
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrolysis of cis- and transplatin: structure and reactivity of the aqua complexes in a solvent free environment†
Davide Corinti a,
Cecilia Colettib,
Nazzareno Reb,
Susanna Piccirilloc,
Marco Giampàad,
Maria Elisa Crestonia and
Simonetta Fornarini*a
a,
Cecilia Colettib,
Nazzareno Reb,
Susanna Piccirilloc,
Marco Giampàad,
Maria Elisa Crestonia and
Simonetta Fornarini*a
aDipartimento di Chimica e Tecnologie del Farmaco, Università degli Studi di Roma “La Sapienza”, P.le A. Moro 5, I-00185 Roma, Italy. E-mail: simonetta.fornarini@uniroma1.it
bDipartimento di Farmacia, Università G. D'Annunzio, Via dei Vestini 31, I-66100 Chieti, Italy
cDip. di Scienze e Tecnologie Chimiche, Università di Roma “Tor Vergata”, via della Ricerca Scientifica, 00133 Rome, Italy
dProteom- und Metabolomforschung, Fakultät für Biologie, Centrum für Biotechnologie (CeBiTec), Universität Bielefeld, Universitätsstraße 27, Bielefeld, Germany
First published on 10th March 2017
Abstract
Singly aquated and diaquated species are key intermediates in the mechanism responsible for the antitumor activity of cisplatin. Aqua complexes [PtX(NH3)2(H2O)]+ (X = Cl, OH), obtained in water by hydrolysis of cisplatin and of the inactive isomer transplatin, are transferred into the gas-phase by electrospray ionization. The so-formed ions, cis- and trans-[PtX(NH3)2(H2O)]+, have been allowed to react with selected ligands, representing platination targets in the biological environment. The reaction kinetics monitored in the gas-phase show consistently higher reactivity for the chloro complexes, [PtCl(NH3)2(H2O)]+, with respect to the hydroxo counterparts, [Pt(OH)(NH3)2(H2O)]+. The latter species, both cis- and trans-isomers, have been assayed by IRMPD spectroscopy in the NH/OH stretching region and their vibrational and geometric features are compared with the ones pertaining to the already described chloro complexes, cis- and trans-[PtCl(NH3)2(H2O)]+.
1. Introduction
Cisplatin, cis-PtCl2(NH3)2, is a leading antitumor drug of impressive efficacy, used both alone and in combination with other drugs for the treatment of various kinds of solid tumors.1–5 The anticancer activity of cisplatin is due to a reaction with cellular DNA, leading to the formation of intrastrand cross-links with a bridging Pt(NH3)2 unit. However, DNA bases do not replace the chloro ligand from platinum(II) directly but predominantly via a solvent assisted pathway. Cisplatin aquation (replacement of Cl− with H2O) yields the singly aquated species, cis-[PtCl(NH3)2(H2O)]+. Also the diaquated cis-[Pt(NH3)2(H2O)2]2+ or its deprotonated form are suggested to be important contributors to DNA platination.6,7 The positive charge on the platinum complex can attract the negatively charged nuclear DNA, thus favoring the formation of DNA lesions.1–5 Interestingly, the charged aqua complexes are directly amenable to mass spectrometric investigation paving the way to an evaluation of their intrinsic properties and hopefully enlightening useful issues for Pt-drug design.8 In this context we have recently undertaken a study of the cis-[PtCl(NH3)2(H2O)]+ complex from the primary aquation reaction.9 The vibrational signatures of the isolated species have been assayed by infrared multiple photon dissociation (IRMPD) spectroscopy,10–15 in parallel with a characterization of the trans isomer, trans-[PtCl(NH3)2(H2O)]+.9 The experimental features have been interpreted by comparison with the calculated IR spectra for the computed stable structures, as typically performed.10–15 Using this approach, the binding motifs in the naked adducts from cisplatin with nucleobases (adenine and guanine) have been shown to involve the N7 position of guanine and N1/N3 positions of adenine, with N7 attack to adenine notably missing.16 Cisplatin binding to a DNA building block (2′-deoxyguanosine-5′-monophosphate) is found to lead to macrochelate species isolated in the gas-phase where IRMPD spectroscopy has shown the metal to be ligated to both N7 of guanine and a phosphate oxygen atom.17 Gas-phase vibrational spectroscopy and ion chemistry have allowed to thoroughly characterize the ligand substitution reaction replacing the aqua ligand of cis-[PtCl(NH3)2(H2O)]+ with simple molecules representing ubiquitous active species in the biological environment faced by the cisplatin drug.18 A simple glycine-linked cisplatin, [(Gly-H)PtCl2]−, has provided a case study for IRMPD experiments combined with a survey of hybrid theoretical approaches for determining structures and IR spectra.19In this contribution we have investigated the aqua-hydroxo cis- and trans-[Pt(OH)(NH3)2(H2O)]+ complexes with regard to both structure and reactivity, in relation to the aqua-chloro cis- and trans-[PtCl(NH3)2(H2O)]+ complexes whose features have already been reported.9,18
Gas-phase studies have provided valuable insight in the mechanistic features of ion-molecule reactions of platinum(I/II) ions, ranging from simple mono- or diatomic species to multiply ligated complexes.20–24 In solution, considerable interest has been devoted to correlations of kinetic and thermodynamic parameters for the ligand substitution reaction of square planar platinum(II) complexes which led to define nucleophilic reactivity constants for various entering nucleophiles.25–27 The best nucleophiles are soft and polarizable and the charge on the substrate is found to affect the order of nucleophilicity. However, it is often difficult to discriminate among the various factors that may affect the reactivity of a metal complex in solution. For example, in examining the reactivities of different PtII–solvento complexes, the influences of the nature of the incoming and leaving ligands and of the trans and cis groups as well as the steric properties of both the substrate complex and the entering nucleophile are not separable.28 In this regards, the gas-phase may represent the ultimate simplified environment.
2. Experimental
2.1 Sample preparation
Cisplatin, cis-PtCl2(NH3)2, and transplatin, trans-PtCl2(NH3)2, used in this work, were commercial products from Sigma-Aldrich and were used as received. Both compounds were dissolved in H2O LC-MS grade at a concentration of 10−3 M. The aqueous solutions were incubated for one night before the analysis effected after dilution in H2O/MeOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to obtain a final concentration of 10−5 M. The end solution contained an appreciable fraction of [PtCl(NH3)2(H2O)]+ ions. In order to favor the formation of hydrolyzed products, both cisplatin and transplatin dilute solutions were incubated two days longer leading to a conspicuous increase of the abundance of [Pt(OH)(NH3)2(H2O)]+ ions, at the expense of [PtCl(NH3)2(H2O)]+.
:1 to obtain a final concentration of 10−5 M. The end solution contained an appreciable fraction of [PtCl(NH3)2(H2O)]+ ions. In order to favor the formation of hydrolyzed products, both cisplatin and transplatin dilute solutions were incubated two days longer leading to a conspicuous increase of the abundance of [Pt(OH)(NH3)2(H2O)]+ ions, at the expense of [PtCl(NH3)2(H2O)]+.
2.2 IRMPD spectroscopy
IRMPD spectroscopy experiments were performed using an Optical Parametric Oscillator/Optical Parametric Amplifier (OPO/OPA) laser system (LaserVision) pumped by a Nd:YAG laser (Continuum Surelite II) operating at 10 Hz, coupled to a Paul ion trap mass spectrometer (Esquire 6000+, Bruker Daltonics) in an already described experimental setup.29 The typical output energy from the OPO/OPA laser was ca. 27 mJ per pulse in the inspected spectral range with a deviation from the average of maximum 2 mJ per pulse. In the trap, ions were mass selected, stored for 30 ms and irradiated for 2 s. The H2O/MeOH solutions of cisplatin and transplatin were directly infused using a syringe pump at a flow rate of 180 μL h−1 to the electrospray ionization (ESI) source. The laser induces the fragmentation of the ions stored in the trap when the IR frequency matches the energy of an IR active mode of the sampled species. The IRMPD spectra are obtained by plotting the photofragmentation yield R = −log(Ip/(Ip + ΣIf)), where Ip and ΣIf are the parent and sum of the fragment ion intensity, respectively, as a function of the radiation wavenumber.2.3 Ion-molecule reactions in FT-ICR mass spectrometry
Ion-molecule reactions have been examined in the cell of a Fourier transform-ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker BioApex) equipped with an Apollo I ESI source and a 4.7 T magnet. Kinetic data were obtained at the nominal cell temperature of 300 K, as described in detail elsewhere.30 The pseudo first order rate constants, obtained from the semilog plot of the parent ion decay with increasing reaction time, were divided by the neutral concentration to give the second order rate constant (kexp). The reaction efficiency (Eff = kexp/kcoll × 100, where kcoll is the collisional rate constant)31 expresses the percentage of reactive collisions.2.4 Density functional theory calculations
Quantum mechanics calculations have been employed to aid in interpreting the experimental data using density functional (DFT) methods as implemented in the Gaussian 09 Rev D.01 package.32 The hybrid functional B3LYP has been used in combination with the aug-cc-pVTZ basis set for the light atoms. Relativistic effects affecting Pt were taken into account employing the pseudopotential aug-cc-pVTZ-PP.33,34 The optimized structures were submitted to harmonic vibrational frequency analysis at the same level of theory in order to characterize the obtained structures as local minima or transition states and to obtain thermodynamic corrections to the electronic energies and IR spectra. The harmonic frequencies were scaled by a factor of 0.957, as used in previous studies.35,36 Calculated linear IR spectra are convoluted with a Lorentzian line shape with a width (fwhm) of 5 cm−1 for convenient comparison with the experimental spectra in the inspected IR region.Structures and energies pertaining to the potential energy surface (PES) for the reaction of cis- and trans-[PtCl(NH3)2(H2O)]+ with thioanisole were calculated at the ω-B97XD/6-311+G** level with the LanL2TZ pseudopotential for the platinum atom, in order to be consistent with previous work.18
3. Results and discussion
3.1 Vibrational features and optimized structures of cis- and trans-[Pt(OH)(NH3)2(H2O)]+ ions
cis-[Pt(OH)(NH3)2(H2O)]+ and trans-[Pt(OH)(NH3)2(H2O)]+ obtained by stepwise hydrolysis of cisplatin in aqueous solution have been assayed by IRMPD spectroscopy in order to unveil vibrational features of the bare mass selected ions. The mass spectrum of trans-[Pt(OH)(NH3)2(H2O)]+ ions isolated in the ion trap mass spectrometer is shown in Fig. 1a. The isotopic pattern is distinctive of a species containing a platinum atom, excluding the contribution of formally isobaric peaks of [PtCl(NH3)2]+ composition. The elemental composition of the ions is thus verified, agreeing with the calculated ion abundances of the isotopic cluster. Further confirmation of the ion composition is obtained by FT-ICR mass spectrometry operated in high resolution mode. As shown in Fig. S1 in the ESI,† the observed m/z values for the isotopic peaks are within ±2 ppm of the theoretical m/z value. The mass spectrum of cis-[Pt(OH)(NH3)2(H2O)]+ is in everything similar. For the sake of simplicity, henceforth isotopic cluster ions are named by the mass of the 194Pt isotope containing species. The mass spectrometric behavior, namely the collision induced dissociation (CID) mass spectrum of the two species, is quite comparable, impeding any structural discrimination (see CID mass spectra reported in Fig. S2 in the ESI†). IR spectroscopy was therefore turned to as potential structural probe. Both cis- and trans-[Pt(OH)(NH3)2(H2O)]+, when submitted to irradiation by IR photons in resonance with an active vibrational mode, undergo water loss yielding ions at m/z 245 (Fig. 1b). Additional NH3 loss is also observed, albeit to a minor extent (m/z 228). The fragment ion profiles present similar features in both m/z 245 and m/z 228 channels and IRMPD spectra are obtained plotting the IRMPD yield as a function of the IR wavenumber.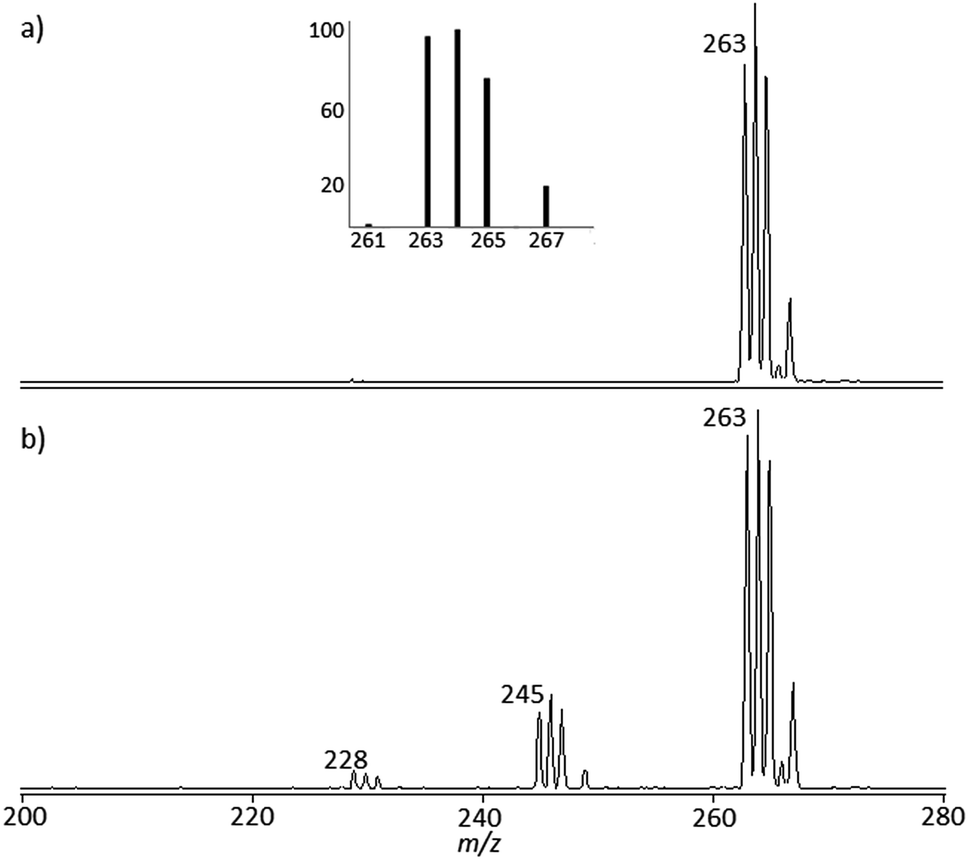 | ||
| Fig. 1 Mass spectrum showing isolated trans-[Pt(OH)(NH3)2(H2O)]+ ions at m/z 263 prior to (a) and after (b) irradiation by IR photons at 3600 cm−1 (Esquire 6000). The inset shows the calculated isotope pattern for the given atom composition. | ||
The experimental spectra shown in blue in Fig. 2a and b are different for the two isomers. To begin with, the photofragmentation yield is different, the process being more efficient in the case of the trans isomer relative to the cis. This behavior is traced to the diverse energy threshold for the loss of water. DFT calculations providing optimized geometries and thermodynamic data (see Scheme S1 and Tables S1–S3 in the ESI†) show that the process is uphill by 149 and 119 kJ mol−1 (ΔH at 298 K) for the cis and trans isomer, respectively.
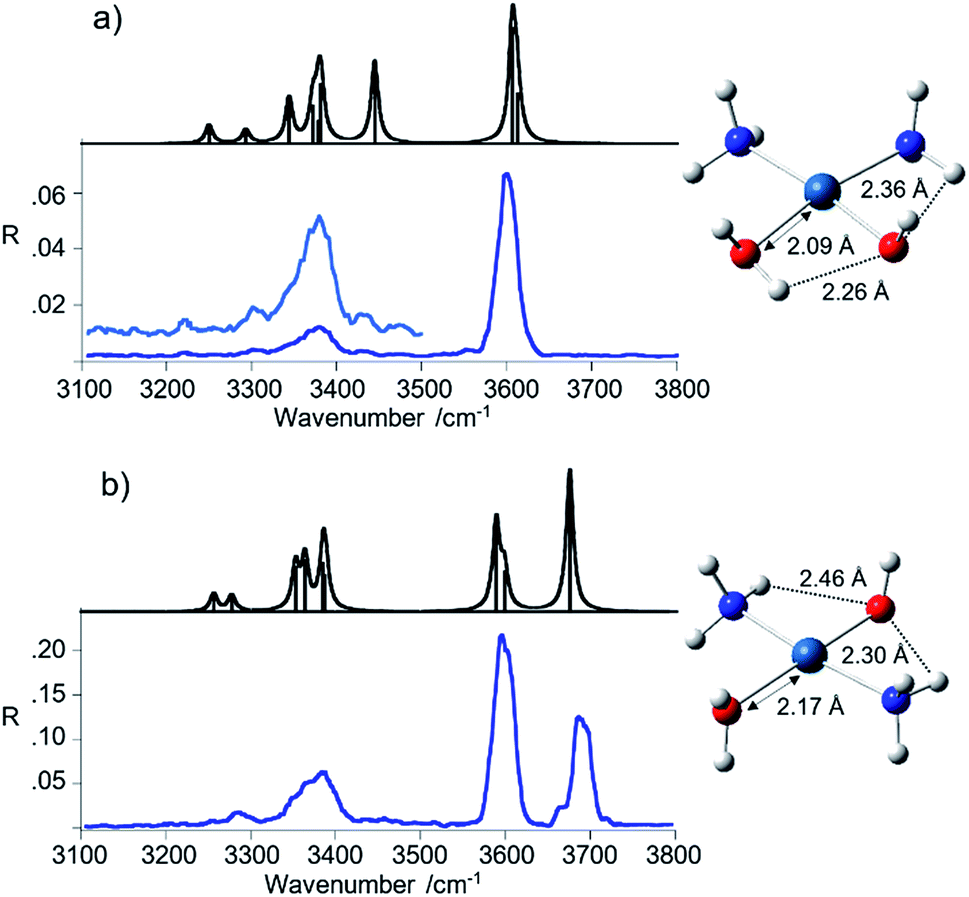 | ||
| Fig. 2 IRMPD spectra of cis-[Pt(OH)(NH3)2(H2O)]+ (a) and trans-[Pt(OH)(NH3)2(H2O)]+ (b) (blue profiles) compared with the calculated linear IR spectra (black profiles). The optimized geometries are shown beside the respective spectra. | ||
The spectrum of the cis isomer presents a strong signal at 3600 cm−1 and less pronounced broad peaks from 3200 to 3450 cm−1, in the range expected for the asymmetric and symmetric N–H stretches of the ammonia ligands. A similar pattern appears in the spectrum of trans-[Pt(OH)(NH3)2(H2O)]+, where the band at 3600 cm−1 is however accompanied by another one at 3683 cm−1 of comparable intensity. In order to gain insight into the observed spectral features, the geometries of cis- and trans-[Pt(OH)(NH3)2(H2O)]+ have been optimized at B3LYP/aug-cc-pVTZ level using the aug-cc-pVTZ-PP pseudopotential for Pt.
The structures are also reported in Fig. 2 while thermodynamic data and Cartesian coordinates are given in Tables S1 and S3 in the ESI,† respectively. Harmonic frequency calculations were performed at the same level of theory and the computed spectra are compared to the experimental ones in Fig. 2, while vibrational frequencies and IR intensities are reported in Table 1.
| Vibrational modea | Exp.b | Theor.b,c |
|---|---|---|
| a νs and νas stand for symmetric and asymmetric stretching modes, respectively.b In cm−1.c Computed harmonic intensities (km mol−1) are reported in brackets. | ||
| cis-[Pt(OH)(NH3)2(H2O)]+ | ||
| ν OH | 3600 | 3612 (61) |
| νas H2O | 3606 (142) | |
| νs H2O | 3442 | 3444 (101) |
| νas NH3 | 3379 | 3381 (72) |
| νas NH3 | 3378 (27) | |
| νas NH3 | 3372 (46) | |
| νas NH3 | 3344 (55) | |
| νs NH3 | 3300 | 3293 (17) |
| νs NH3 | 3222 | 3250 (22) |
|
||
| trans-[Pt(OH)(NH3)2(H2O)]+ | ||
| νas H2O | 3683 | 3674 (166) |
| ν OH | 3600 | 3598 (46) |
| νs H2O | 3588 (103) | |
| νas NH3 | 3384 | 3387 (42) |
| νas NH3 | 3385 (57) | |
| νas NH3 | 3363 (60) | |
| νas NH3 | 3352 (52) | |
| νs NH3 | 3284 | 3277 (19) |
| νs NH3 | 3256 (21) | |
The calculated linear IR spectra account well for the major signatures observed in the IRMPD spectra of both cis- and trans-[Pt(OH)(NH3)2(H2O)]+ isomers. The matching is underlined in the data summarized in Table 1. Few structural and vibrational features may be highlighted. A notable difference between the two geometries is in the length of the Pt–OH2 bond. The shorter distance in the cis isomer may be related to the presence of a H-bond interaction between the hydrogen of the water ligand lying on the molecular plane and the adjacent hydroxyl oxygen. This feature bears consequence in the vibrational spectrum.
The IRMPD spectrum of the trans-[Pt(OH)(NH3)2(H2O)]+ isomer shows two bands in the OH stretching region at 3683 and 3600 cm−1 corresponding to the H2O asymmetric stretching and a combination of the OH and H2O symmetric stretching modes, respectively. Interestingly, the O–H stretching modes in the spectrum of trans-[Pt(OH)(NH3)2(H2O)]+ well reproduce the situation observed in the IRMPD spectrum of trans-[PtCl(NH3)2(H2O)]+, where absorbances at 3683 and 3596 cm−1 have been reported.9 This finding highlights the comparable effect of either chloro or hydroxo substituents on the vibrational modes of the trans water ligand. A difference in relative intensity, enhancing the band at 3600 cm−1 compared to the one at 3683 cm−1 in trans-[Pt(OH)(NH3)2(H2O)]+, is proportional to the additive contribution in the former by the OH stretching and H2O symmetric stretching oscillators.
In the cis isomer, one hydrogen atom of the water ligand on the molecular plane is oriented towards the adjacent hydroxyl oxygen, as already noticed. This interaction causes an important red shift of the water stretching modes. In particular, the asymmetric stretching shifts to 3606 cm−1, coalescing with the OH stretching calculated at 3612 cm−1, accounting for the band observed at 3600 cm−1. The symmetric H2O stretching is calculated at 3444 cm−1 and barely emerges at 3442 cm−1 in the IRMPD spectrum. However, it is not unusual to find an unexpectedly low intensity associated to stretching modes involved in hydrogen bonding. Possible reasons for this behavior include anharmonicity effects as well as effects related to the stepwise excitation in the IRMPD process, disrupting the hydrogen bond and removing the affected mode from resonance.36–39
Stretching modes of the ammonia ligands appear below 3400 cm−1. These modes are not particularly influenced by the relative position of the other ligands. Indeed, only few differences emerge from the analysis of the NH3 stretching vibrations. The asymmetric stretching modes give rise to a broad feature with maximum at ca. 3380 cm−1 in the spectra of both isomers. The presence of multiple absorptions close in frequency is compatible with the broad signal, well interpreted by the calculations. The symmetric stretching modes, instead, are poorly active and are observed at lower frequencies. It is interesting to note that in the IRMPD spectrum of cis-[Pt(OH)(NH3)2(H2O)]+ the small band at 3300 cm−1 matches the NH3 symmetric stretching calculated at 3293 cm−1, while in the spectrum of the trans isomer the corresponding band is found at 3284 cm−1 (calculated value 3277 cm−1). This slightly lower frequency is likely due to the interaction of both NH3 ligands with the hydroxyl oxygen, a stronger binding motif that the one with water. In the cis isomer only one ammonia molecule is allowed to interact with the OH group while the second one may interact only with H2O, yielding resonances calculated at 3250 cm−1 and 3293 cm−1, respectively.
3.2 Reactivity of [PtX(NH3)2(H2O)]+ (X = Cl, OH) complexes in ligand substitution reactions
cis- and trans-[PtX(NH3)2(H2O)]+ (X = Cl, OH) complexes, formed in solution by stepwise hydrolysis of the respective precursors, cisplatin and transplatin, cis- and trans-PtCl2(NH3)2, have been allowed to react with selected ligands of appropriate volatility introduced at stationary concentration in the cell of an FT-ICR mass spectrometer. The ligands are endowed with functional groups that are well established biomolecular platination sites, for example the thioether functional group of methionine, the aza group present in pyridine and in nucleobases, the oxygen atoms of phosphoric acid derivatives.4,29The reaction common to all complexes [PtX(NH3)2(H2O)]+ is ligand exchange whereby a water molecule is replaced by the incoming L, with L equal to trimethylphosphate (TMP), pyridine (Py), thioanisole (TA), and dimethylsulfide (DMS) (reaction (1)). Fig. 3 shows an example of kinetic plot for the reaction of cis-[PtCl(NH3)2(H2O)]+ with DMS.
| cis/trans-[PtX(NH3)2(H2O)]+ + L → cis/trans-[PtX(NH3)2(L)]+ + H2O | (1) |
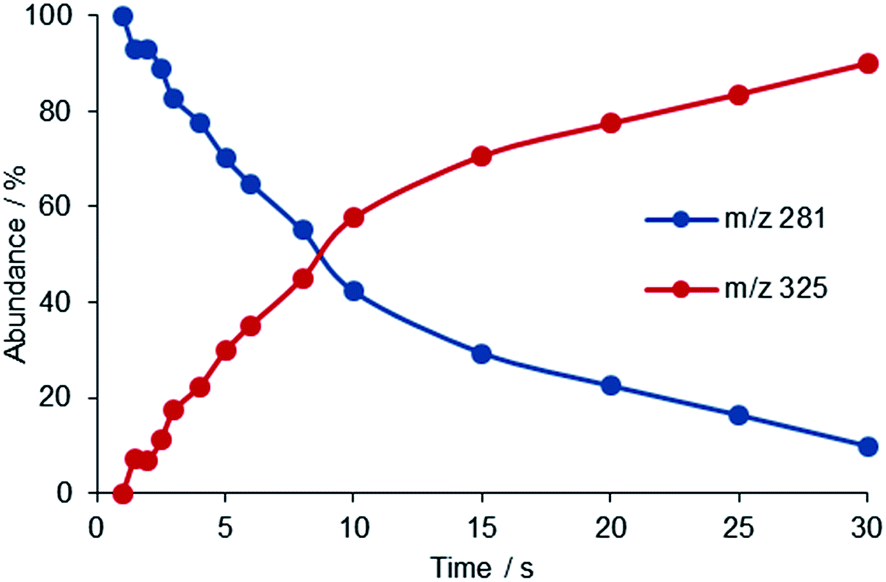 | ||
| Fig. 3 Time dependence of ion abundancies for the reaction of cis-[PtCl(NH3)2(H2O)]+ (m/z 281) with DMS forming [PtCl(NH3)2(DMS)]+ at m/z 325 in the FT-ICR cell at the DMS pressure of 1.5 × 10−7 mbar. | ||
Table 2 summarizes the second order rate constants and corresponding efficiencies, representing the fraction of reactive collisions. Pyridine, the most basic among the selected ligands, displays also proton transfer reactivity and ligand addition to some extent. All reactions show rather low efficiencies. This finding is in line with the behavior of platinum(II) complexes, characterized by typically slow ligand exchange kinetics.25
| Reagent ion | Neutral | kexpa | Effb (%) |
|---|---|---|---|
| a Second order rate constant in units of 10−11 cm3 s−1 at 298 K, estimated error ±30%. Other sampled compounds proved to be unreactive: H218O, H2S.b Ref. 18.c The ligand substitution reaction is accompanied by ligand addition yielding [PtCl(NH3)2(Py)(H2O)]+ (Eff = 0.13%) and by proton transfer to pyridine yielding [pyridine H]+ (Eff = 1.6%).d The ligand substitution reaction is accompanied by proton transfer to pyridine yielding [pyridine H]+ (Eff = 12%).e The ligand substitution reaction is accompanied by ligand addition (Eff = 0.10%) and by proton transfer (Eff = 0.29%).f n. r. stands for non-reactive.g The ligand substitution reaction is accompanied by proton transfer (Eff = 0.64%). | |||
| cis-[PtCl(NH3)2(H2O)]+ | TMP | 3.7 | 2.5b |
| Pyridine | 0.6 | 0.41b,c | |
| Thioanisole | 1.4 | 1.1b | |
| Dimethylsulfide | 0.034 | 0.026 | |
| trans-[PtCl(NH3)2(H2O)]+ | TMP | 3.3 | 2.3 |
| Pyridine | 1.4 | 0.93d | |
| Thioanisole | 7.7 | 6.3 | |
| Dimethylsulfide | 2.4 | 1.7 | |
| cis-[Pt(OH)(NH3)2(H2O)]+ | TMP | 0.96 | 0.66 |
| Pyridine | 0.46 | 0.31e | |
| Thioanisole | 0.1 | 0.08 | |
| Dimethylsulfide | n. r.f | ||
| trans-[Pt(OH)(NH3)2(H2O)]+ | TMP | 1.1 | 0.78 |
| Pyridine | 0.05 | 0.03g | |
| Thioanisole | 0.46 | 0.37 | |
| Dimethylsulfide | n. r.f | ||
The most notable feature emerging from the kinetic data in Table 2 is the consistently higher reactivity of [PtCl(NH3)2(H2O)]+ complexes with respect to the hydroxo counterparts [Pt(OH)(NH3)2(H2O)]+. For example, reaction efficiency ratios for the reaction of TA with cis-[PtX(NH3)2(H2O)]+ and trans-[PtX(NH3)2(H2O)]+ are 14 and 17, respectively. TA is a soft ligand expected to show high efficiency toward the soft platinum center.25–27,40–45 However, TMP, an O-donor nucleophile, shows relatively high reactivity. It should be noticed, though, that the ligand exchange reaction outlined in eqn (1) is in fact a stepwise process in the gas-phase involving: (i) formation of a reactant collision complex (RC); (ii) ligand substitution yielding the product complex (PC) and (iii) separation of the product pair, as depicted in eqn (2).
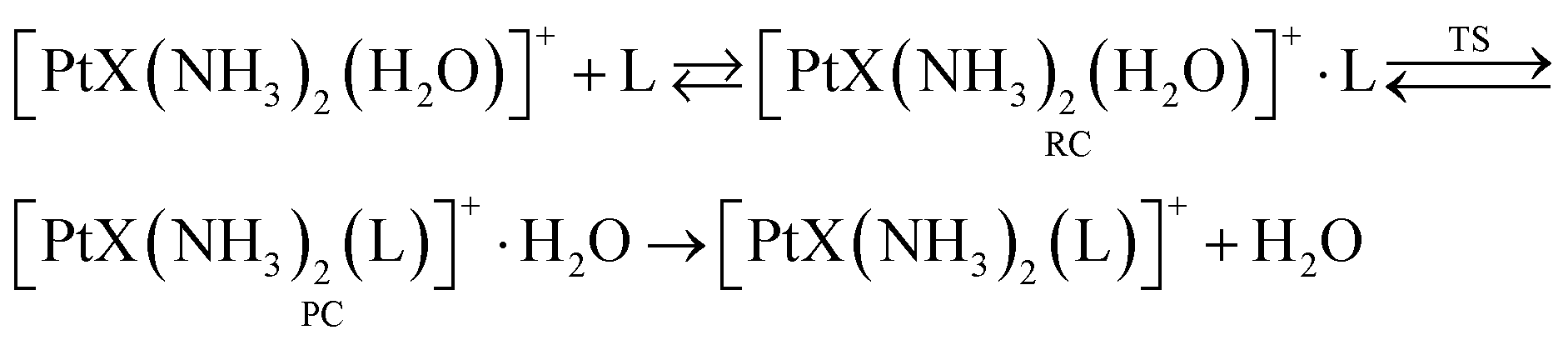 | (2) |
The efficiency of the overall process is determined by the branching of RC which may either proceed to the ligand substitution product (PC) or revert to separated reactants.46,47 The kinetic data are well interpreted by the computed profile of the potential energy surface (PES). A detailed discussion can be found in a recent report regarding results that include the first three entries in Table 1.18 It is worth noting that the relatively high efficiency for the TMP reaction with cis-[PtCl(NH3)2(H2O)]+ is to a large extent determined by the high threshold energy for back dissociation of RC compared with the activation energy for ligand exchange. In the TA reaction the activation barrier is lower but the energy released in the RC formation is sensibly diminished, increasing the competition of the back dissociation channel and thus leading to lower reaction efficiency.18 The energy profile for reaction (2) is further discussed in the following paragraph.
S-Donor nucleophiles like TA and DMS do not engage in strong hydrogen bonding interactions and the binding energy of RC is not sizeable. Ligand exchange is kinetically highly favored in the trans- with respect to the cis-isomers for the chloro complexes with a 5.7 times higher efficiency for TA and a 65 fold higher efficiency for DMS. This finding may be ascribed to the trans effect operated by a resident ligand on the rate of substitution of the ligand trans to it.25–27 In the present case, the water leaving group is displaced by S-nucleophiles more easily when the trans-site is occupied by a chloro ligand than by NH3, in agreement with the qualitative trans-directing sequence Cl− > NH3. However, this effect is not apparently clear in other instances.
To shed light on this point, the potential energy surface for reaction (2) involving the substitution of water in both cis and trans-[PtCl(NH3)2(H2O)]+ with TA was explored and is reported in Fig. 4, while thermodynamic data and Cartesian coordinates for the partaking species are reported in Tables S2 and S4 in the ESI.† The square planar Pt complexes conform to a substitution mechanism of prevailing associative character and this is clearly verified in the gas-phase reaction.25 The substitution process typically occurs with stereoretention by way of a trigonal bipyramidal transition state where the formerly trans ligand and the entering and leaving ligands are placed on the equatorial plane. The energy released in the formation of the RC complexes is comparable in the two species, 115 and 105 kJ mol−1 for cis-RC and trans-RC respectively. Both RC complexes present interactions between the π-system of the aryl group and the NH3 ligands.
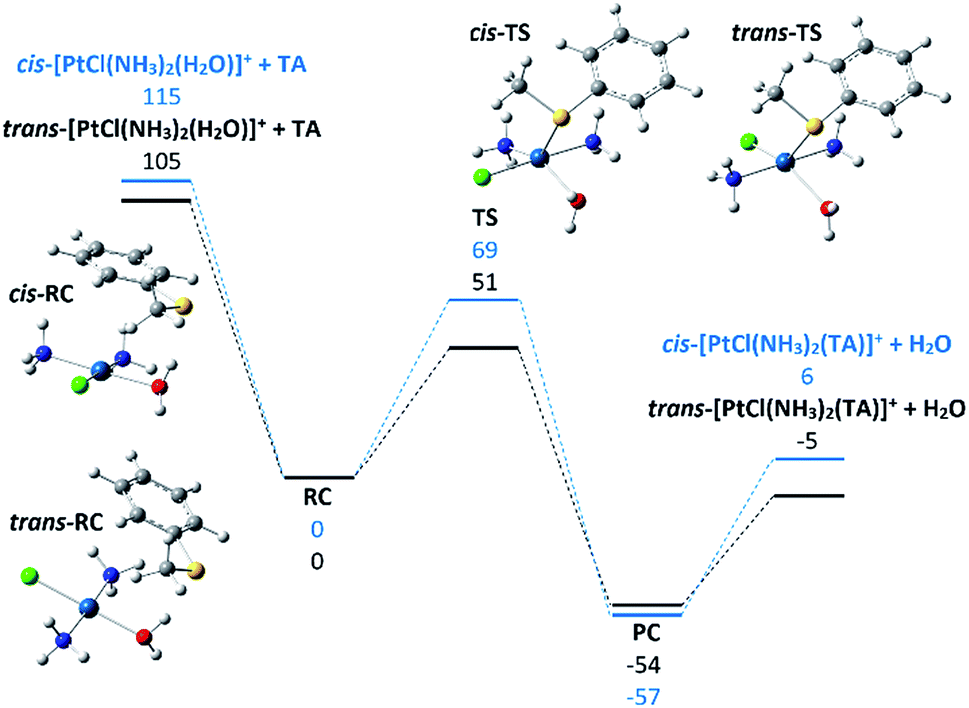 | ||
| Fig. 4 Computed profile for the reaction of cis- and trans-[PtCl(NH3)2(H2O)]+ with TA. Optimized geometries of the reactant collision complexes (cis- and trans-[PtCl(NH3)2(H2O)]+·TA) and of the transition states TS are shown. Relative enthalpies at 298 K (kJ mol−1) are reported. | ||
The observed difference in energy of ca. 10 kJ mol−1 is ascribable to the possibility for the cis-isomer to entangle interactions with both ammonia molecules due to their favorable arrangement. In contrast, the activation barrier involved to reach the TS for ligand exchange has a more pronounced variation. The two calculated TS structures are very similar, making the interpretation of the observed difference a difficult task. Two points should be highlighted: the presence of the chlorine atom in trans to the exchanging ligands leads to slightly longer bonds by ca. 0.07 Å suggesting the effect of the trans ligand not to be negligible. Both transition states present multiple interactions between the two ligands engaged in the substitution and the ligands in the cis-sites that can be implicated in the calculated difference. The listed elements probably contribute to give the final results that are anyway in fair agreement with the experiments. The reaction efficiency of the trans-isomer being 5.7 times higher with respect to the cis-one (6.3 versus 1.1, see Table 2) is, in fact, well interpreted by the higher energy difference between the separated reactants and the transition state for the trans with respect to the cis configuration (54 and 46 kJ mol−1, respectively). The trans isomer is thus more prone to undergo ligand substitution in view of the relatively more energy demanding back dissociation to reactants. The latter process remains, however, entropically favored which accounts for the low values of reaction efficiency, well below collision control.
Few other tested ligands proved to be unreactive, including H218O, which suggests a sizeable activation barrier for the water exchange process. Fig. 5 shows the symmetrical profile for reaction (2) with cis-[Pt(OH)(NH3)2(H2O)]+ as reactant and product ion (thermodynamic data and coordinates of all species are given in Tables S1 and S3 in the ESI,† respectively). Here the collision complexes RC and PC are symmetrical, apart from the 18O-isotope distribution. Their geometry is characterized by two hydrogen bonds linking a hydrogen atom of ligated water with an O atom of the external water and a H atom of the external H2O with the hydroxyl oxygen. In forming the RC complex, however, the hydrogen bond interaction between the in plane water hydrogen and the hydroxyl oxygen in cis-[Pt(OH)(NH3)2(H2O)]+ is impaired, which may account for the relatively small binding energy of 72 kJ mol−1. The activation barrier for the ligand exchange (RC → TS) is 91 kJ mol−1, protruding above the energy level of the isolated reactant pair. This feature implies that, in the single collision regime prevailing in the FT-ICR cell, the reaction will not occur, as experimentally observed. The RC complex would require an additional source of ΔE = 91 − 72 = 19 kJ mol−1 for the reaction to proceed in the isolated, low pressure environment.
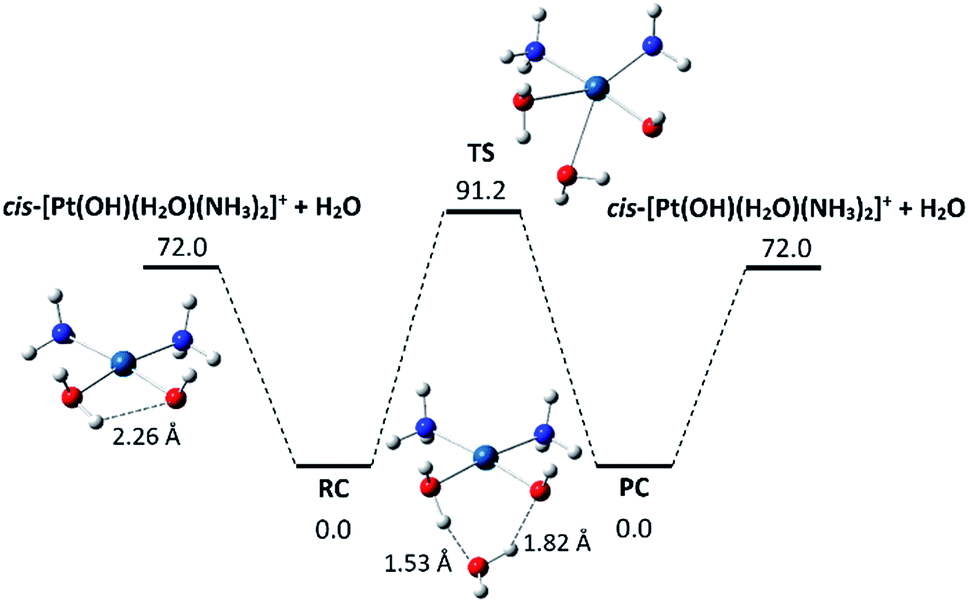 | ||
| Fig. 5 Computed profile for the degenerate reaction of cis-[Pt(OH)(NH3)2(H2O)]+ with H2O. Optimized geometries of cis-[Pt(OH)(NH3)2(H2O)]+, of the collision complex RC/PC (cis-[Pt(OH)(NH3)2(H2O)]+·H2O) and of the transition state TS are shown. Relative enthalpies at 298 K (kJ mol−1) are reported. | ||
For comparison purposes, the transition state for the substitution of water from cis-[PtCl(NH3)2(H2O)]+ by TMP, TA and Py has been found to lie 67, 46, and 43 kJ mol−1 below the energy of the reactants, respectively, giving rise to productive collisions as shown in Table 2.18 However, entropic factors account for reaction efficiencies (2.5, 1.1, and 0.41, for the tested series of ligands, TMP, TA, and Py, respectively) that are considerably below 100.
The whole set of calculations performed is thus consistent in interpreting the experimental results as a function of multiple factors. The reactivity of square planar complexes is often related to the presence of certain ligated atoms in trans position. The evidence presently obtained points in a different direction showing a remarkable importance of the cis-sites in entangling interactions with the reacting ligands all along the entire reaction. Significantly, groups possessing multiple binding sites, e.g. TMP, present the worst agreement with the qualitative indications suggested by the trans effect.
4. Conclusions
The early hydrolysis products from cis and transplatin, cis- and trans-[PtX(NH3)2(H2O)]+ (X = Cl, OH), have been obtained by ESI and isolated in the gas-phase where their structure and reactivity properties have been comparatively examined. The gaseous environment permits to unambiguously characterize the selected ions. This task is hard to achieve in water solution where prototropic equilibria occur and the formation of hydroxo-bridged polynuclear complexes is an important competing process.48 Even the use of buffered solutions presents the drawback of interferences due to reaction of hydrolyzed cisplatin with buffer components.7,44IRMPD spectroscopy in the NH/OH stretching range has revealed the vibrational signatures of bare cis- and trans-[Pt(OH)(NH3)2(H2O)]+ ions. While the IR spectrum of trans-[Pt(OH)(NH3)2(H2O)]+ is very similar to the one of the chloro complex, the spectra of the cis complexes are rather different. The major variation in frequency affects the water OH stretching modes. Asymmetric and symmetric resonances are at 3624 and 3531 cm−1 in the chloro complex and 3600 and 3442 cm−1 in the hydroxo complex, respectively. Hydrogen bonding interactions are responsible for the different behavior that is manifested in the orientation of the water ligand, establishing a bisected hydrogen bonding interaction with the adjacent Cl in cis-[PtCl(NH3)2(H2O)]+ while privileging a single H-bond between an in plane water hydrogen atom and the hydroxyl oxygen in cis-[Pt(OH)(NH3)2(H2O)]+.
Both hydrolyzed complexes, [PtCl(NH3)2(H2O)]+ and [Pt(OH)(NH3)2(H2O)]+, have been allowed to react with model compounds of biological ligands. All reactions show rather low efficiencies. This finding is in line with the behavior of platinum(II) complexes, characterized by relatively slow ligand exchange kinetics.25 This feature is critically related to antitumor activity because it allows cisplatin and related drugs to reach the cell intact and effectively bind to cellular DNA rather than to extracellular binding sites.25,49
The reactivity of the chloro complexes, [PtCl(NH3)2(H2O)]+, is consistently higher than for the hydroxo-ligated counterparts, [Pt(OH)(NH3)2(H2O)]+, the chloro substituent likely enhancing the electrophilicity of the metal. This finding may be related to the observation that the majority of the binding of cisplatin to DNA occurs via the monoaquated species cis-[PtCl(NH3)2(OH2)]+.1–7 In comparing the reactivity of different ligands, caution is due to the contribution of the reactant pair binding energy that affects the reaction efficiency together with the ligand exchange activation barrier. As expected from the soft character of platinum(II) an S-nucleophile like TA is fairly reactive, in accord with the function of S-donor compounds both as Pt-reservoirs in the body and as “rescue agents”.40–45 Notably, also TMP, an O-donor ligand, shows relatively high efficiency in ligand exchange, thus supporting a role for phosphate groups in cisplatin binding and delivery.
It may be underlined here that the present report on the gas phase reactivity of cis- and trans-[PtX(NH3)2(H2O)]+ complexes does not take into account the effect of the solvent that has to be included, for example, also when theoretical work is applied to interpret reactions occurring in solution, as in the case of cisplatin hydrolysis itself.50 Still, it is interesting to observe here a manifestation of the higher intrinsic reactivity of transplatin towards sulfur nucleophiles at the level of the naked species. This result is reminiscent of the more extensive reaction of transplatin with glutathione in red blood cells.51 In this regard, further efforts will be devoted to gain insight into the structural and reactivity features that affect the distinct behavior of cis and trans platinum(II) complexes in a variety of contexts,52,53 most importantly in binding to DNA within the cell nucleus, using the tool of isolating from solution the key intermediates to be examined as long lived species in the gas phase.
Acknowledgements
Thanks are due to Dr Annito Di Marzio for expert technical assistance. This work was supported by the Università degli Studi di Roma “La Sapienza” (Project no. C26H15MHLB).References
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- B. W. Harper, A. M. Krause-Heuer, M. P. Grant, M. Manohar, K. B. Garbutcheon-Singh and J. R. Aldrich-Wright, Chem.–Eur. J., 2010, 16, 7064–7077 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS PubMed.
- J. D. White, M. M. Haley and V. J. DeRose, Acc. Chem. Res., 2016, 49, 56–66 CrossRef CAS PubMed.
- J. Kozelka, Inorg. Chim. Acta, 2009, 362, 651–668 CrossRef CAS.
- M. S. Davies, S. J. Berners-Price and T. W. Hambley, Inorg. Chem., 2000, 39, 5603–5613 CrossRef CAS PubMed.
- A. Springer, C. Buergel, V. Boehrsch, R. Mitric, V. Bonacic-Koutecky and M. W. Linscheid, ChemPhysChem, 2006, 7, 1779–1785 CrossRef CAS PubMed.
- A. De Petris, A. Ciavardini, C. Coletti, N. Re, B. Chiavarino, M. E. Crestoni and S. Fornarini, J. Phys. Chem. Lett., 2013, 4, 3631–3635 CrossRef CAS.
- J. Oomens, B. G. Sartakov, G. Meijer and G. Von Helden, Int. J. Mass Spectrom., 2006, 254, 1–19 CrossRef CAS.
- N. C. Polfer, Chem. Soc. Rev., 2011, 40, 2211–2221 RSC.
- J. Roithova, Chem. Soc. Rev., 2012, 41, 547–559 RSC.
- T. D. Fridgen, Mass Spectrom. Rev., 2009, 28, 586–607 CrossRef CAS PubMed.
- J. R. Eyler, Mass Spectrom. Rev., 2009, 28, 448–467 CrossRef CAS PubMed.
- L. MacAleese and P. Maitre, Mass Spectrom. Rev., 2007, 26, 583–605 CrossRef CAS PubMed.
- B. Chiavarino, M. E. Crestoni, S. Fornarini, D. Scuderi and J.-Y. Salpin, J. Am. Chem. Soc., 2013, 135, 1445–1455 CrossRef CAS PubMed.
- B. Chiavarino, M. E. Crestoni, S. Fornarini, D. Scuderi and J.-Y. Salpin, Inorg. Chem., 2015, 54, 3513–3522 CrossRef CAS PubMed.
- D. Corinti, C. Coletti, N. Re, B. Chiavarino, M. E. Crestoni and S. Fornarini, Chem.–Eur. J., 2016, 22, 3794–3803 CrossRef CAS PubMed.
- C. C. He, B. Kimutai, X. Bao, L. Hamlow, Y. Zhu, S. F. Strobehn, J. Gao, G. Berden, J. Oomens, C. S. Chow and M. T. Rodgers, J. Phys. Chem. A, 2015, 119, 10980–10987 CrossRef CAS PubMed.
- D. Schröder and H. Schwarz, Can. J. Chem., 2005, 83, 1936–1940 CrossRef.
- B. Butschke and H. Schwarz, Chem.–Eur. J., 2012, 18, 14055–14062 CrossRef CAS PubMed.
- M.-E. Moret and P. Chen, Organometallics, 2007, 26, 1523–1530 CrossRef CAS.
- R. Liyanage, M. L. Styles, R. A. J. O'Hair and P. B. Armentrout, J. Phys. Chem. A, 2003, 107, 10303–10310 CrossRef CAS.
- S. Wee, R. A. J. O'Hair, A. J. Richard and W. D. McFadyen, Rapid Commun. Mass Spectrom., 2004, 18, 1221–1226 CrossRef CAS PubMed.
- D. T. Richens, Chem. Rev., 2005, 105, 1961–2002 CrossRef CAS PubMed.
- U. Belluco, L. Cattalini, F. Basolo, R. G. Pearson and A. Turco, J. Am. Chem. Soc., 1965, 87, 241–246 CrossRef CAS.
- B. Pitteri, L. Canovese, G. Chessa, G. Marangoni and P. Uguagliati, Polyhedron, 1992, 11, 2363–2373 CrossRef CAS.
- S. Elmroth, Z. Bugarcic and L. I. Elding, Inorg. Chem., 1992, 31, 3551–3554 CrossRef CAS.
- R. K. Sinha, P. Maître, S. Piccirillo, B. Chiavarino, M. E. Crestoni and S. Fornarini, Phys. Chem. Chem. Phys., 2010, 12, 9794–9800 RSC.
- F. Angelelli, B. Chiavarino, M. E. Crestoni and S. Fornarini, J. Am. Soc. Mass Spectrom., 2005, 16, 589–598 CrossRef CAS PubMed.
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183–5185 CrossRef CAS.
- M. J. Frisch, et al., Complete reference is given in the ESI data.†.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108–164119 CrossRef PubMed.
- B. Pinter, V. Van-Speybroeck, M. Waroquier, P. Geerlings and F. De-Proft, Phys. Chem. Chem. Phys., 2013, 15, 17354–17365 RSC.
- P. Sinha, S. E. Boesch, C. Gu, R. A. Wheeler and A. K. Wilson, J. Phys. Chem. A, 2004, 108, 9213–9217 CrossRef CAS.
- A. Bouchet, M. Schütz, B. Chiavarino, M. E. Crestoni, S. Fornarini and O. Dopfer, Phys. Chem. Chem. Phys., 2015, 17, 25742–25754 RSC.
- F. Turecek, C. L Moss, I. Pikalov, R. Pepin, K. Gulyuz, N. C. Polfer, M. F. Bush, J. Brown, J. Williams and K. Richardson, Int. J. Mass Spectrom., 2013, 354, 249–256 CrossRef.
- N. Heine, T. I. Yacovitch, F. Schubert, C. Brieger, D. M. Neumark and K. R. Asmis, J. Phys. Chem. A, 2014, 118, 7613–7622 CrossRef CAS PubMed.
- B. Chiavarino, M. E. Crestoni, M. Schütz, A. Bouchet, S. Piccirillo, V. Steinmetz, O. Dopfer and S. Fornarini, J. Phys. Chem. A, 2014, 118, 7130–7138 CrossRef CAS PubMed.
- Z. D. Bugarcic, J. Bogojeski, B. Petrovic, S. Hochreuther and R. van Eldik, Dalton Trans., 2012, 41, 12329–12345 RSC.
- A. Casini and J. Reedijk, Chem. Sci., 2012, 3, 3135–3144 RSC.
- G. Ferraro, L. Messori and A. Merlino, Chem. Commun., 2015, 51, 2559–2561 RSC.
- H. Li, Y. Zhao, H. I. A. Phillips, Y. Qi, T. Y. Lin, P. J. Sadler and P. B O'Connor, Anal. Chem., 2011, 83, 5369–5376 CrossRef CAS PubMed.
- J. Vinje, E. Sletten and J. Kozelka, Chem.–Eur. J., 2005, 11, 3863–3871 CrossRef CAS PubMed.
- O. Pinato, C. Musetti, N. P. Farrell and C. Sissi, J. Inorg. Biochem., 2013, 122, 27–37 CrossRef CAS PubMed.
- M. L. Chabinyc, S. L. Craig, C. K. Regan and J. I. Brauman, Science, 1998, 279, 1882–1886 CrossRef CAS PubMed.
- S. Gronert, Chem. Rev., 2001, 101, 329–360 CrossRef CAS PubMed.
- S. J. Berners-Price and T. G. Appleton, The Chemistry of Cisplatin in Aqueous Solution, in Platinum Based Drugs in Cancer Therapy, ed. L. R.Kelland and N. Farrell, Humana Press Inc., Totowa, N. J., U. S. A., 2000, ch. 1 Search PubMed.
- J. Reedijk, Eur. J. Inorg. Chem., 2009, 10, 1303–1312 CrossRef.
- Y. Zhang, Z. Guo and X.-Z. You, J. Am. Chem. Soc., 2001, 123, 9378–9387 CrossRef CAS PubMed.
- S. J. Berners-Price and P. W. Kuchel, J. Inorg. Biochem., 1990, 38, 327–345 CrossRef CAS PubMed.
- Z. Tao, Y. Xie, J. Goodisman and T. Asefa, Langmuir, 2010, 26, 8914–8924 CrossRef CAS PubMed.
- G. McGowan, S. Parsons and P. J. Sadler, Inorg. Chem., 2005, 44, 7459–7467 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01182b |
| This journal is © The Royal Society of Chemistry 2017 |