H), 7.54–7.57 (m, 3H), 7.32 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 4.03 (s, 1H), 3.41 (dd, J = 13.0, 4.5 Hz, 1H), 3.32 (t, J = 13.0 Hz, 1H), 2.82 (d, J = 10.5 Hz, 1H), 2.72 (d, J = 11.5 Hz, 1H), 2.47 (s, 2H), 1.87–2.13 (m, 6H), 1.58–1.75 (m, 4H), 1.39–1.56 (m, 5H); HRMS (ESI): calcd for C26H31O2N2 ([M + H]+), 403.2380; found, 403.2377.
Data for 4k
Yield: 49%, pale yellow solid, mp 162–164 °C; [α]20D = 32 (c 3.0 mg mL−1, CHCl3); IR cm−1 (KBr): 3111, 3029, 2771, 1675, 1650, 1613, 1588, 1553, 1513, 1494; 1H NMR (500 MHz, CDCl3) δ: 8.21 (d, J = 8.5 Hz, 2H), 7.16 (d, J = 8.5 Hz, 2H), 6.60 (s, 1H), 4.46 (dd, J = 12.5, 3.5 Hz, 1H), 3.89–3.94 (m, 1H), 3.09 (t, J = 13.0 Hz, 1H), 2.78–2.86 (m, 2H), 2.27–2.41 (m, 3H), 2.10 (s, 1H), 1.87–2.01 (m, 3H), 1.62–1.75 (m, 4H), 1.39–1.55 (m, 6H); HRMS (ESI): calcd for C22H28O4N3 ([M + H]+), 398.2074; found, 398.2073.
Data for 4l
Yield: 33%, pale yellow solid, mp 152–154 °C; [α]20D = 18 (c 3.2 mg mL−1, CHCl3); IR cm−1 (KBr): 3070, 3026, 2774, 1697, 1672, 1596, 1505; 1H NMR (500 MHz, CDCl3) δ: 9.92 (s, 1H), 7.85 (d, J = 8.5 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 6.63 (s, 1H), 4.48 (dd, J = 12.5, 3.0 Hz, 1H), 3.89–3.92 (m, 1H), 3.08 (t, J = 12.5 Hz, 1H), 2.78–2.86 (m, 2H), 2.26–2.40 (m, 3H), 2.10 (s, 1H), 1.87–2.01 (m, 3H), 1.63–1.74 (m, 4H), 1.42–1.55 (m, 6H); HRMS (ESI): calcd for C23H29O3N2 ([M + H]+), 381.2173; found, 381.2172.
Biological assay
Oral toxicity of 1 and 4a, d, e, g, i, k against Plutella xylostella. Thirty 3rd-instar larvae of P. xylostella were chosen as the test insects for each compound. Solutions of 1; 4a, d, e, g, i, k; and toosendanin (a positive control) were prepared in acetone at 20 mg mL−1. The corresponding solution (1 μL) was added to a fresh Brassica oleracea leaf disc (0.5 × 0.5 cm), and dried. A fresh Brassica oleracea leaf disc was treated by acetone alone as the blank control group (CK). One piece of the above discs was offered to and consumed by each insect, which was raised in each well of 12- or 24-well culture plates for 48 h (temperature: 25 ± 2 °C; relative humidity (RH): 65–80%; photoperiod: light/dark (L/D) = 16/8 h). Their corrected mortality rate values were calculated as follows: corrected mortality rate (%) = (T − C) × 100/(100% − C); C is the mortality rate of CK, and T is the mortality rate of the treated P. xylostella.23,24
Growth inhibitory activity of 1, 2, and 4a–l against Mythimna separata. Thirty early 3rd-instar larvae of M. separata were chosen as the test insects for each compound. Solutions of 1, 2, 4a–l, and toosendanin (a positive control) were prepared in acetone at 1 mg mL−1. After dipping into the corresponding solution for 3 s, wheat leaf discs (1 × 1 cm) were taken out, and dried. Wheat leaf discs were treated by acetone alone as the blank control group (CK). Several of the above discs were added to each culture dish (ten insects per dish). Once the discs had been consumed, additional ones were added. After 48 h, the rest of the compound-soaked discs were cleaned out, and untreated ones were added until the end of pupation (temperature: 25 ± 2 °C; RH: 65–80%; photoperiod: L/D = 12/12 h). Their corrected mortality rate values were calculated as follows: corrected mortality rate (%) = (T − C) × 100/(100% − C); C is the mortality rate of CK, and T is the mortality rate of the treated M. separata.25,26
Results and discussion
Synthesis
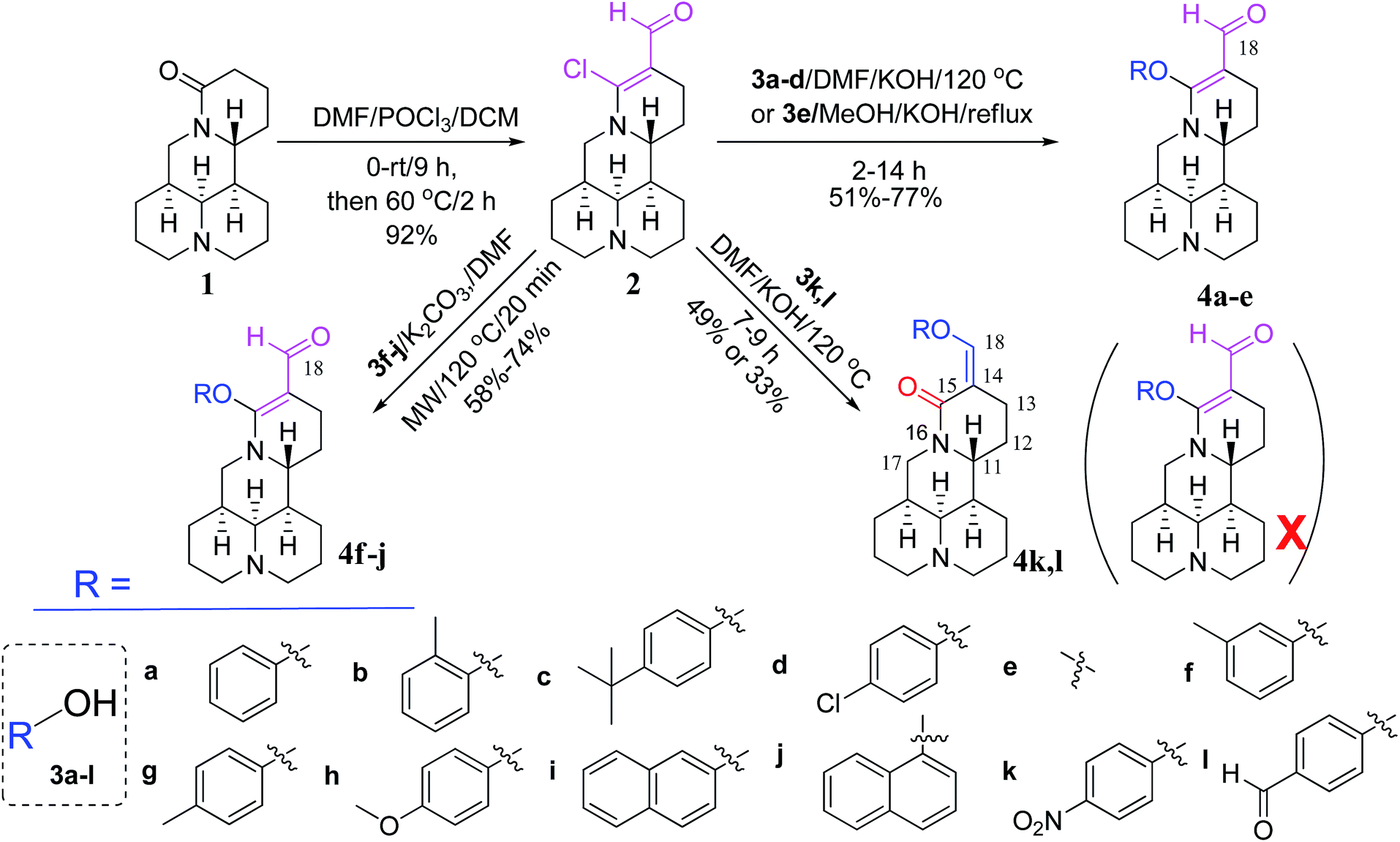
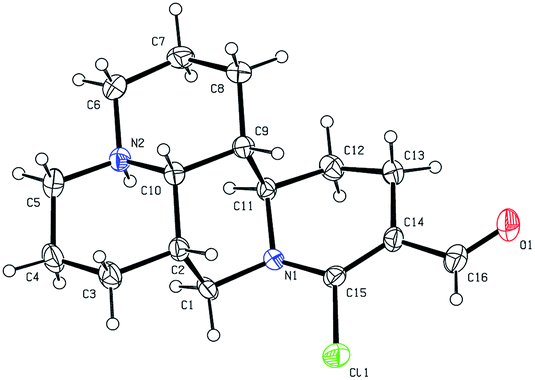
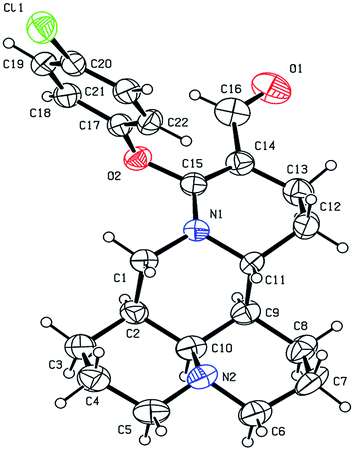
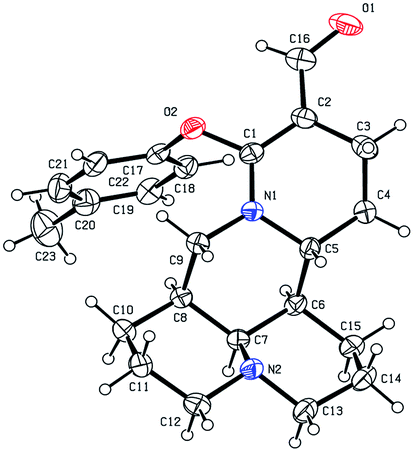
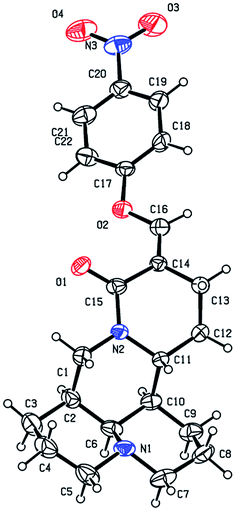
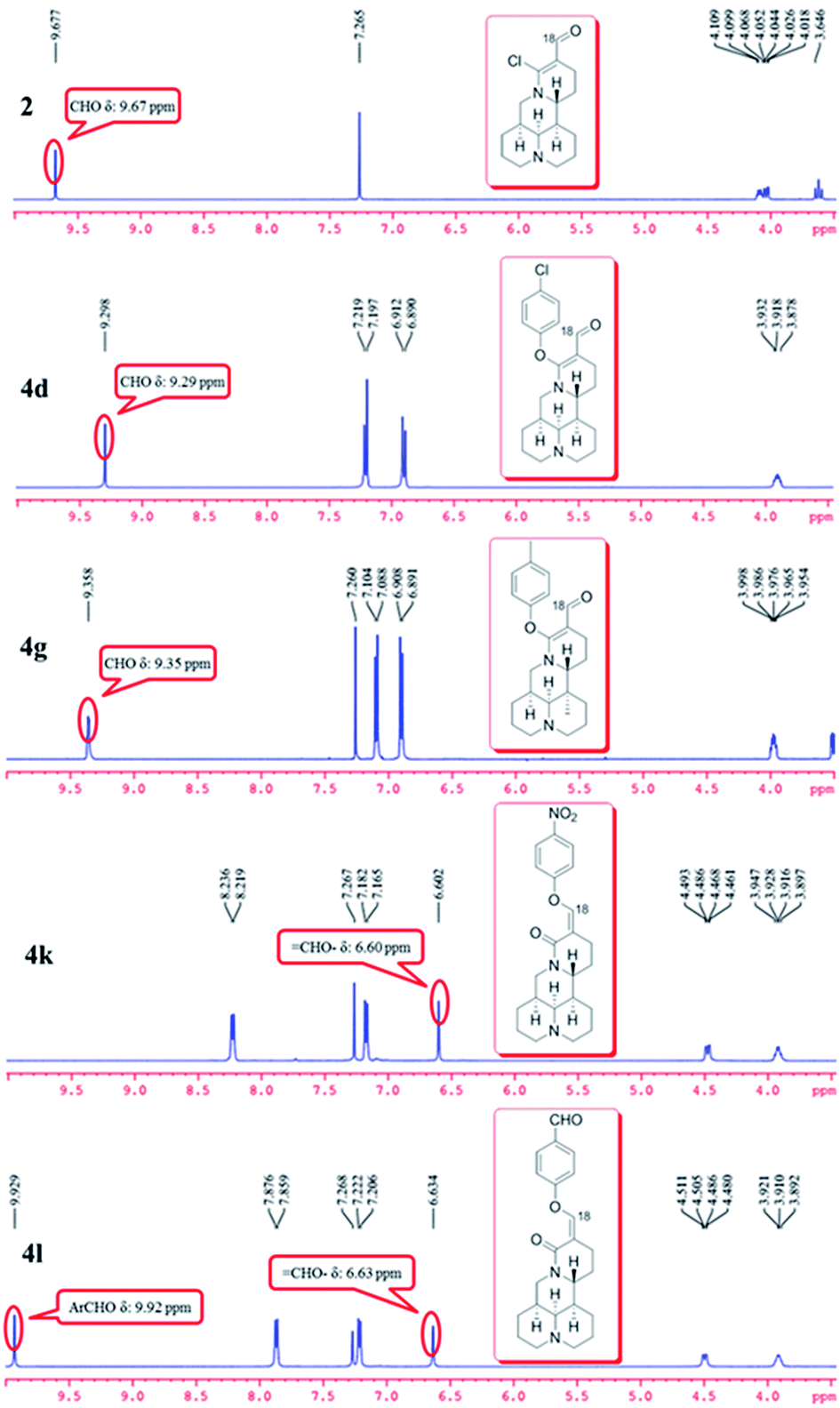
As shown in Scheme 1, first, the key intermediate, 14-formyl-15-chloromatrine (2), was smoothly synthesized by the Vilsmeier–Haack reaction of matrine (1) with DMF in the presence of POCl3. Then, 14-formyl-15-aryloxy/methoxymatrine derivatives (4a–e) were easily prepared by the reaction of 2 with corresponding phenols (3a–d) or methanol (3e) in the presence of KOH at 120 °C or under reflux.27 However, when 2 reacted with 3f–j under the above reaction conditions, it was very difficult to separate 14-formyl-15-aryloxymatrine derivatives (4f–j) from their by-products. It is noteworthy that when 2 reacted with 3f–j under microwave irradiation at 120 °C for 20 min, compounds 4f–j were smoothly obtained. Interestingly, when 2 reacted with 3k, l in the presence of KOH at 120 °C, only 14-aryloxymethylidenylmatrines (4k, l) were produced. Their structures were well determined by melting points, optical rotation, IR, 1H NMR, and HRMS. In particular, three-dimensional structures of compounds 2, 4d, 4g and 4k were determined by X-ray crystallography (Fig. 2–5). Crystallographic data (excluding structure factors) of 2, 4d, 4g and 4k were deposited at the Cambridge Crystallographic Data Centre (CCDC) with deposition numbers of 1522361, 1524882, 1524886 and 1524887, respectively. Assignment of the chemical structure for 4l was based on the chemical shift of H-18. As shown in Fig. 6, the chemical shifts of H-18 of 2, 4d, and 4g were at 9.67, 9.29, and 9.35 ppm, respectively. Whereas the chemical shift of H-18 of 4k was at 6.60 ppm. Here the chemical shift of H-18 of 4l was at 6.63 ppm, so the steric structure for 4l was the same as that of 4k.
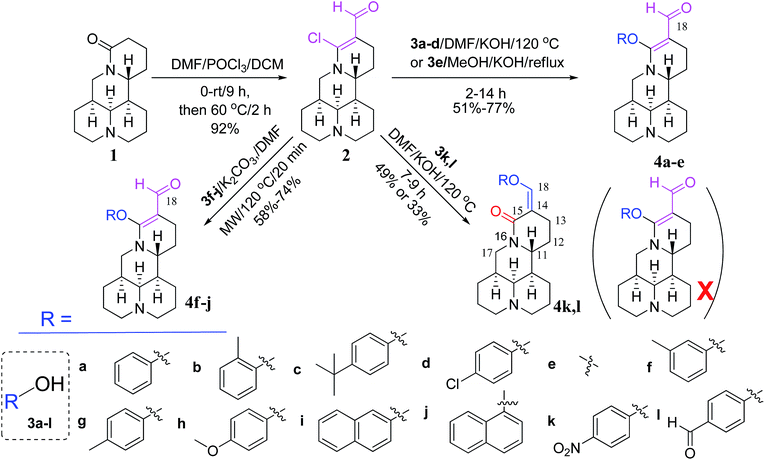 |
| | Scheme 1 Synthesis of 14-formyl-15-aryloxy/methoxymatrines (4a–j) and 14-aryloxymethylidenylmatrines (4k, l) from matrine. | |
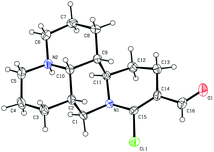 |
| | Fig. 2 X-ray crystal structure of compound 2. | |
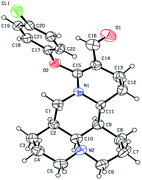 |
| | Fig. 3 X-ray crystal structure of compound 4d. | |
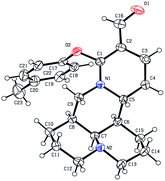 |
| | Fig. 4 X-ray crystal structure of compound 4g. | |
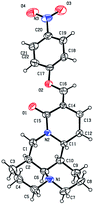 |
| | Fig. 5 X-ray crystal structure of compound 4k. | |
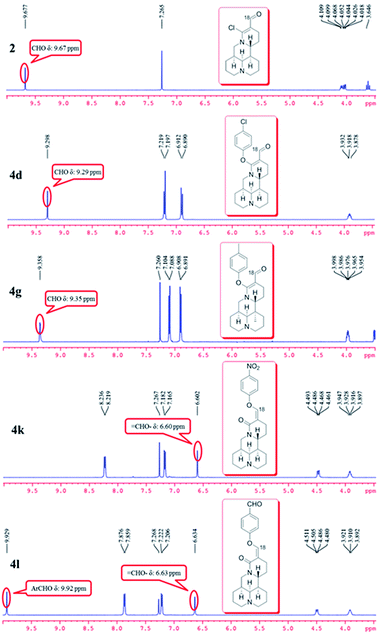 |
| | Fig. 6 Comparison of partial 1H NMR spectra of compounds 2, 4d, 4g, 4k, and 4l. | |
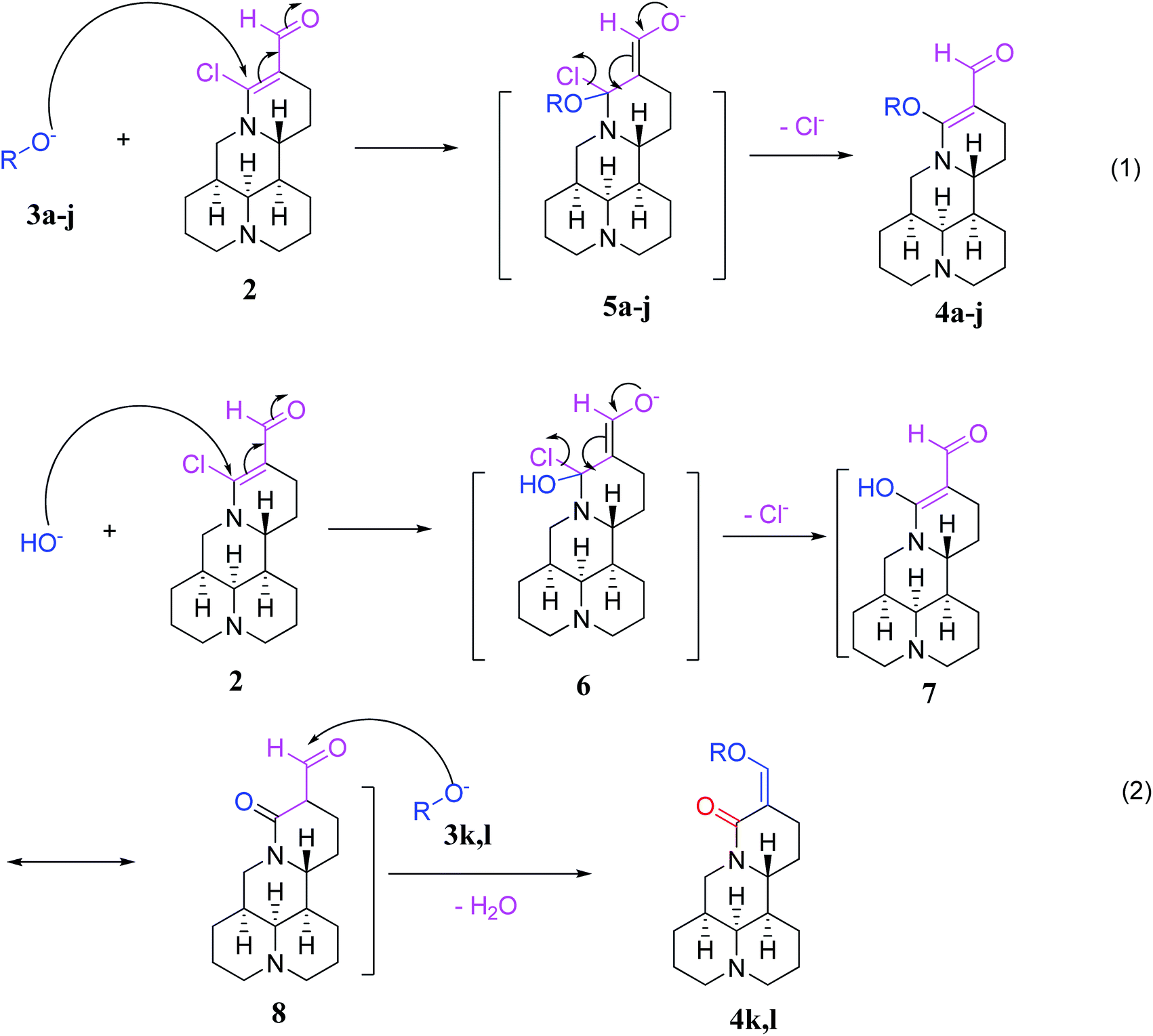
The possible mechanism for 3a–l reacting with 2 is described in Fig. 7. Compounds 3a–j reacted with 2 via the intermediates 5a–j to afford 14-formyl-15-aryloxy/methoxymatrine derivatives (4a–j) by the 1,4-addition–elimination reaction (eqn (1)). However, for compounds 3k, l, first, the hydroxyl ion reacted with 2 via the intermediate 6 to afford 14-formyl-15-hydroxylmatrine (7) by the 1,4-addition–elimination reaction. Then, compound 7 was converted into intermediate 8. Finally, compound 8 reacted with 3k, l, followed by removal of a molecule of water to give 14-aryloxymethylidenylmatrines (4k, l) (eqn (2)).
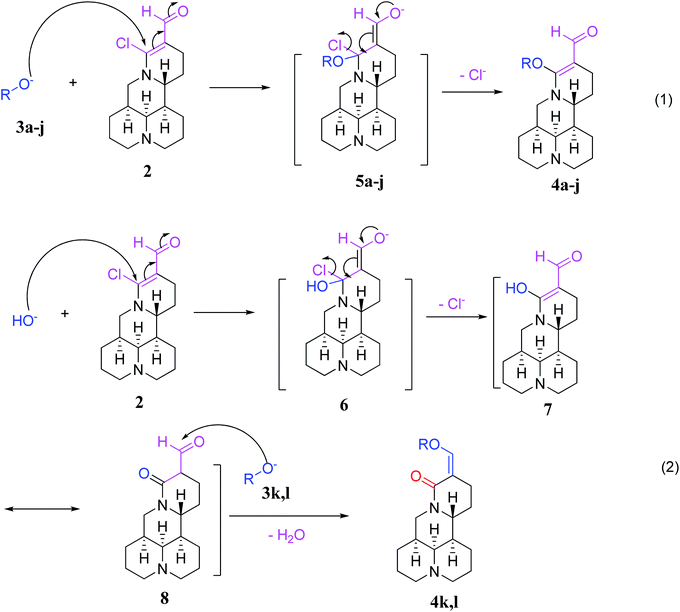 |
| | Fig. 7 Possible mechanism for 3a–l reacting with 2. | |
Insecticidal activity
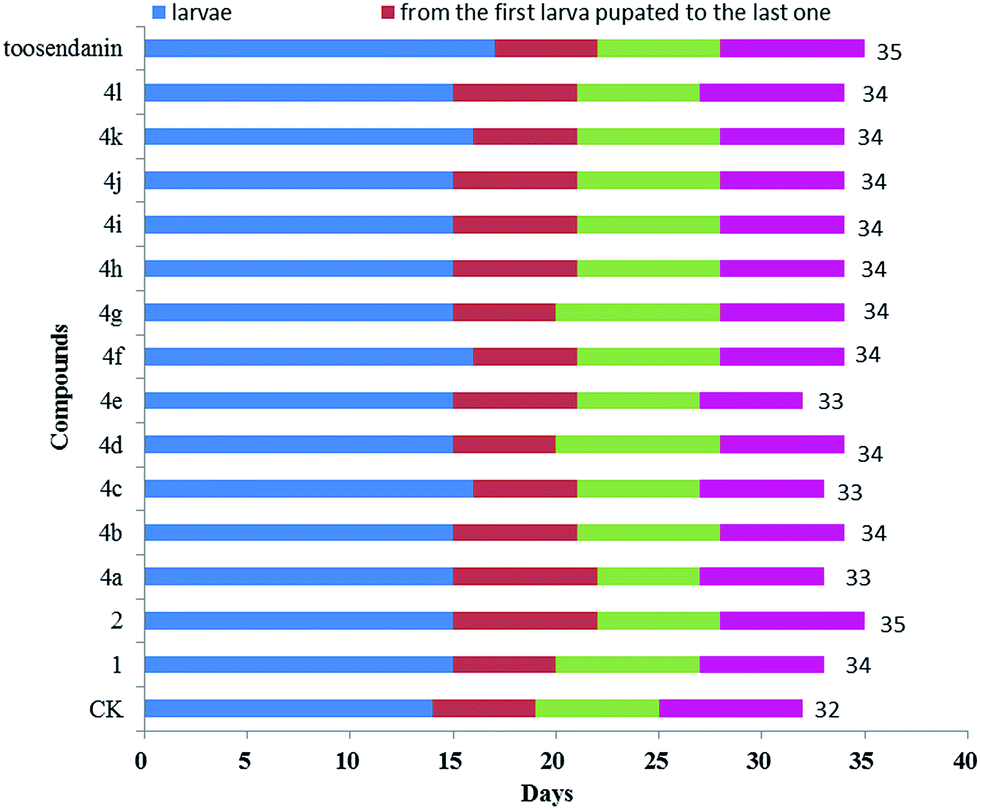
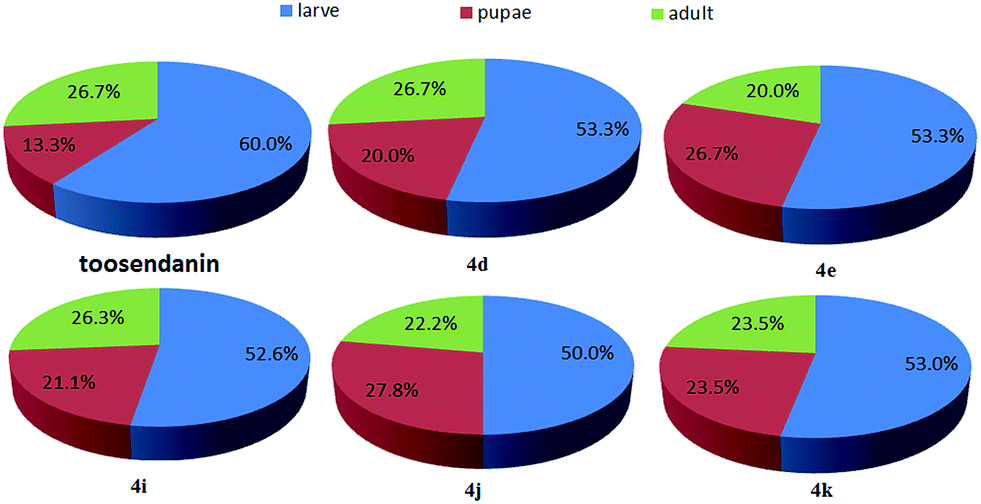
The oral toxicity of compounds 1 and 4a, d, e, g, i, k against P. xylostella treated at 20 μg/larvae is described in Table 1. The mortality rates after 48 h of 1, 4a, 4d, 4e, 4g, 4i, and 4k were 28.5%, 32.1%, 35.7%, 35.7%, 32.1%, 39.3%, and 42.8%, respectively. Among them, compounds 4d, 4e, 4i, and 4k exhibited potent oral toxicity when compared with toosendanin. The growth inhibitory activity of compounds 1, 2, and 4a–l against M. separata was tested at 1 mg mL−1. As shown in Table 2, compounds 4i, 4j and 4k showed higher insecticidal activity than toosendanin. For example, the final mortality rates (FMRs) of 4i, 4j and 4k were 62.1%, 58.6%, and 55.2%, respectively; whereas the FMR of 1 was only 24.2%. In particular, compound 4i exhibited the most potent insecticidal activity. The symptoms for the treated M. separata during the larval, pupation and adult periods were observed. Fig. 8 shows the dead larvae with thin and wrinkled bodies at the larval stage; Fig. 9 shows some malformed and dead pupae during the pupation stage; Fig. 10 shows some malformed moths during the adult emergence stage. This demonstrates that matrine derivatives probably affected the insect molting hormone. On the other hand, the times for three growth periods of M. separata treated with 1, 2, and 4a–l are shown in Fig. 11. This suggests that the times from the larvae to the adult in the treated groups were generally prolonged (33–35 days vs. 32 days for CK). Finally, the percentages of FMRs at three different growth stages of compounds 4d, 4e, 4i, 4j, 4k and toosendanin were investigated, as displayed in Fig. 12. More than/equal to half of FMRs for compounds 4d, 4e, 4i, 4j, 4k and toosendanin were at the larval stage. This result was the same with those of esters of fraxinellone C4/10-oxime.24
Table 1 Oral toxicity of compounds 1 and 4a, d, e, g, i, k against P. xylostella treated at 20 μg/Larvae
| Compound |
Corrected mortality rate (mean ± SD, %) |
| 24 h |
48 h |
| Multiple range test using Duncan's test (p < 0.05). The same letters denote treatments not significantly different from each other. |
| 1 |
10.4 ± 0 |
28.5 ± 3.3 ca |
| 4a |
13.8 ± 3.3 |
32.1 ± 3.3 bc |
| 4d |
10.4 ± 3.3 |
35.7 ± 5.8 abc |
| 4e |
13.8 ± 3.3 |
35.7 ± 0 abc |
| 4g |
10.4 ± 3.3 |
32.1 ± 6.7 bc |
| 4i |
10.4 ± 3.3 |
39.3 ± 3.3 abc |
| 4k |
17.3 ± 0 |
42.8 ± 3.3 ab |
| Toosendanin |
0 ± 3.3 |
46.4 ± 0 a |
Table 2 Growth inhibitory activity of compounds 1, 2, and 4a–l against M. separata on leaves treated with a concentration of 1 mg mL−1
| Compound |
Corrected mortality rate (mean ± SD, %) |
| 10 days |
25 days |
35 days |
| Multiple range test using Duncan's test (p < 0.05). The same letters denote treatments not significantly different from each other. |
| 1 |
3.3 ± 3.3 |
16.7 ± 3.3 |
24.2 ± 3.3 ea |
| 2 |
6.7 ± 3.3 |
23.3 ± 3.3 |
31.1 ± 6.7 de |
| 4a |
10.0 ± 5.8 |
26.7 ± 6.7 |
41.4 ± 3.3 cd |
| 4b |
6.7 ± 6.7 |
23.3 ± 6.7 |
38.0 ± 5.8 cd |
| 4c |
10.0 ± 5.8 |
23.3 ± 3.3 |
31.1 ± 3.3 de |
| 4d |
13.3 ± 6.7 |
30.0 ± 5.8 |
48.3 ± 0 bc |
| 4e |
20.0 ± 0 |
33.3 ± 3.3 |
48.3 ± 5.8 bc |
| 4f |
13.3 ± 6.7 |
33.3 ± 3.3 |
41.4 ± 3.3 cd |
| 4g |
10.0 ± 0 |
26.7 ± 3.3 |
38.0 ± 5.8 cd |
| 4h |
10.0 ± 5.8 |
30.0 ± 5.8 |
41.4 ± 3.3 cd |
| 4i |
13.3 ± 3.3 |
40.0 ± 0 |
62.1 ± 3.3 a |
| 4j |
13.3 ± 3.3 |
36.7 ± 3.3 |
58.6 ± 0 ab |
| 4k |
20.0 ± 0 |
33.3 ± 6.7 |
55.2 ± 3.3 ab |
| 4l |
6.7 ± 3.3 |
23.3 ± 3.3 |
38.0 ± 5.8 cd |
| Toosendanin |
16.7 ± 6.7 |
33.3 ± 3.3 |
48.3 ± 0 bc |
 |
| | Fig. 8 The representative abnormal larvae pictures of 4d (HJL-39), 4k (HJL-40), 4i (HJL-62), 4j (HJL-63), and 4e (HJL-72) during the larval period (CK: blank control group). | |
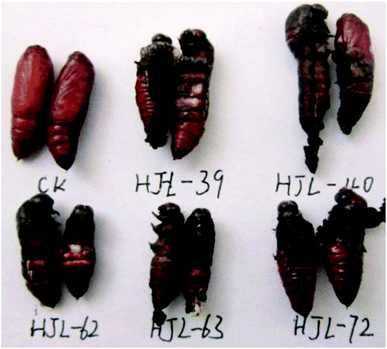 |
| | Fig. 9 The representative malformed pupae pictures of 4d (HJL-39), 4k (HJL-40), 4i (HJL-62), 4j (HJL-63), and 4e (HJL-72) during the pupation period (CK: blank control group). | |
 |
| | Fig. 10 The representative malformed moth pictures of 4a (HJL-32), 4b (HJL-45), 4f (HJL-67), 4h (HJL-68), and 4e (HJL-72) during the emergence period (CK: blank control group). | |
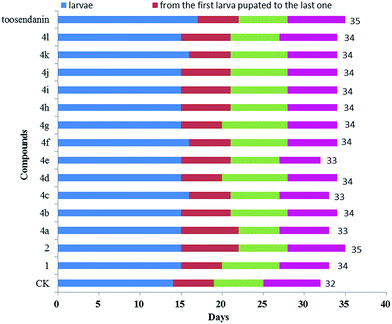 |
| | Fig. 11 Times for different developmental stages of M. separata treated with 1, 2, and 4a–l. | |
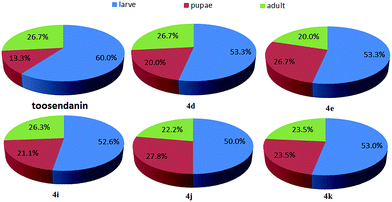 |
| | Fig. 12 The percentages of FMRs at three different growth stages of compounds 4d, 4e, 4i, 4j, 4k and toosendanin. | |
Additionally, some interesting results of structure–activity relationships of the tested compounds were also obtained. When the chlorine atom of 2 was substituted by other aryloxy/methoxy groups such as 4-chlorophenyloxy, methoxy, 2-naphthyloxy, and 1-naphthyloxy, the corresponding products 4d (FMR: 48.3%), 4e (FMR: 48.3%), 4i (FMR: 62.1%), and 4j (FMR: 58.6%) showed more potent insecticidal activity than 1 (FMR: 24.2%) and 2 (FMR: 31.1%). Interestingly, one 14-formyl-15-alkyloxymatrine, 4e (containing a methoxy group), exhibited promising insecticidal activity, so in the future other alkyloxy groups could be considered for introduction at the C-15 position of 2. In general, the introduction of an electron-withdrawing group on the phenyl ring of 4a (FMR: 41.4%) could result in a more potent compound. For example, the FMR of 4d (containing 4-chlorophenyl) was 48.3%; whereas the FMRs of 4b (containing 2-methylphenyl), 4c (containing 4-t-butylphenyl), 4f (containing 3-methylphenyl), 4g (containing 4-methylphenyl), and 4h (containing 4-methoxyphenyl), were 38.0%, 31.1%, 41.4%, 38.0%, and 41.4%, respectively. The introduction of a polycyclic aromatic hydrocarbon oxy at the C-15 position of 2 led to more potent products 4i and 4j. For example, the FMR of 4a was 41.4%; whereas the FMRs of 4i and 4j were 62.1%, and 58.6%, respectively.
Conclusions
In summary, we prepared a series of 14-formyl-15-aryloxy/methoxymatrine and 14-aryloxymethylidenylmatrine derivatives as pesticidal agents by structural modification of matrine. A possible reaction mechanism for 3a–l reacting with 2 was also proposed. Their pesticidal activities were evaluated against two crop-threatening agricultural insect pests, M. separata and P. xylostella. In particular, compounds 4i and 4k exhibited more promising pesticidal activities than matrine against M. separata and P. xylostella. This demonstrated that the introduction of an electron-withdrawing group on the phenyl ring of 14-formyl-15-phenyloxymatrine, and the introduction of an alkyloxy or polycyclic aromatic hydrocarbon oxy at the C-15 position of 14-formyl-15-chloromatrine could result in more potent compounds. This will lay the foundations for further structural modification and application of matrines as biorenewable pesticidal agents for agriculture.
Acknowledgements
The present research was partly supported by National Natural Science Foundation of China (No. 31672071), and Special Funds of Central Colleges Basic Scientific Research Operating Expenses (No. 2452015096) to H. X.
Notes and references
- X. F. Jiang, L. Z. Luo, L. Zhang, T. W. Sappington and Y. Hu, Environ. Entomol., 2011, 40, 516–533 CrossRef CAS PubMed.
- J. Y. Sun, P. Liang and X. W. Gao, Pest Manage. Sci., 2012, 68, 285–289 CrossRef CAS PubMed.
- J. Zeng, Y. Jiang and J. Liu, Plant Prot., 2013, 39, 117–121 Search PubMed.
- H. Wei, J. Liu, B. Li, Z. X. Zhan, Y. X. Chen, H. J. Tian, S. Lin and X. J. Gu, Crop Prot., 2015, 76, 68–74 CrossRef CAS.
- D. G. Heckel, Science, 2012, 337, 1612–1614 CrossRef PubMed.
- K. Etebari, M. J. Furlong and S. Asgari, Sci. Rep., 2015, 5, 14642 CrossRef CAS PubMed.
- J. K. Kim, S. R. Choi, J. Lee, S. Y. Park, S. Y. Song, J. Na, S. W. Kim, S. J. Kim, I. S. Nou and Y. H. Lee, J. Agric. Food Chem., 2013, 61, 11222–11230 CrossRef CAS PubMed.
- Q. Fu, Q. Fang, B. L. Feng, S. J. Sun, W. Du, A. Enijian, A. P. Xiao and C. Chang, Journal of Chromatography B, 2011, 879, 894–900 CrossRef CAS PubMed.
- Z. J. Wu, D. M. Sun, D. M. Fang, J. Z. Chen, P. Cheng and G. L. Zhang, Int. J. Mass Spectrom., 2013, 341–342, 28–33 CrossRef CAS.
- Y. Liu, Y. Xu, W. D. Ji, X. Y. Li, B. Sun, Q. G. Gao and C. Q. Su, Tumor Biol., 2014, 35, 5111–5119 CrossRef CAS PubMed.
- B. Zhang, Z. Y. Liu, Y. Y. Li, Y. Luo, M. L. Liu, H. Y. Dong, Y. X. Wang, Y. Liu, P. T. Zhao, F. G. Jin and Z. C. Li, Eur. J. Pharm. Sci., 2011, 44, 573–579 CrossRef CAS PubMed.
- H. G. Hu, S. Z. Wang, C. M. Zhang, L. Wang, L. Ding, J. P. Zhang and Q. Y. Wu, Bioorg. Med. Chem. Lett., 2010, 20, 7537–7539 CrossRef CAS PubMed.
- L. M. Gao, Y. X. Han, Y. P. Wang, Y. H. Li, Y. Q. Shan, X. Li, Z. G. Peng, C. W. Bi, T. A. Zhang, N. N. Du, J. D. Jiang and D. Q. Song, J. Med. Chem., 2011, 54, 869–876 CrossRef CAS PubMed.
- L. Q. He, H. X. Gu, D. K. Yin, Y. H. Zhang and X. S. Wang, Chemical Journal of Chinese Universities, 2010, 31, 1541–1547 CAS.
- L. S. Wang, Y. J. You, S. Q. Wang, X. Liu, B. M. Liu, J. N. Wang, X. Lin, M. S. Chen, G. Liang and H. Yang, Bioorg. Med. Chem. Lett., 2012, 22, 4100–4102 CrossRef CAS PubMed.
- J. L. Huang and H. Xu, Curr. Top. Med. Chem., 2016, 16, 3365–3378 CrossRef CAS PubMed.
- Z. Z. Odimar, D. P. R. Leandro, F. A. Thiago, S. S. Monica, P. B. Gabriela, T. Y. Pedro and D. V. Jose, Crop Prot., 2015, 67, 160–167 CrossRef.
- B. Z. Zhong, C. J. Lu, X. D. Sun, W. Q. Qin and Z. Q. Peng, Agrochemicals, 2010, 49, 924–926 CAS.
- J. Yuan, L. Z. Lu, B. Cong, Z. J. Zhang and F. Y. Wang, Chin. J. Pestic., 2016, 16, 3365–3378 Search PubMed.
- M. Garrido, M. Cabeza, F. Cortes, J. Gutierrez and E. Bratoeff, Eur. J. Med. Chem., 2013, 68, 301–311 CrossRef CAS PubMed.
- A. Sanchez-Marquez, Y. Arellano, E. Bratoeff, Y. Heuze, K. Cordova, G. Nieves, J. Soriano and M. Cabeza, J. Enzyme Inhib. Med. Chem., 2016, 31, 1170–1176 CrossRef CAS PubMed.
- E. Bratoeff, M. Garrido, T. Ramirez-Apan, Y. Heuze, A. Sanchez, J. Soriano and M. Cabeza, Bioorg. Med. Chem., 2014, 22, 6233–6241 CrossRef CAS PubMed.
- M. Lv, W. J. Wu and H. X. Liu, Pestic. Biochem. Physiol., 2008, 90, 114–118 CrossRef.
- Q. Li, X. B. Huang, S. C. Li, J. C. Ma, M. Lv and H. Xu, J. Agric. Food Chem., 2016, 64, 5472–5478 CrossRef CAS PubMed.
- R. Wang, X. Y. Zhi, J. Li and H. Xu, J. Agric. Food Chem., 2015, 63, 6668–6674 CrossRef CAS PubMed.
- X. Yu, D. Shi, X. Zhi, Q. Li, X. Yao and H. Xu, RSC Adv., 2015, 5, 31700–31707 RSC.
- A. L. LaFrate, J. R. Gunther, K. E. Carlson and J. A. Katzenellenbogen, Bioorg. Med. Chem., 2008, 16, 10075–10084 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*