
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrathin Co–Fe hydroxide nanosheet arrays for improved oxygen evolution during water splitting†
Tingting Zhouab,
Zhen Caoa,
Heng Wanga,
Zhen Gaoa,
Long Lia,
Houyi Mab and
Yunfeng Zhao *a
*a
aTianjin Key Laboratory of Advanced Functional Porous Materials, Institute for New Energy Materials and Low-Carbon Technologies, Tianjin University of Technology, Tianjin 300384, China. E-mail: yfzhao@tjut.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
First published on 26th April 2017
Abstract
The Fe-doping of hierarchical Co hydroxide nanosheet arrays (CoyFe1−y(OH)x NSAs) integrated on a three-dimensional electrode is shown to contribute to both increasing the available surface area and number of active sites. Ultrathin secondary nanosheets with different Co to Fe ratios that are subsequently grown on these primary nanoarrays are found to exhibit high oxygen evolution reaction (OER) activity. The optimal composition of CoyFe1−y(OH)x NSAs turns out to be Co0.7Fe0.3(OH)x NSAs, which allows for an OER onset overpotential as low as 220 mV and a small Tafel slope at 62.4 mV dec−1, while also providing excellent long-term durability (>100 h) and a high turnover frequency (TOF) of 0.172 s−1 at an overpotential of 380 mV. The specific activity of Fe-doped Co0.7Fe0.3(OH)x NSAs at an overpotential of 350 mV (0.37 mA cmBET−2) is also twice as high as that of undoped Co(OH)2 NSAs.
Introduction
The excessive consumption of fossil fuels and the increasingly serious environmental concerns associated with this have stimulated intense research into alternative energy conversion and storage systems that can provide high efficiency, environmental safety and economic feasibility.1–4 The electrochemical splitting of water into O2 and H2 is widely accepted as one of the most promising technologies for producing fuel from a renewable and abundant source (i.e., water), but the kinetically slow oxygen evolution reaction (OER) restricts the overall reaction.5,6 This has led in recent years to considerable interest in electrocatalysts capable of promoting the OER process, with RuO2 and IrO2 being found to provide excellent performance.7 Their practical application, however, has been limited by the high price of these scarce elements.More recently, first-row transition metal compounds8,9 have been investigated as more abundant and economical OER catalysts, with hydroxides receiving particular attention due to their excellent OER performance.10–13 Of these, there are a number of Co-based14–16 and Ni-based17,18 catalysts work as attractive alternative for OER thanks to their relative abundance, low cost and competitive OER activity. Their catalytic performance can be further improved by modulating their 3d orbital electron state through heterogeneous doping to create more active sites and greater electrical conductivity.19 For example, Fe-doping has been found to improve the water oxidation activity of both Ni-based20–23 and Co-based24,25 catalysts. A well-known example of Co–Fe as an OER catalyst was CoFe-layered double hydroxide (LDH),26 which exhibited higher OER activity than CoNi-LDH. Qiu et al. propose that Fe-doping provide enlarged LDH interlayer space, which is beneficial to the absorption of OH−,27 while other researchers have suggested that the addition of Fe in Co1−xFex(OOH) films provides more active sites.28 A more recent study by Feng et al. suggested that the FeOOH on FeOOH/Co/FeOOH nanotube host can significantly lower the energy barrier of products and intermediates, thereby promoting catalytic reactions.29 However, the addition of heterogeneous ions can also efficiently increase the geometric roughness.30,31 Furthermore, in ensuring the electro-catalytic performance of these powder nanomaterials, various additives are often used to enhance their conductivity and film-forming ability.32 Addition of polymers or the aggregation of catalyst particles bring about a deteriorate electrode performance, making the direct construction of a hierarchically structured and high-performance nanocatalysts a more attractive approach.33,34
In this work, we present a unique Co–Fe hydroxide nanosheet arrays (CoyFe1−y(OH)x NSAs) supported on Cu foam electrode that exhibits high OER performance due to optimization of both its intrinsic activity and geometric roughness. The low Tafel slope and Rct of Co0.70Fe0.30(OH)x NSAs indicate an improved charge transfer kinetic, indicating doping of Fe is a sufficient solution to eliminate the kinetically slow of OER. The specific activity of Co0.7Fe0.3(OH)x NSAs at an overpotential of 350 mV (0.37 mA cmBET−2) is twice as high as that of undoped Co(OH)2 NSAs, further prove the positive effect of Fe doping on OER activity. The Cu foam plays a role as both electron collector and source to grow the Cu2O nanoarrays that serve as a sacrificial template to guide the formation of the Co–Fe hydroxide nanosheets. Those self-supported electrodes possess good conductivity and sturdiness structure, bring about excellent stability, with no obvious decline in current density after 100 h. This three-dimensional hierarchically porous electrode design offer the advantages of low working potential, large current density, high turnover frequency (TOF) and good durability.
Results and discussion
To provide a sacrificial template to induce the growth of CoyFe1−y(OH)x NSAs, Cu2O nanoarrays were first produced on Cu foam by a simple anodic oxidation route.35 As shown in Fig. 1a, the CoyFe1−y(OH)x NSAs were then fabricated by a solution-phase cation exchange method at room temperature by simply dipping the Cu foam loaded with Cu2O nanoarrays into a aqueous solution of CoCl2 and FeCl2.36 During the cation exchange process, the Cu2O nanowires were etched by S2O32−, releasing OH−. Then the CoyFe1−y(OH)x NSAs precipitated, and these new CoyFe1−y(OH)x NSAs structures inherited the geometry of the Cu2O template. Secondary CoyFe1−y(OH)x NSAs nanostructures formed depending on the solubility of the products and the pH of the reaction system. Therefore, we can regulate the surface area of CoyFe1−y(OH)x NSAs by change the ratio of Co–Fe. As shown in Fig. S1a,† the initial Cu foam had a 3D skeleton structure with a smooth microscopic surface, Cu2O nanoarrays with a mean diameter of 30 nm were observed on its surface after anodic oxidation (Fig. 1b). The XRD pattern obtained from the Cu2O nanowires (Fig. S4†) exhibited distinct diffraction peaks that indexed well to Cu2O and Cu phases. | ||
| Fig. 1 (a) Schematic illustration of fabrication process of the CoyFe1−y(OH)x NSAs; (b) SEM image of the Cu2O nanoarrays; (c) SEM image of the Co0.70Fe0.30(OH)x NSAs. | ||
In Fig. 1c and S1b–f,† we see that the CoyFe1−y(OH)x NSAs inherit the morphology of the Cu2O arrays, and so align well on the surface of the Cu foam. The average diameter of these Co(OH)2 NSAs were estimated to be ∼100 nm. A number of secondary nanosheets grow perpendicularly to form cross-like nanosheets that attenuate after Fe doping, resulting a dramatic change in the morphology of the nanoarrays into something with a mace-like appearance and fluffy surface. These structural characteristics are convenient for the expose of active sites, which should bring about an improvement in catalytic performance. Changing the Co to Fe ratio from 75/25 to 70/30 and 55/45 caused a slight increase in the diameter of the CoyFe1−y(OH)x NSAs, but only within a narrow range of 100–200 nm. Interestingly, the top portion of the Co0.41Fe0.59(OH)x and Fe(OH)x NSAs increased more dramatically in diameter and transformed into a dendritic morphology.
Selected-area electron diffraction (SAED) patterns were employed to investigate the crystal structure of the CoyFe1−y(OH)x NSAs catalysts. The SAED patterns of single metal hydroxides were found to exhibit characteristic traits of a blurry hexagonal matrix (inset of Fig. 2a and S2d†), indicating a predominantly crystal structure. Only blurred rings were observed in the SAED patterns of change to a predominantly amorphous structure. Amorphous hybrid metal hydroxides (inset of Fig. 2b & S2a–c†), indicating materials with the great number of under-coordinated metal atoms provide more reactive sites at the catalyst surface, thus facilitating adsorb of bindings, like hydroxyl for OER.17,37 The high-resolution TEM (HRTEM) image of Co0.70Fe0.30(OH)x NSAs in Fig. 2d lacks discernible lattice fringe, further confirms the amorphous structure. The ultrathin (∼3 nm) nature of the Co0.70Fe0.30(OH)x NSAs can also be clearly observed from Fig. 2d and S12.† The XRD patterns of the CoyFe1−y(OH)x NSAs indicates the presence of Cu and a small amount of Cu2O (Fig. S4†). The two faint peaks in 17° and 23° for CoyFe1−y(OH)x NSAs (y > 0) can be indexed Co layer double hydroxide (Co LDH),38,39 and the faint peaks suggests a predominantly amorphous structure. Additionally, there was no peak in 17° and 23° for Fe2O3 NSAs. It should be noted that the amorphous nature of the Co(OH)2 NSAs observed by XRD did not conflict with the crystal structures obtained from HRTEM, because the faint crystal lattice the Co(OH)2 NSAs indicated a low crystallinity.36,38 The reduction in strength of the Cu2O peak compared to the XRD pattern for the Cu2O nanoarray indicates a scarcity of Cu2O phase in the CoyFe1−y(OH)x NSAs.
 | ||
| Fig. 2 Low-magnification TEM images and SAED patterns (inset) of (a) Co(OH)2 and (b) Co0.70Fe0.30(OH)x NSAs; high-magnification (c) and high resolution (d) TEM images of Co0.70Fe0.30(OH)x NSAs. | ||
The genuine atomic ratios of Co to Fe in the CoyFe1−y(OH)x NSAs obtained by ICP were found to be roughly analogous to the reactant ratios (Table S1†), from which the specific value of y in CoyFe1−y(OH)x was determined. The EDS mapping images of Co0.70Fe0.30(OH)x NSAs in Fig. S3† show that Co, Fe and Cu were uniformly distributed in the nanostructure. The XPS results for the chemical state of Co, Fe and O species in the as-prepared CoyFe1−y(OH)x NSAs shown in Fig. 3 and S5–S7.† Spectra of Co 2p were split into 2p3/2 and 2p1/2 doublets due to spin–orbit coupling. Peak fitting analysis identified two Co states, with the characteristic peaks at binding energies of 797.4 and 781.5 eV identified as Co2+, while the other two peaks at 796.1 and 780.5 eV are assigned to Co3+.39,40 Peak fitting analysis of the Co 2p spectra for the other Co–Fe bimetal NSAs hydroxides produced similar results to those of the Co0.70Fe0.30(OH)x NSAs. Without Fe doping, the main peaks of Co 2p3/2 and Co 2p1/2 were at higher energy regions and only one chemical state of Co (Co2+ at 782.2 and 797.8 eV) was detected. This can therefore be considered to be a Co(OH)2 NSAs structure. Additional evidence for the existence of Co2+ is provided by the satellite peaks (803.3 and 786.9 eV). The fact that the main Co 2p peak shifts to a higher energy with increased Fe doping implies that the oxidation of Co2+ is facilitated by the introduction of Fe.41 Peak fitting analyses of the Fe 2p spectra for the four bimetal NSAs hydroxides identified only one chemical state of Fe, i.e., Fe3+ at 724.6 and 711.9 eV.42,43 The peak in the Fe 2p spectra of Fe NSAs hydroxides are blue-shifted by about 0.6 eV relative to those of Co–Fe bimetal NSAs hydroxides, which suggests that the pure Fe NSAs prepared using the same protocol are oxide rather than hydroxide.44 These results confirm that strong electron interactions involving Fe and Co occur in the CoyFe1−y(OH)x NSAs. The O 1s spectra of the CoyFe1−y(OH)x NSAs could be fitted with two peaks at binding energies of 530.6 and 531.6 eV, which prove the presence of lattice oxygen and hydroxide oxygen, respectively.45,46 The O 1s spectra shifted to a lower binding energy following the addition of Fe, which indicates that O was in a more oxide-like environment due to the partial conversion of Co/Fe(OH)x to Fe/Co-oxide phases.28,47 The O 1s spectrum of the Fe(OH)x NSAs shifted to 530.1 eV, thereby proving the dominance of oxide ions over oxygen atoms. Combined with the blue-shift of Fe 2p spectra, we can confirm that the Fe(OH)x more likely to be Fe2O3. The status of the Co–Fe based hydroxide can be verified by the decrease in strength in the O–H stretching vibration peaks (≈3297 cm−1) and scissoring vibration peaks (1700–1500 cm−1 from water, 1400–1300 cm−1 from structure hydroxyl groups) with increasing Fe ratio that is evident in the infrared (IR) spectra of the samples (Fig. S8†), which is consistent with the XPS results.48,49 The broadening of the M–O scissoring vibration peaks (500–700 cm−1) with increasing Fe doping indicate an increase of Fe–O.
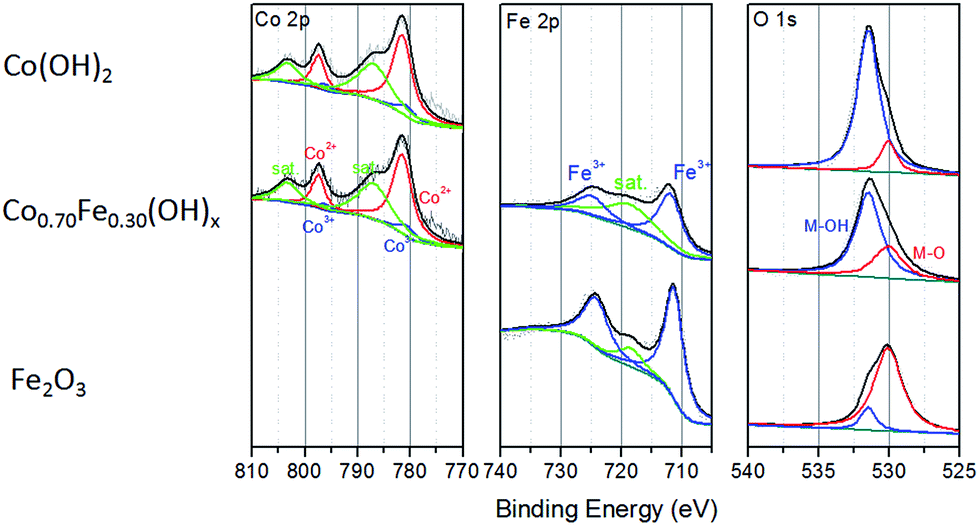 | ||
| Fig. 3 High-resolution XPS spectra of Co(OH)2, Co0.70Fe0.30(OH)x, and Fe2O3 NSAs. | ||
The electrocatalytic performance of the CoyFe1−y(OH)x NSAs with different ratios of Co and Fe was investigated by linear sweep voltammetry (LSV) between 1.3 and 1.65 V (versus RHE) in 1 M KOH using a three-electrode system (Fig. 4a). The Cu2O nanoarrays on Cu foam show negligible activity, as these were simply used as a sacrificial template for the CoyFe1−y(OH)x NSAs. In contrast, the Co0.70Fe0.30(OH)x and Co0.55Fe0.45(OH)x NSAs exhibit a sharp onset potential that is indicative of OER current at 1.45 V vs. RHE, with a small overpotential (η, η = E − 1230 mV)50 of 220 mV. The other two hybrid samples (i.e., the Co0.75Fe0.25(OH)x and Co0.41Fe0.59(OH)x NSAs) also exhibit high performance, with a slightly positive-shift onset η. Remarkably, all of the samples possess lower onset η values than single-metal catalysts. The Co0.70Fe0.30(OH)x NSAs also exhibit a markedly faster increase in anodic current density when compared to the other samples. In addition to the onset potential, the η required for a current density of 10 mA cm−2 is also commonly used as a criterion for evaluating the OER activity, and this can be seen to exhibit a similar trend. It is interesting that the as-made Co0.70Fe0.30(OH)x NSAs shows a dramatically increased peak current density and reduce of η in comparison to that of the Co(OH)2 NSAs, which mainly attributed to the synergistic effects between Co and Fe species in the hydroxide structure.27 In addition to providing excellent performance under typical OER testing conditions, the Co0.70Fe0.30(OH)x NSAs provide a useable working ability under much more stringent conditions, which allows a large amount of high-performance active catalyst to be directly grown on its surface. This results in a current density as high as 1200 mA cm−2 at a η as low as 410 mV (Fig. 4a). The wavy line on the high potential part of Co0.70Fe0.30(OH)x NSAs owing to the violent release of gas bubbles. The electron conductivity between the collector and active catalyst is also more favourable than in a traditional nanoparticle-cast electrode. Moreover, as shown in Fig. 4b, the improved reaction kinetics of Co0.70Fe0.30(OH)x NSAs is reflected by its low Tafel slope of 62.4 mV dec−1 compared to that of Co(OH)2 (74.9 mV dec−1), Co0.75Fe0.25(OH)x (67.3 mV dec−1), Co0.55Fe0.45(OH)x (65.4 mV dec−1) and Co0.41Fe0.59(OH)x (68.9 mV dec−1) NSAs. It is worth noticing that Fe2O3 NSAs possess a much lower Tafel slope of 50.9 mV dec−1 and a positive onset η of 270 mV, which suggests more favourable kinetics yet poor thermodynamics. This is because Fe catalyst only has measurable conductivity at η larger than 400 mV, therefore, its outstanding kinetics property only show in high η regions.28 However, the addition of Co efficiently enhance the electrical conductive and result in enhanced OER activity. Therefore, doping of Fe is a sufficient solution to eliminate the kinetically slow of OER.
 | ||
| Fig. 4 (a) Polarization LSV curves of CoyFe1−y(OH)x NSAs, Cu2O nanoarrays and IrOx; (b) Tafel and (c) TOF plots of CoyFe1−y(OH)x NSAs; (d) chronoamperometric measurements (η = 300 mV) of Co0.70Fe0.30(OH)x NSAs. | ||
The improvement in OER activity can be quantified by the turnover frequency (TOF), which is defined as the number of moles of O2 per mole of metal catalyst per second. If it is assumed that all metal ions (Co and Fe) in the hydroxide NSAs are available for OER, then an apparent TOF value can be calculated at different values of η. As shown in Fig. 4c, the TOF of the Co0.70Fe0.30(OH)x NSAs increases at a markedly faster rate than the other samples; and so not surprisingly, produces a higher TOF (0.172 s−1) than Co0.75Fe0.25(OH)x NSAs (0.103 s−1) and Co0.55Fe0.45(OH)x NSAs (0.126 s−1) at a working potential of 380 mV. This TOF is also much higher than that achieved with the non-doped NSAs, i.e., Co(OH)2 NSAs (0.081 s−1) and Fe2O3 NSAs (0.045 s−1). The OER performance of the CoyFe1−y(OH)x NSAs is summarized in Table S2,† from which we see that the critical values of Co0.70Fe0.30(OH)x NSAs are comparable to the state-of-the-art materials for OER catalysts (Table S4†). The durability of the electrode was evaluated by means of the chronoamperometry method. In Fig. 4d, the as-prepared Co0.70Fe0.30(OH)x NSAs electrode clearly exhibits greater stability, with no obvious decline in current density after 100 h. This remarkable operational stability can be ascribed to the excellent intrinsic stability and enhanced activity of the Co0.70Fe0.30(OH)x catalyst, as well as the sturdiness of the entire electrode and reduced coverage of gas bubbles.51
The electrode kinetics were further analyzed by simulating the charge transfer resistance obtained through electrochemical impedance spectroscopy (EIS) during oxygen evolution (η = 300 mV) in a solution of 1.0 M KOH (Fig. 5). The Zsimpwin 3.5 was used to fit the resistance values, and the results obtained are shown in Table S3.† As illustrated in Fig. S9,† the equivalent circuit consists of three main parts: the solution resistance Rs, the charge transfers resistance Rct and the constant phase resistance Rcp. The Rct of the six electrodes was in the order of Co0.70Fe0.30(OH)x < Co0.55Fe0.45(OH)x < Co(OH)2 < Co0.25Fe0.75(OH)x < Co0.41Fe0.59(OH)x < Fe2O3 NSAs, indicating a marked increase in Rct with increasing Fe content, especially at >40 at% Fe.52 A much smaller Rct suggesting a much faster electron transfer and a higher faradaic efficiency during reaction.53,54 Therefore, good conductivity and faster reaction speed of materials can improve the charge transfer kinetics. The Co(OH)2 NSAs possess a small Rct, yet only provide a relatively unromantic electrocatalytic activity on account of the inherent electrical conductivity and inferior activity of Co hydroxide compared to Co–Fe hydroxide. This again proves that Co0.70Fe0.30(OH)x NSAs provides the appropriate conductivity and good electrocatalytic activity and bring about optimal performance for OER. These results consistent with the kinetics results of Tafel plots, which further prove the electronic interaction between Fe and Co in the hydroxides. Similar optimal of Co to Fe ratios being found in previous studies: e.g., CoFe-LDH with a Co to Fe ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 has a greater OER activity than Co(OH)2 and mixed-phase samples of Co(OH)2 and FeOOH.27 The intrinsic OER activity of Co1−xFex(OOH) is also known to be ∼100-fold higher when x ≈ 0.6–0.7 than when x = 0 on a per-metal TOF basis.28
:1 has a greater OER activity than Co(OH)2 and mixed-phase samples of Co(OH)2 and FeOOH.27 The intrinsic OER activity of Co1−xFex(OOH) is also known to be ∼100-fold higher when x ≈ 0.6–0.7 than when x = 0 on a per-metal TOF basis.28
 | ||
| Fig. 5 Nyquist plots of CoyFe1−y(OH)x NSAs, the insert is magnification Nyquist plots of Co(OH)2, Co0.75Fe0.25(OH)x, Co0.70Fe0.30(OH)x and Co0.55Fe0.45(OH)x NSAs. | ||
Based on the SEM and TEM results, it would seem that Co–Fe based NSAs expose more surface area than Co(OH)2 NSAs. To confirm this hypothesis, type-IV nitrogen adsorption–desorption isotherms were obtained for the Co0.70Fe0.30(OH)x NSAs and Co(OH)2 NSAs, as this provides an indication of the mesoporous nature of the nanosheet (Fig. 6a). A H3-type hysteresis loop was also identified in the isotherms, which provides further evidence of nanosheet aggregation.55 The BET surface area of Co0.70Fe0.30(OH)x NSAs was much higher than that of Co(OH)2 NSAs (89 vs. 67 m2 g−1), which is consistent with the electrochemical surface area (ECSA) of Co0.70Fe0.30(OH)x (105 cm2) and Co(OH)2 (75 cm2) NSAs calculate by double-layer capacitance (Fig. S10†). However, as the structural disorder in these amorphous materials may create more defect sites capable of serving as efficient reaction centers,56,57 the specific activity (current per BET area) was used to specify the density of active sites. Fig. 6b shows the LSV curves after normalizing the current to the BET surface area. It is evident from this that the specific activity of Co0.70Fe0.30(OH)x was 0.37 mA cmBET−2 at a η value of 350 mV, which is twice as high as that of Co(OH)2 NSAs (0.19 mA cmBET−2). This confirms that Fe doping contributes to creating more active sites, and as the performance of a catalyst is strongly influenced by its geometric roughness,34,58 the excellent activity of Co0.70Fe0.30(OH)x can be attributed to its high surface area and greater number of active sites. Previous studies have also demonstrated that the large surface area of ultrathin nanosheet array is beneficial to the mass transfer and utilization of catalysts, as well as providing a sturdy and close-knit current collector that effectively reduces the size of any gas bubbles and their associated adverse effects.51
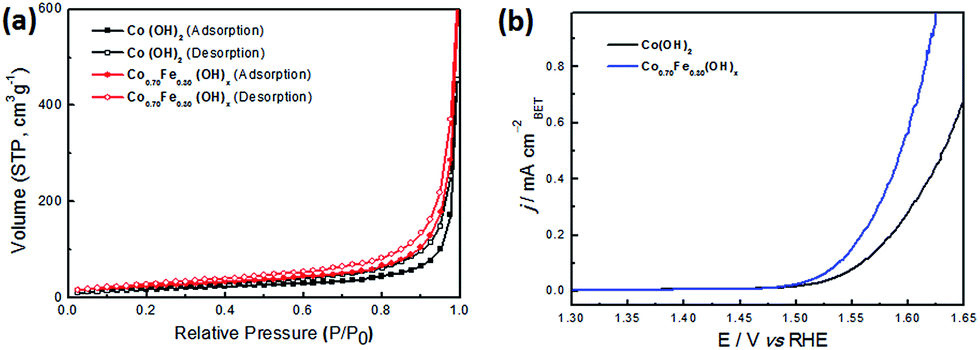 | ||
| Fig. 6 (a) Nitrogen adsorption–desorption isotherms and (b) polarization LSV curves normalized to the BET surface area of Co(OH)2 and Co0.70Fe0.30(OH)x NSAs. | ||
The above results clearly demonstrate that compared with Co(OH)2 NSAs, the introduction of Fe significantly improves the catalytic activity. Firstly, hybrid Co–Fe hydroxides (or oxides) are well-known excellent active materials with high activity and stability in OER applications.28 The low Tafel slope and Rct of Co0.70Fe0.30(OH)x NSAs indicate an improved charge transfer kinetic, indicating doping of Fe is a sufficient solution to eliminate the kinetically slow of OER. The specific activity of Co0.7Fe0.3(OH)x NSAs at an overpotential of 350 mV (0.37 mA cmBET−2) is twice as high as that of undoped Co(OH)2 NSAs, further prove the positive effect of Fe doping on OER activity. Secondly, nanoarray structures with high specific surface area not only provide large reaction interface but also ensure efficient charge conductivity, which is benefit for utilization in areas of electrochemical catalyst. The BET surface area of Co0.70Fe0.30(OH)x NSAs was much higher than that of Co(OH)2 NSAs (89 vs. 67 m2 g−1). The empty space between nanoarrays would facilitate the transfer of the electrolyte thus benefit the charge transfer kinetic. Thirdly, integrated electrode can provide good conductivity and sturdiness electrode structure, bring about excellent stability, with no obvious decline in current density after 100 h. Most of all, Co0.70Fe0.30(OH)x NSAs provide an optimum Fe-doping ratio, increased geometric roughness, more active sites and a sturdy electrode structure, all of which contribute to greater overall electrocatalytic performance.
Conclusions
This study has succeeded in synthesizing a series of hierarchical CoyFe1−y(OH)x NSAs integrated on a three-dimensional electrode and shown that the Fe-doping helps to increase both the geometric roughness and density of active sites. Of all the samples tested, Co0.7Fe0.3(OH)x NSAs delivered the highest anodic current for OER at a low η, and exhibited excellent stability with no obvious decline in current density after 100 h. This electrode also possessed a large surface area of 89 m2 g−1 and a specific activity of 0.37 mA cmBET−2 at a η value of 350 mV that is twice as much as that of Co(OH)2 NSAs. The outstanding electrocatalytic performance of Co0.7Fe0.3(OH)x NSAs can be attributed to an optimization of the effects of Fe-doping and an open 3D electrode design. These results open a new avenue for the rational design and controllable preparation of hierarchical Co–Fe based nanoelectrodes capable of providing the high catalytic activity and excellent stability needed for clean energy technologies.Experimental
Materials
Cobalt(II) dichloride (CoCl2·6H2O, AR), iron(II) dichloride (FeCl2·4H2O, AR), potassium hydroxide (KOH, AR), sodium hyposulfite (Na2S2O3·5H2O, AR), oxalic acid (H2C2O4, AR), ethanol (CH3CH2OH, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification. Milli-Q water (resistivity > 18 MΩ cm) was used throughout the experiments.Synthesis of the CoyFe1−y(OH)x NSAs
Copper foam (100 pores per inch, 98% porosity, and ∼1.5 mm thick) was cut into squares (2.0 cm × 2.0 cm) and cleaned with Milli-Q water and ethanol before use. It was then anodized in a 0.4 M H2C2O4 solution for 20 min at 36 V using a graphite plate as the cathode electrode. Electro-oxidation was then conducted using a potentiostat (760D, CHI Instruments) with a three-electrode system, wherein the anodized Cu foam was used as a working electrode, platinum wire as the counter electrode and an Ag/AgCl electrode as the reference electrode. Cyclic voltammetry was performed over a potential range from −0.3 to 0.1 V at a scanning rate of 1 mV s−1 in a 1 M KOH aqueous solution to accomplish the in situ growth of Cu2O nanowire arrays on the surface of the Cu foam.35 These Cu2O nanowire arrays were subsequently sacrificed to fabricate CoyFe1−y(OH)x NSAs36 by first dissolving set amounts of CoCl2·6H2O and FeCl2·4H2O in a mixed solution of 17.5 mL Milli-Q water and 17.5 mL ethanol to give a molar ratios of CoCl2·6H2O to FeCl2·4H2O of 1:0, 3:1, 2:1, 1:1, 1:2 and 0:1. The total amount of CoCl2·6H2O and FeCl2·4H2O was 8 × 10−5 mol. The samples of Cu foam decorated with Cu2O nanowire arrays were immersed into one of the above suspensions, and then 10 mL of Na2S2O3 (1 M) solution was added dropwise with magnetic stirring for 1 h. Finally, the substrate was taken out and washed in ethanol and Mill-Q water several times, followed by drying at 60 °C in a vacuum oven for 4 h.
Material characterizations
Scanning electron microscopy (SEM) was performed using a ZEISS MERLIN and microstructure investigations were carried out using a JEOL JEM-2100 at 200 kV. Element mappings were obtained with a FEI Tecnai G2F2O operating at 200 kV, while X-ray diffraction (XRD) patterns were recorded using a Rigaku Ultima IV. The chemical valence state of the elements was determined by X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe), with all spectra being corrected by the C 1s binding energy of 284.8 eV. The genuine atomic ratios of Co to Fe were evaluated by inductively coupled plasma (ICP) emission spectrometry (VISTA-MPX). Brunauer–Emmett–Teller (BET) measurements were performed on a Quadrasorb SI analyzer at 77 K.Electrochemical measurements
Electrochemical measurements were performed in an O2-saturated 1 M KOH electrolyte with an electrochemical analyzer (CHI 760D Instruments) and a three-electrode system that consisted of Hg/HgO (1 M KOH) with a double salt bridge as a reference electrode, platinum wire as a counter electrode, and the CoyFe1−y(OH)x NSAs (0.5 cm × 0.5 cm) on Cu foam as the working electrode. The Hg/HgO electrode was calibrated against a reversible hydrogen electrode (RHE) in a 1 M KOH solution (see Fig. S11†) that was bubbled with hydrogen for 30 min prior to calibration to ensure it was saturated with hydrogen. Two platinum wires were used as the working electrode and counter electrode. Cyclic voltammetry (CV) curves were recorded at a scan rate of 2 mV s−1 and the average of the positive and negative potentials at which the current crossed zero was taken to be the thermodynamic potential for the hydrogen electrode reaction. All polarization measurements were performed at a scanning rate of 5 mV s−1, from which the potentials were calculated relative to the (RHE) according to the following equation: E(RHE) = E(Hg/HgO) + 0.098 + 0.0591 × pH. The calibrated potential of the Hg/HgO (0.9 V vs. RHE) was found to be consistent with the calculated result (0.896 V vs. RHE). The durability was assessed using the controlled potential electrolysis method, in which all of the cyclic electrochemical measurements are 75% iR-compensated. Electrochemical impedance spectroscopy was performed over a frequency range of 10−2 to 104 Hz with an amplitude of 5 mV at the Princeton PMC 1000 electrochemical workstation. All electrochemical tests were carried out at 25 °C.The TOF values were calculated from the followed equation:
| TOF = (j × a)/(4 × n × F). |
485 C mol−1).
The electrochemically active surface area (ECSA) was estimated by the double-layer capacitance at a potential range (0.896–0.946 V vs. RHE) with no faradaic current from cyclic voltammetry (CV). The electrochemical double-layer capacitance (CDL) was measured by scan-rate dependent CVs, as given by the followed equation:
| i = vCDL |
The ECSA was calculated according to the followed equation:
| ECSA = CDL/Cs |
Acknowledgements
This work was financially supported by the National 973 Program Project of China (2012CB932800), National Natural Science Foundation of China (51572016, 21402136), Natural Science Foundation of Tianjin City (16JCYBJC17000), and the Specialized Research Fund for the Doctoral Program of Higher Education of China (20133201120004). YFZ thank the support provided by the “Talent Program” of Tianjin University of Technology. YFZ thanks the financial support received from the “Youth Thousand Talents Program” of Tianjin City.Notes and references
- M. R. Shaner, S. Hu, K. Sun and N. S. Lewis, Energy Environ. Sci., 2015, 8, 203–207 Search PubMed.
- X. Lang, L. Zhang, T. Fujita, Y. Ding and M. Chen, J. Power Sources, 2012, 197, 325–329 Search PubMed.
- B. Li, M. Zheng, H. Xue and H. Pang, Inorg. Chem. Front., 2016, 3, 175–202 Search PubMed.
- W. Yu, X. Jiang, F. Meng, Z. Zhang, H. Ma and X. Liu, RSC Adv., 2016, 6, 34501–34506 Search PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. Ren, M. Antonietti and T.-P. Fellinger, Adv. Energy Mater., 2015, 5, 1401660 CrossRef.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- J. Jiang, A. Zhang, L. Li and L. Ai, J. Power Sources, 2015, 278, 445–451 CrossRef CAS.
- H. Yin and Z. Tang, Chem. Soc. Rev., 2016, 45, 4873–4891 RSC.
- X. Yu, M. Zhang, W. Yuan and G. Shi, J. Mater. Chem. A, 2015, 3, 6921–6928 CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 1600621 CrossRef.
- G. B. B. Varadwaj and V. O. Nyamori, Nano Res., 2016, 9, 3598–3621 CrossRef CAS.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araujo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed.
- X. Li, G. Guan, X. Du, A. D. Jagadale, J. Cao, X. Hao, X. Ma and A. Abudula, RSC Adv., 2015, 5, 76026–76031 RSC.
- K. He, Z. Cao, R. Liu, Y. Miao, H. Ma and Y. Ding, Nano Res., 2016, 9, 1856–1865 CrossRef CAS.
- J. Nai, H. Yin, T. You, L. Zheng, J. Zhang, P. Wang, Z. Jin, Y. Tian, J. Liu, Z. Tang and L. Guo, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- M. Wei, M. Renzhi, W. Chengxiang, L. Jianbo, L. Xiaohe, Z. Kechao and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef PubMed.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- T. Grewe, X. Deng and H. Tüysüz, Chem. Mater., 2014, 26, 3162–3168 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- G. Abellán, J. A. Carrasco, E. Coronado, J. Romero and M. Varela, J. Mater. Chem. C, 2014, 2, 3723–3731 RSC.
- X. Han, C. Yu, J. Yang, C. Zhao, H. Huang, Z. Liu, P. M. Ajayan and J. Qiu, Adv. Mater. Interfaces, 2016, 3, 15000782 Search PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- J. X. Feng, H. Xu, Y. T. Dong, S. H. Ye, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2016, 55, 3694–3698 CrossRef CAS PubMed.
- X. Liu, Z. Chang, L. Luo, T. Xu, X. Lei, J. Liu and X. Sun, Chem. Mater., 2014, 26, 1889–1895 CrossRef CAS.
- H.-Y. Wang, Y.-Y. Hsu, R. Chen, T.-S. Chan, H. M. Chen and B. Liu, Adv. Energy Mater., 2015, 5, 1500091 CrossRef.
- T. Tian, L. Ai and J. Jiang, RSC Adv., 2015, 5, 10290–10295 RSC.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- R. Chen, H.-Y. Wang, J. Miao, H. Yang and B. Liu, Nano Energy, 2015, 11, 333–340 CrossRef CAS.
- C. Dong, Y. Wang, J. Xu, G. Cheng, W. Yang, T. Kou, Z. Zhang and Y. Ding, J. Mater. Chem. A, 2014, 2, 18229–18235 CAS.
- J. Nai, Y. Tian, X. Guan and L. Guo, J. Am. Chem. Soc., 2013, 135, 16082–16091 CrossRef CAS PubMed.
- J. Xie and Y. Xie, ChemCatChem, 2015, 7, 2568–2580 CrossRef CAS.
- S. Li, Y. Wang, S. Peng, L. Zhang, A. M. Al-Enizi, H. Zhang, X. Sun and G. Zheng, Adv. Energy Mater., 2016, 6, 1501661 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- Q. Zhang, H. Chen, X. Han, J. Cai, Y. Yang, M. Liu and K. Zhang, ChemSusChem, 2016, 9, 186–196 CrossRef CAS PubMed.
- R. Ma, J. Liang, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2011, 133, 613–620 CrossRef CAS PubMed.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Graya and A. M. Müller, Energy Environ. Sci., 2016, 9, 1734–1743 CAS.
- S. Helmut, S. Shamaila, W. Lorenz, K. Karsten, D. Stephan, W. Joachim, S. Lilli, S. Martin, H. Jörg and D. Diemo, Energy Environ. Sci., 2015, 8, 2685–2697 Search PubMed.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- T. Sun, L. Xu, Y. Yan, A. A. Zakhidov, R. H. Baughman and J. Chen, ACS Catal., 2016, 6, 1446–1450 CrossRef CAS.
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, ACS Catal., 2016, 6, 2416–2423 CrossRef CAS.
- J. Qi, W. Zhang, R. Xiang, K. Liu, H.-Y. Wang, M. Chen, Y. Han and R. Cao, Adv. Sci., 2015, 2, 1500199 CrossRef PubMed.
- Y. Qiu, L. Xin and W. Li, Langmuir, 2014, 30, 7893–7901 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, C.-V. Federico, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Z. Lu, Y. Li, X. Lei, J. Liu and X. Sun, Mater. Horiz., 2015, 2, 294–298 RSC.
- S. Wang, J. Nai, S. Yang and L. Guo, ChemNanoMat, 2015, 1, 324–330 CrossRef CAS.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- Y. Li, L. Zhang, R. Liu, Z. Cao, X. Sun, X. Liu and J. Luo, ChemCatChem, 2016, 8, 2765–2770 CrossRef CAS.
- K. S. W. Sing and R. T. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- M. Y. Song, D.-S. Yang, K. P. Singh, J. Yuan and J.-S. Yu, Appl. Catal., B, 2016, 191, 202–208 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01202k |
| This journal is © The Royal Society of Chemistry 2017 |