
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Paper-based patterned 3D neural cultures as a tool to study network activity on multielectrode arrays†
Harald Dermutz‡
,
Greta Thompson-Steckel‡,
Csaba Forró,
Victoria de Lange,
Livie Dorwling-Carter,
János Vörös and
László Demkó *
*
Laboratory of Biosensors and Bioelectronics, Institute for Biomedical Engineering, ETH Zurich, CH-8092 Zurich, Switzerland. E-mail: demko@biomed.ee.ethz.ch
First published on 11th August 2017
Abstract
Cells in vitro behave differently if cultured in a 2D or 3D environment. In spite of the continuous progress over the recent years, methods available for realizing 3D cultures of primary neurons are still fairly complex, limited in throughput and especially limited in compatibility with other techniques like multielectrode arrays (MEAs) for recording and stimulating the network activity with high temporal precision. In this manuscript, a paper-based approach is presented using cellulose filter paper as a mobile substrate for 3D cultures of primary rat hippocampal and cortical neurons. Acting as 3D scaffolds for network development, filter membranes with different surface treatments were prepared to control network homogeneity and laser cut to change the network topology through spatial confinement. The viability of the prepared cultures was comparable to that of reference 2D cultures for over 4 weeks, and the mechanical stability of the paper substrates made it possible to transfer the cultures to MEA chips in an on-demand manner. Once the cultures were successfully transduced with a gene-encoded calcium indicator and transferred to a MEA chip, the optical and electrical signals of neuronal activity were simultaneously recorded and combined to study the different activity patterns with high spatiotemporal resolution. The high-throughput nature of the presented approach makes it a valuable tool for investigating the intimate relationship between topology and function, by studying the intrinsic parameters influencing network synchronization and signal propagation through the different activity patterns of 3D neural cultures with arbitrary topology. The developed platform provides a robust and simple alternative to existing 3D culturing technologies for neurons.
Introduction
It is becoming increasingly evident that classical 2D in vitro cell cultures have their limitations.1 Organs are inherently 3D systems, therefore there is a great need to improve the classical models in order to better mimic the complex cellular interactions present in in vivo environments, especially in the case of neural networks.2,3 In vitro models have the advantage of being potentially high-throughput, less complex, and more reproducible compared to in vivo animal experiments. Because of these benefits, there is a high potential for using 3D in vitro assays to study cell and network development, and to identify the relevant factors leading to diseases.4 Recognizing the fundamental differences between 2D and 3D cultures increased the demand for a transition to 3D culturing systems and accelerated the progress of related technologies, contributing to improvements, for example in survival rate,5 gene expression,6 and ion channel formation.7Synthetic and biohybrid hydrogels,8–10 as well as biologic-derived materials based on extracellular matrix proteins,11–13 methacrylated gelatin,14 alginate,15–17 and collagen18 have all been tested as potential substrates for 3D cell cultures. While neural tissues could be fabricated without any scaffold material,19 neurons in particular were successfully combined with silk,20,21 glass microbeads,22,23 and bacterial nanocellulose scaffolds24,25 to provide a higher mechanical stability accommodating network development. Cellulose is already a promising substrate for medical applications,26–28 such as bone regeneration,29–31 wound healing,32,33 or vascular reconstruction,34 and related techniques are well-established in pharmacology, especially for drug delivery due to their associated low cost and ease of processing.35–37 In addition, cellulose-based analytical devices have already been used for many years38 and gained a special interest in modern high-throughput approaches.39 The review of Akram et al. provides a great summary of the key properties of cellulose-based substrates, their advantages and disadvantages, as well as a detailed comparison with other substrate types.40 Using cell types other than neurons, different assays have been created by stacking single paper sheets on top of each other,41–44 or introducing hydrophobic barriers to create wells within the sheets, for chemical surface patterning or realizing microreactors.45–48
In spite of the various techniques available, there is no single method that incorporates all the features required for a breakthrough towards achieving realistic 3D neural cultures in vitro. In order to have the potential for becoming widely adopted, a method has to be high-throughput, compatible with primary neurons and co-culturing techniques, and the 3D nature and long-term viability of the prepared cultures have to be verified as well. Although the presented 3D cell culture systems are promising, they mostly require extensive cell-type-dependent experience and a delicate fine-tuning of the different parameters to reach the right encapsulation and stiffness optimal for development and long-term cell survival. Especially in the case of neural cultures, culturing conditions and structural stability are particularly important to promote network development and reach maximum viability over long periods, because neurons are sensitive to culture conditions and develop in a highly dynamic manner.
In addition to the long-term stability, the possibility to control the network topology is also required to successfully mimic in vivo environments. The anatomical structure of the brain is highly organized, and several studies proved that a spatial separation is also often reflected in functional differences.49 However, boundaries of such functional units are often not easy to distinguish in vivo. An in vitro approach with a simplified structure of discrete and identifiable units, implementing the complexity of in vivo networks within these 3D units, could bring further insights into the details of the relationship between network structure and function.
Furthermore, neurological diseases may manifest in highly complex network behaviors, for which conventional random cultures cannot serve as realistic models. Neurons plated and adhered without any topological restrictions form random networks and develop activity patterns highly sensitive to the only parameter under control, the initial seeding concentration and its homogeneity. Although such networks already provide valuable information for pharma- and toxicological studies,50 the extremely rich repertoire of activity patterns makes their interpretation mostly only possible in terms of global, network-wide properties.51 The high number of degrees of freedom can be radically reduced by introducing topological restrictions in order to reach improved reproducibility, increased target specificity in drug design, and eventually to lead to neurocomputational models in a deterministic rather than statistical way.52
Finally, studies investigating the network dynamics of the 3D cultures prepared with the available techniques, which could be the main driving force behind all the related developments, are limited and mostly based on methods using calcium imaging with low temporal resolution and signal-to-noise ratio. As a possible extension, compatibility with multielectrode arrays (MEAs) would be advantageous for both stimulation and readout, even though 3D MEA chips for in vitro studies are only scarcely available at the moment and need further development.53
Here, we propose a platform based on filter membranes used as 3D substrates for primary neuronal cultures. Within the substrates, neurons form 3D networks according to the surface treatment and porosity of the membranes, similarly to neurons in hydrogels and in other scaffold materials. Applying the developed technology is simple and cheap and requires no previous experience or further fine-tuning of the protocol parameters. Paper is structurally stable and easy to handle, resulting in an on-demand availability of the prepared cultures for measurements and providing the platform with high-throughput capabilities. In addition to the different surface treatments, we present two simple patterning approaches, wax printing and laser cutting, to further control the network topology of the neuronal connections formed during maturation. Our study demonstrates the long-term high viability of the prepared cultures, as well as their compatibility with both optogenetic sensors for calcium and conventional MEA technology for electrical readout. Finally, the developed platform includes a setup and protocol for the simultaneous measurement of large-scale network activity and local activity profiles for studying the network dynamics of the prepared patterned 3D neural cultures with high spatiotemporal resolution. As the main novelty, to the best of our knowledge, this work is the first to present a method that successfully use filter membranes as substrates for 3D neural cultures, and combines the proposed platform with the conventional MEA technology.
Materials and methods
Substrate preparation
Fig. 1 illustrates a typical workflow for preparing the paper-based 3D cultures. Cellulose filter membranes (Whatman Grade 602 h, Grade 2, and Grade 1 qualitative filter papers; Sigma-Aldrich, Switzerland) were manually cut with sterile scissors or scalpel into square shapes, which fit into the wells of standard 24-well plates. As an alternative to this step, the lateral topology of the paper cultures was optionally fine-tuned by either printing hydrophobic wax patterns or high-resolution laser cutting (see below). After this, the pieces were autoclaved (at 120 °C for 20 min followed by 15 min cool-down with a Varioklav 25T; Sterico, Switzerland) or plasma treated (oxygen plasma for 2 min at 18 W with a PDC-32G; Harrick Plasma, USA) for sterilization. If an adhesive coating was used, the pieces were always plasma treated and subsequently immersed in either a 20 µg ml−1 laminin (L2020; Sigma-Aldrich) or 100 µg ml−1 poly-D-lysine (PDL, P7280; Sigma-Aldrich) solution, both diluted in phosphate-buffered saline (PBS, pH 7.4; Thermo Fisher), for 45 min. After coating, the paper substrates were intensively rinsed with purified Milli-Q water (from an Elix 5 water purification system; Merck Millipore, USA), transferred into the well plates, covered with culture medium (see below), and placed into an incubator (37 °C, 5% CO2) to adjust the pH and temperature before cell plating. The nominal thicknesses of Grade 602 h, Grade 2, and Grade 1 filter membranes are 160, 190, and 180 µm, respectively, while the corresponding particle retention sizes are <2, 8, and 11 µm, further referred as the characteristic pore sizes.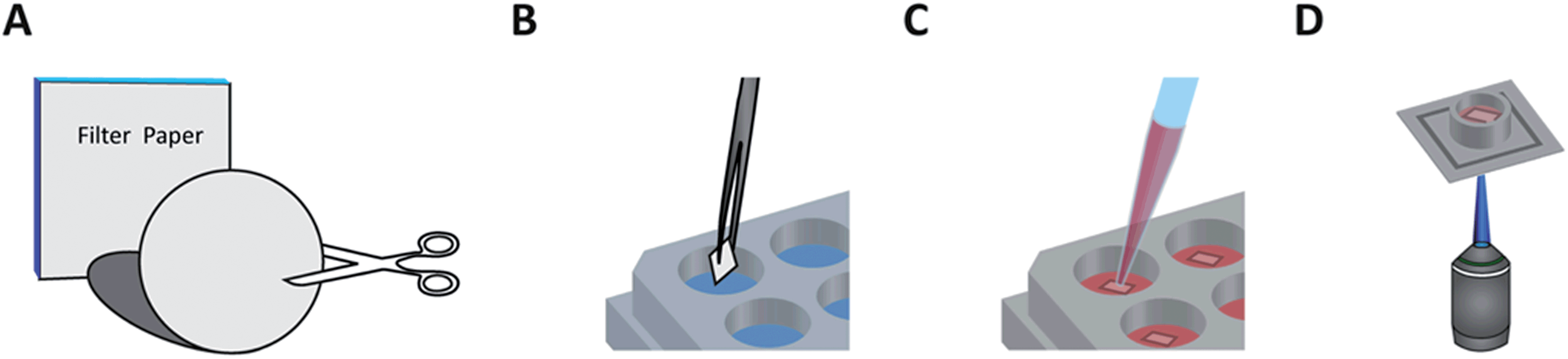 | ||
| Fig. 1 A typical workflow for preparing the paper-based 3D cultures for high-throughput studies. (A) Standard filter papers were cut to the desired size or patterned (optional, see the text for details), then autoclaved and/or O2-plasma treated for sterilization and surface activation (optional). (B) Surface treatment with e.g. laminin (optional). (C) Following a short incubation period with culture medium in multi-well plates, dissociated neurons were pipetted directly on top of the paper pieces. After the necessary incubation time, the pieces were flipped over and the plating step was repeated for double-sided cultures (optional). Later on, the cultures were transduced with a gene-encoded calcium indicator (optional). (D) On the day of measurements the cultures were transferred to a multielectrode array chip one by one and positioned under a fluorescent microscope to observe the network activity. | ||
Primary neuronal cultures
Both the cell types were plated and cultured in serum-free medium, referred to from hereon as culture medium, in order to suppress astroglia proliferation and optimize cell survival.55 For this, Neurobasal medium (21103-049) was supplemented with 2% B-27 serum free supplement (17504-044), 1% penicillin–streptomycin (15140-148), and 1% GlutaMAX (61965-026; all from Thermo Fisher). After dilution, cells were plated in concentrations of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000 cells per cm2, calculated for the bottom surface area of the well plates, and cultured at 37 °C and 5% CO2, exchanging half of the medium with fresh medium once a week. In the case of double-sided plating, paper substrates seeded on one side were flipped over after an incubation period of 60 min, and the plating procedure was repeated according to the experimental protocol.
000–200000 cells per cm2, calculated for the bottom surface area of the well plates, and cultured at 37 °C and 5% CO2, exchanging half of the medium with fresh medium once a week. In the case of double-sided plating, paper substrates seeded on one side were flipped over after an incubation period of 60 min, and the plating procedure was repeated according to the experimental protocol.
Changing from the commonly used DAPI nuclei stain to DRAQ5 greatly improved the imaging quality by eliminating the autofluorescence of the cellulose fibers seen in Fig. S5 of the ESI.†
:1000 dilution of the AAV stock solution (batch number CS0473DL). The cells were incubated with the virus for 3 days, after which the whole medium was replaced with fresh medium and the cells were cultured normally.Experimental setup for recording the neuronal activity
Results and discussion
Cell adhesion, network formation, and viability using different surface treatments
Cellulose of both plant and bacterial origin have been shown to cause only a mild inflammatory response.58–60 Although upscaling the production process of bacterial cellulose is targeted by several groups,33,61–63 and it is becoming commercially available as a wound dressing material, the availability of plant-cellulose-based filter paper is much higher due to its simple preparation and low cost.The commercially available Whatman filter papers are made of eucalyptus and spruce sulfate cellulose, and has already been characterized and used extensively in the literature for biological applications.64–68 As shown in Fig. 2, with a live/dead viability kit we assessed the cell adhesive properties and toxicity of such filter membranes when used as substrates with different surface treatments for neuronal cultures. Neurons do not stay and grow on bare plasma treated glass or polystyrene surfaces due to the lack of cell adhesion, and therefore different surface coating protocols have been established to anchor the cells to the surface.69,70 Interestingly, cells cultured with paper substrates that were heat sterilized only, stayed adhered to the substrate and showed constant viability over 4 weeks as presented in Fig. S2A of the ESI.† Non-specific adsorption of proteins related to the extracellular matrix might be responsible for this effect by providing the minimal adhesion forces on the cellulose surface required for the initial cell survival and network formation.71 Once adhered, neurons formed strong connections with each other assumingly via cadherin–catenin complexes, and probably the attractive forces acting through these connections were that have eventually led to the formation of microspheres similar to those visible in Fig. 2A at 14 DIV.72
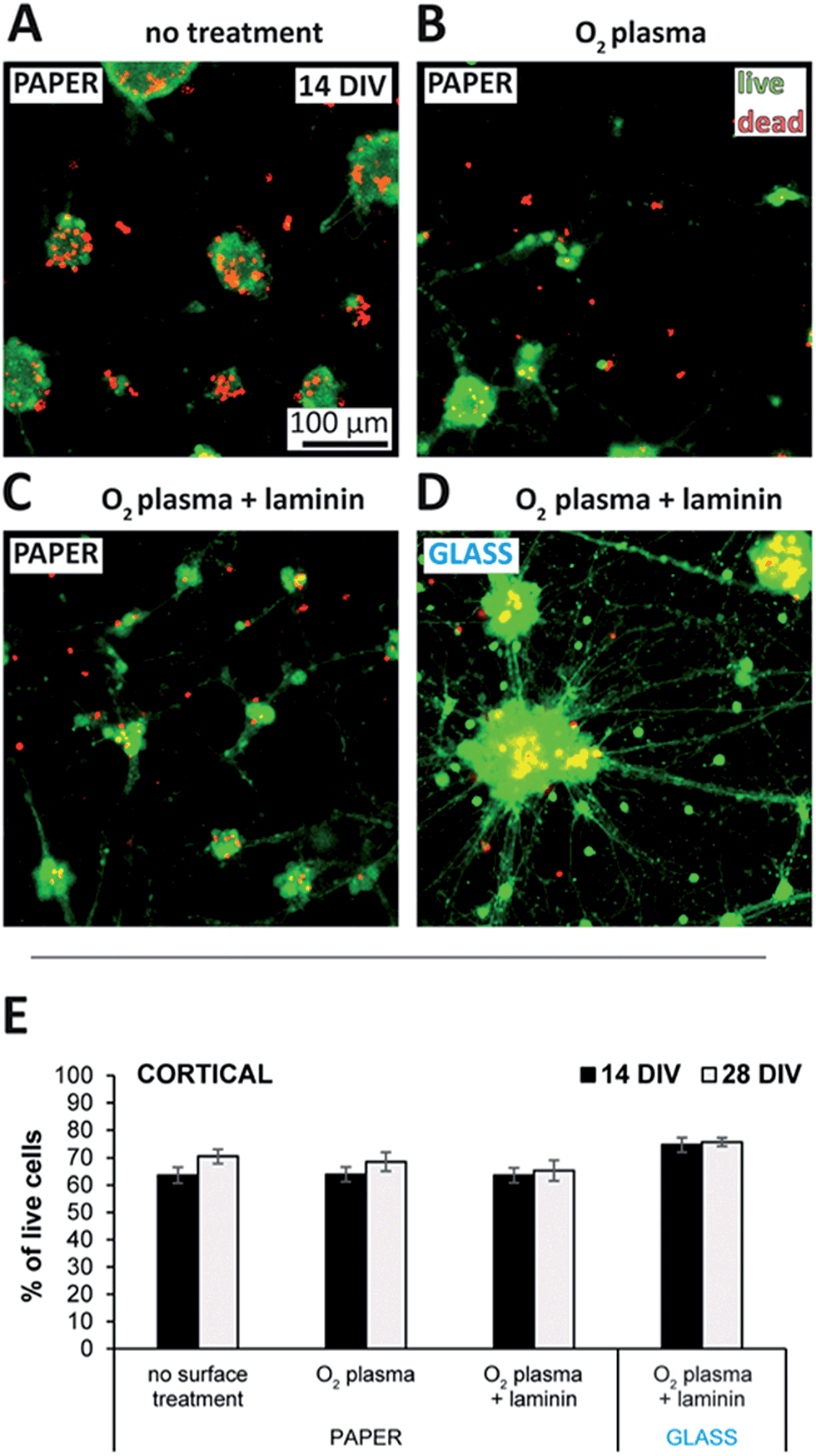 | ||
| Fig. 2 Viability of cortical paper cultures with different surface treated substrates. (A–D) Merged fluorescence signals of the live/dead viability assay at 14 days in vitro (DIV). Green and red indicate live and dead cells, respectively. The representative images correspond to cortical neurons seeded onto (A) non-treated, (B) O2 plasma treated, (C) plasma treated and laminin coated Grade 602 h cellulose membrane, and (D) plasma treated and laminin coated glass coverslip. (E) Comparison of the viability with different surface treated substrates at 14 and 28 DIV. Data is presented as mean ± the standard error. | ||
Such sphere formation could be reduced by O2-plasma treatment as indicated by the appearance of smaller cell clusters similar to those visible in Fig. 2B, implying a higher cell adhesion. The expected reduction of carbon contaminations, increase in surface roughness,73 or changes in the chemical structure and adhesion properties of cellulose74 induced by the plasma could all be responsible for the effect. The scanning electron microscopy (SEM) pictures of Fig. S3† do not show any significant difference in the surface topography of non-treated membranes to those treated the same way as in the cell-related experiments (2 min at 18 W). However, even though only a prolonged plasma treatment at high power (10 min at 100 W) induced changes clearly visible in the surface structure, minor differences in the nanotopography caused by the low-power treatment might still have influenced the adhesion of neurons.75
Promoting cell adhesion by the adsorption of the extracellular matrix protein laminin decreased sphere formation further. As represented in Fig. 2C, the enhanced cell spreading and neurite outgrowth resulted in small clusters mostly, with network topologies more homogeneous than those of the reference cultures grown on glass using the same surface treatment (Fig. 2D). Therefore, network topology of the presented paper-based 3D cultures could be controlled via cell adhesion using different surface treatments. Network homogeneity could be improved even more by increasing cell adhesion via crafting antibodies,76 or using proteins that attach to the cellulose fibers.77,78
The level of clumping and sphere formation as a function of the different surface treated substrates has been quantified by a method introduced via Fig. S4A of the ESI.† The resulting clump size distributions plotted in Fig. S4B† confirm the visual observations deducted from Fig. 2A–D, identifying the combination of O2-plasma treatment and laminin coating with the smallest clump sizes, where close to 70% of all the neurons were in clusters with diameters less than 40 µm. The reference cultures grown on glass using the same surface treatment showed a significantly more homogeneous clump size distribution and inhomogeneous network topology, with more than 40% of all the neurons in clusters with diameters of 80–200 µm.
In order to estimate the neurite lengths and characterize the local network topologies descriptive of the cultures, nearest neighbor inter-clump distance distributions have also been determined and plotted in Fig. S4C.† Nearest neighbor distance is a good measure, since every cluster was found to be connected to at least its neighboring clusters. The results of the analysis show that short nearest neighbor cluster distances are dominant in the case of all the substrates studied, with more than 80% of the clusters having at least one neighboring cluster within 40 µm.
Fig. 2E compares cell viability using the different surface treatments and that of the reference cultures on glass at 14 and 28 DIV. While there was no significant difference in these cases, neurons with poly-D-lysine (PDL) treated paper substrates showed significantly reduced viability values throughout the testing period as plotted in Fig. S2A of the ESI.† Poly-L-Lysine (PLL) and its artificial enantiomer PDL are commonly used to coat culture substrates for enhanced neuron adhesion and survival, because the positive charge of the lysine molecule is thought to induce the adsorption of negatively charged, neuron-adhesive glycoproteins from the media.71 However, since unbound PLL is known to be toxic for the cells,79 the observed reduced viability with PDL could be explained by its slow detachment from the cellulose surface as a result of the surface equilibration processes within the medium.80
All the experimental results presented so far were achieved using cortical neurons. This type has the highest availability in laboratories working with primary cell cultures, mostly because of their higher number. Repeating the experiments with hippocampal neurons showed similar results but with higher, close to 90%, viability in all cases except the one with PDL coating. The toxic effect of PDL-coated paper substrates was found to be independent of the neuron type used. Fig. S2B of the ESI† summarizes the results comparing the two cell types at 14 DIV. Here, the viability values of the glass reference cultures were also representative for the respective cell viabilities prior to seeding. The overall higher viability in the case of hippocampal neurons might have originated from their earlier dissection and plating times reducing the stress on the cells.
3D network structure within the paper membranes
We investigated the effect of the fiber structure on neurite guidance and 3D network formation by immunolabeling with specific markers for axons, dendrites, glial cells, and nuclei. As demonstrated with the representative images of Fig. S5A of the ESI† showing a cortical culture at 44 DIV, the neurons within the paper were highly interconnected with each other. While many of the neurites followed the fiber structure, they also bridged the gaps in between, similar to cultures grown on microfabricated 3D structures.81,82 Although the neurons were plated and grown in serum free media, proliferation of glial cells was not fully suppressed according to the expression of the glial marker GFAP (yellow) visible throughout the right pane of Fig. S5A.†The 3D structure of the paper-based neuronal networks was mapped by combining images from different substrate depths. The main disadvantage of using cellulose filter membranes over hydrogels is their lack of transparency in normal medium, caused by scattering at the fiber-medium interfaces. A possible solution could be to chemically modify the cellulose fibers,83 but such an extra process would complicate both the preparation and the evaluation of the cell experiments. Instead, we followed the approach of using refractive index matching oils first described by Saarela et al.84 and adapted for paper-based devices by Ellerbee et al.,85 according to the description in the Materials and Methods section. Although the method increased the transparency considerably, reaching more depths through the filter membranes while imaging, the full thickness of the substrate as a whole still remained partly opaque.
In order to study the degree of cell invasion as a function of substrate porosity, three different filter paper grades have been compared with fiber densities designed to filter particles larger than <2, 8, and 11 µm. All substrates were plasma-treated, laminin-coated, and seeded at an equal cell density. As represented in Fig. 3A, cultures with substrates of the smallest porosity (602 h) formed networks of small-sized neural nodes, with the interconnecting neurites spanning on top and within the paper matrix, but just minimally interacting with the fibers. According to the dimensions typical for the neurons studied here, 8–10 µm in length and a few micrometers in height once adhered, the gap distribution between the fibers allowed cell bodies to penetrate into the paper substrate to a certain extent, but prevented them from passing through to the other side.
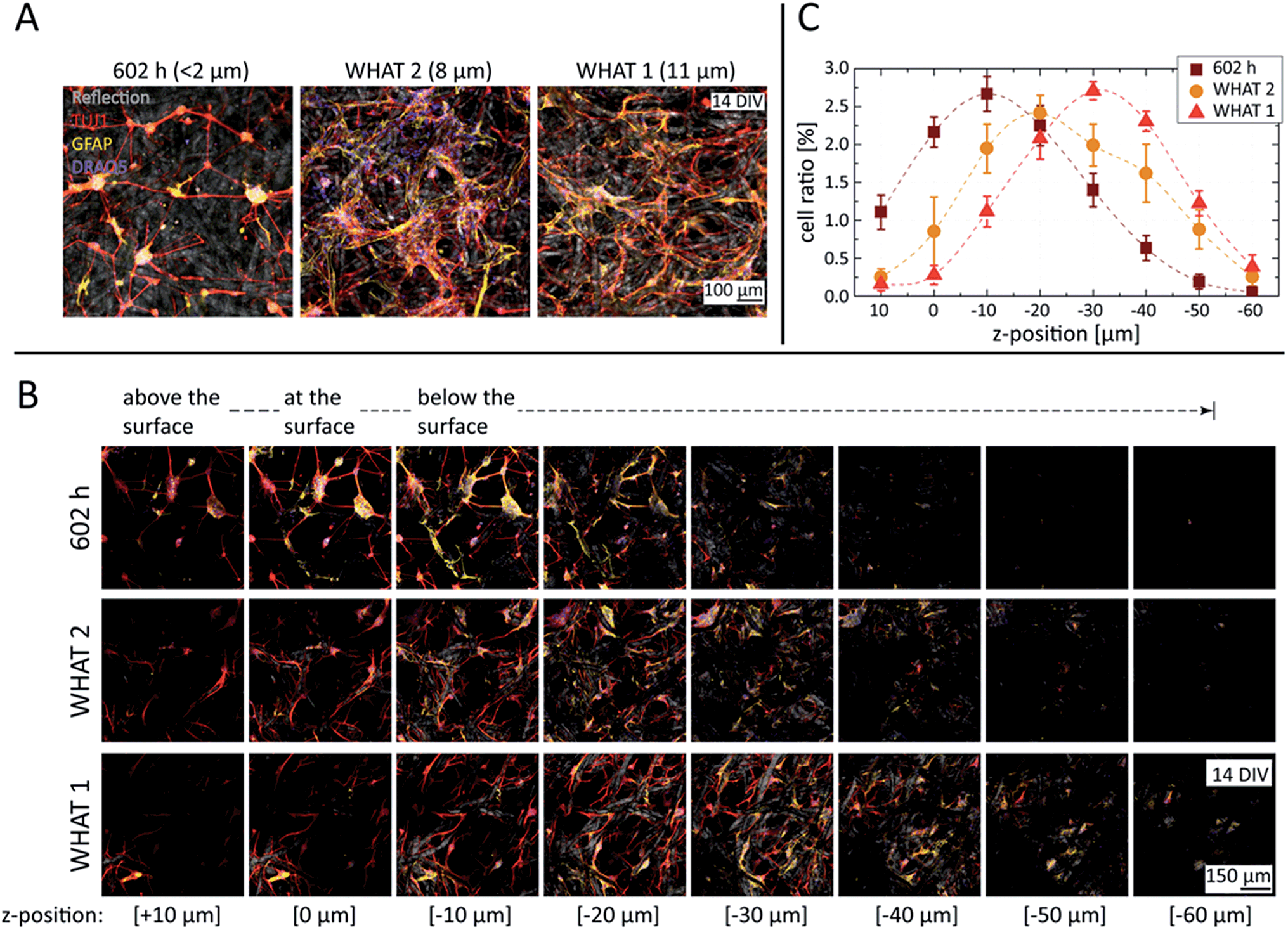 | ||
| Fig. 3 3D network structure of paper-based neuronal cultures as a function of paper porosity. (A and B) Representative fluorescent images of cortical cultures with different substrates at 14 DIV, merging the channels of neurites (TUJ1, red), glial cells (GFAP, yellow), and nuclei (DRAQ5, blue), complemented with the reflection channel in (A) visualizing the cellulose structure (grey). (A) Images taken with open pinhole for an extended z range. (B) z stacks taken with closed pinhole. (C) Cell invasion as a function of paper porosity. Cell nuclei counts were normalized by the total counts of the image stacks, and the plotted cell ratio values correspond to slices with a z-thickness of 1 µm at the given z-positions. Data is presented as mean ± the standard error, while dashed curves serve as visual guides only. | ||
Substrates with higher porosities (WHAT 2 & 1) resulted in more widespread and homogeneous cell invasion, reaching also the deeper regions of the paper matrix, as visualized by the z stacks of Fig. 3B. With 8 µm porosity especially (WHAT 2), large 3D glial cell patches were detected, where neurites could integrate into the glial matrix without the additional support of the cellulose fibers. Further increasing the porosity to 11 µm (WHAT 1), the larger inter-fiber distances caused the neurites to follow the fibers more, using them as guidance for their outgrowth. As neurites have significantly smaller diameters than cells, in the range of a few micrometers, they could extend freely throughout the substrate, reaching over 60 µm trackable penetration depth in the case of the largest pore size studied.
Nucleus cell counts and cell invasion as a function of the paper porosity have been assessed in the 3D image stacks using the cell counting algorithm and method described in the Materials and Methods section. The results, presented in Fig. 3C, reveal a monotonous trend for cell nuclei distribution shifting towards deeper z-positions with increasing porosities. In addition, the neurite, glial cell, and nuclei distributions have also been estimated directly from the total fluorescent intensity values calculated for each channel and all the slices of the 3D stacks. The results, plotted in Fig. S6 of the ESI,† also show monotonous trends for all the channels similar to that of the cell counting analysis, but with less pronounced differences. However, the cell counting algorithm should be more robust against background fluctuations, providing a better measure and description of the 3D network structure of paper-based neuronal cultures.
Fig. S5B† depicts two 3D renderings of sample image stacks, reconstructing cortical networks with Grade 602 h (left) and Grade 2 (right) filter membranes at 14 DIV. In order to test the possibility of creating compartmentalized neuronal cultures, cortical neurons were also plated on both sides of paper substrates with the smallest pore size (602 h). The cultures were immunostained and imaged at 21 DIV, flipping them over at the half of the image stack. As demonstrated by the corresponding z stack of Fig. S5C of the ESI† (second row), cell bodies and neurites invaded the paper substrate from both sides when seeded accordingly, but no neurite interactions have been established between the subcultures of the opposite sides.
For all the following experiments, the filter paper type with the smallest porosity (602 h) was chosen as substrate because of the more discernible 3D networks closer to the surface, resulting also in higher signal-to-noise ratios in the multielectrode array recordings.
Patterned 3D neural networks
In order to keep our paper-based platform simple, widely available, and high-throughput, the techniques of wax printing and laser cutting were selected to investigate their compatibility for controlling the topology of the prepared 3D neural cultures.Although the technique of wax printing has been successfully combined with various cell types, in the case of primary neurons wax-modified cultures showed a reduced viability compared to that of the reference, unmodified ones. Additionally, the total number of cells within the membrane was found to decrease over time. All these observations implied that the toxicity of the printed wax, especially its ink component, affected primary neurons via a slow diffusion process into the medium. To investigate this, cultures with wax-printed patterns were created where the duration of the cleaning step in Milli-Q water introduced in the Materials and Methods section was prolonged from 10 s to 2 weeks. Even though this slightly improved the viability, the long-term toxic effect still remained, and the cultures were empty/dead within typically 1 week. While the problem could be further investigated by testing different ink types, our focus shifted to another technique, which improved both cell survival and resolution.
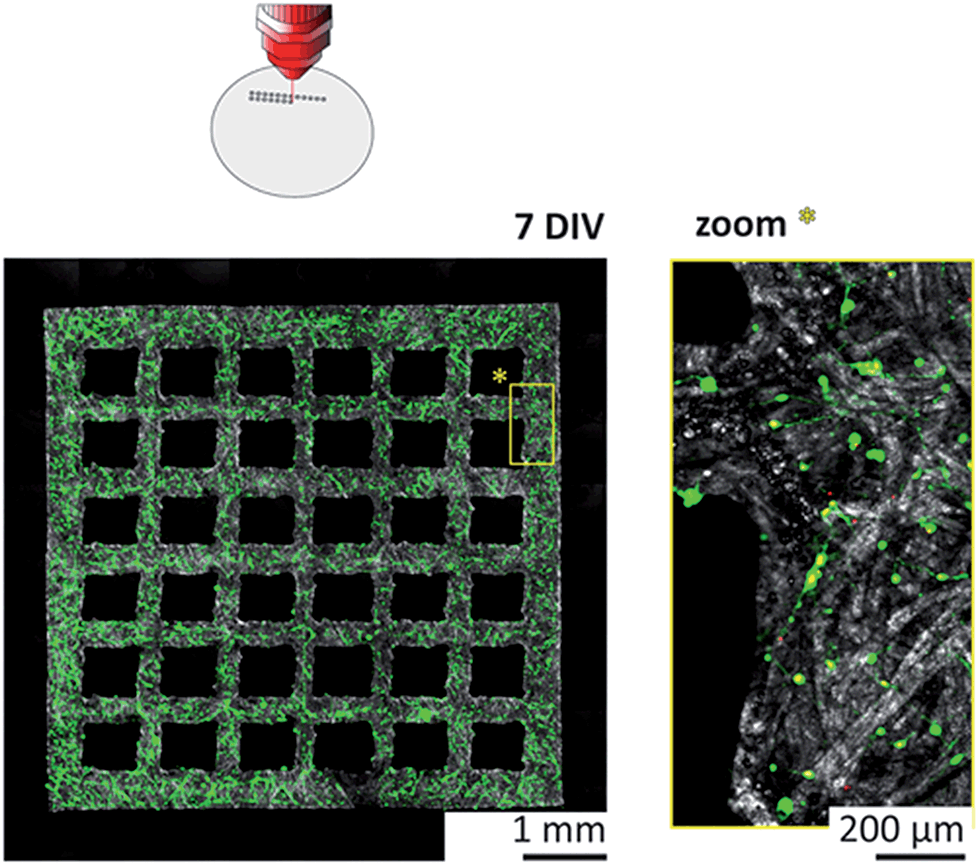 | ||
| Fig. 4 High-resolution laser cutting for creating patterned 3D neural networks tested by live/dead viability assays. The merged fluorescent signals indicate the live and dead cells in green and red, respectively, while the shades of grey represent the reflection channel. The paper substrate was laser cut before cell plating to physically confine network formation. The images show a cortical culture at 7 DIV using Grade 602 h cellulose membrane. | ||
The more than fivefold reduction in both minimal line width and average boundary roughness compared to wax printing have been accompanied with improved survival rates as well. Cortical cultures grown in laser-cut substrates showed viability values with no significant difference compared to that of control cultures with non-structured substrates. The neurons invaded the paper substrate according to the initial plating density, and formed complex 3D networks throughout the fiber matrix over time, as demonstrated in Fig. S7B of the ESI† at 14 DIV.
Simultaneous recording of intra- and extracellular neural activity
While several recording techniques have been established to measure signals related to neuronal activity, there is no such technique that could access direct, intracellular information with both maximal spatial and temporal resolution at the same time, and also keep the complexity at minimum.87 Keeping the focus on simplicity and high-throughput, and also maintaining compatibility, the two complementary techniques of calcium imaging and electrical recording were selected for studying the neural activity of the presented paper-based cultures. Neuron-specific calcium indicators provide a network-wide overview of the intracellular activity with high spatial resolution limited mainly by the optical setup, but their low temporal resolution and signal-to-noise ratio restrict the detailed analysis of signal propagation.57 Multielectrode arrays on the other hand measure signals with high temporal resolution, but their spatial resolution is limited by the interelectrode distances and they provide access to the distance-weighted extracellular activity only.88Using the optimized protocol described in the Materials and Methods section, the paper-based cultures have been successfully transduced with the genetically encoded calcium indicator GCaMP6, making the neural network and its activity visible 5 days after transduction. Fig. 5A shows a representative snapshot of a transduced cortical paper-based culture at 22 DIV, with average time traces of the selected regions of interest plotted in Fig. 5B. Since the signal intensity was found to be high enough for a pixel-based analysis, the spatial resolution of the presented example using a 20× objective was below 2 µm, comparable to the scale of neurites.
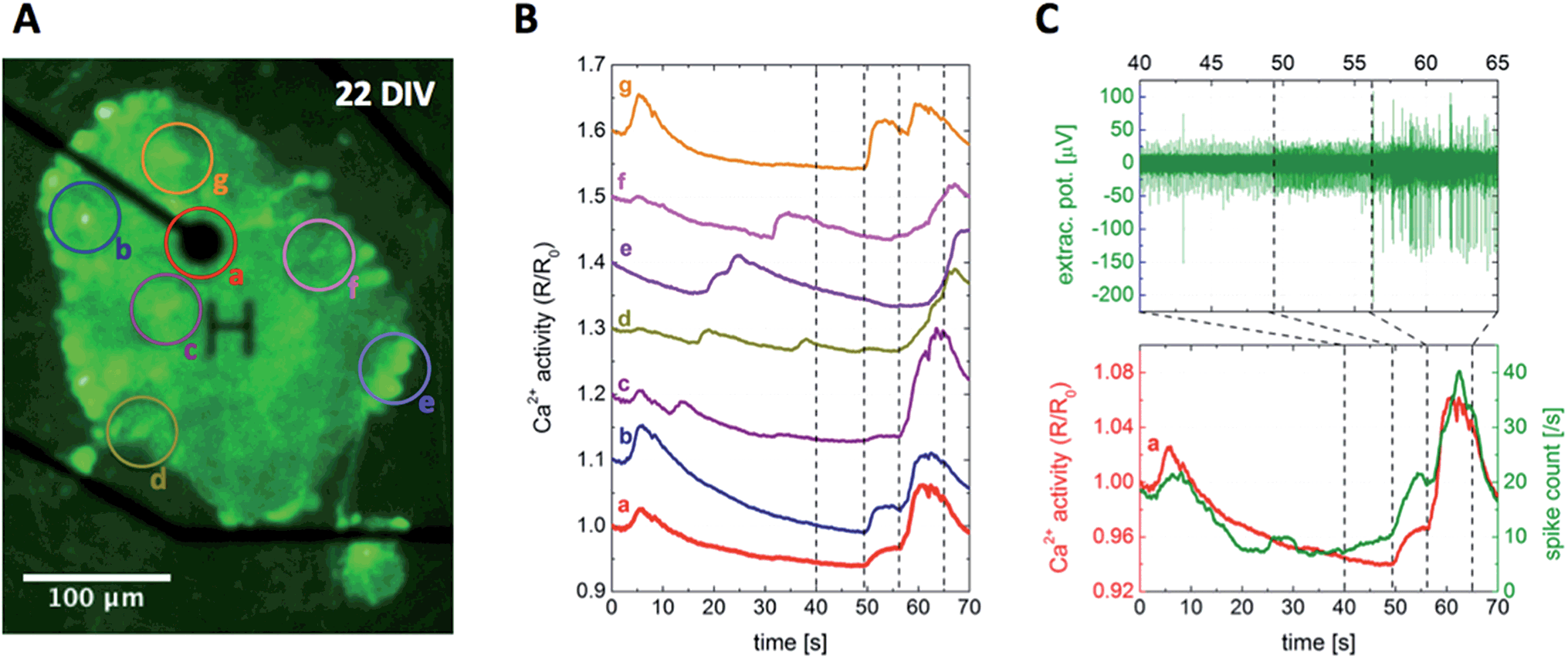 | ||
| Fig. 5 Simultaneous recording of intra- and extracellular neuronal activity. (A) Snapshot of the intracellular Ca2+ level visualized by the fluorescent calcium indicator GCaMP6, in a cortical paper-based culture using Grade 602 h cellulose membrane positioned on top of a multielectrode array chip at 22 DIV. The recording electrode with 30 µm in diameter, denoted by the character H, and insulated tracks appear in black, while the circles labeled from (a) to (g) indicate the regions of interest analyzed further. (B) Average fluorescent intensity time traces of the regions marked in (A). The corresponding curves were proportionally shifted along the vertical axis to increase visibility. (C) The combined analysis of intra- and extracellular signals of region (a), showing a clear correlation between the intracellular Ca2+ level and extracellular spike count. The simultaneous recording can be followed in ESI Movie M1.† | ||
In typical protocols used for studying the network dynamics and information processing of neural cultures in vitro, recording of neural activity starts only 1–2 weeks after plating to assure that the neurons have already developed stable synaptic connections and formed a mature network. During this period, the chips of MEA-based cultures are occupied unnecessarily, while also reducing their lifetime due to the extra incubation. In contrast, the structure of the filter membranes provided enough mechanical stability to transfer the presented paper-based cultures between the multiwell plates used for incubation, and MEA chips used for recording, in an on-demand manner and without affecting the paper substrate or changing the neural activity. As such, the developed technique provides a simple platform for both optical and electrical high-throughput screening of patterned 3D neural cultures.
While electrical recordings of neurons cultured directly on MEA surfaces have already been validated by optical measurements89 and also combined with two-photon calcium imaging,90 the electrical recording of the paper-based cultures required further developments. Immersed in media, the hydrated cellulose membranes float at the MEA surface, but not close enough to detect neural activity. Thus, a pushdown device based on a harp slice grid was constructed to bring the paper-based cultures close to the chip surface and precisely control their vertical position relative to the electrodes. A simple weight-based method without control proved to be inadequate, because the precise force needed to keep the cultures at a certain position depended on their exact weight, and any deviation from this value resulted in either an increased neuron – electrode distance spoiling the readout, or an extra mechanical stress squeezing the neurons. In addition to the pushdown device, a custom-made moving stage has been developed to position the MEA headstage on the microscope within the incubator box, together with a cooling system to dissipate the extra heat produced by the MEA amplifier and avoid overheating the cultures. See the Materials and Methods section, as well as S1 of the ESI† for further details on the experimental setup.
Network dynamics of the 3D neural networks
Once the paper-based mature cultures were pushed down to the MEA surface such that the neuron – electrode distance fell below 10 µm, extracellular signals of the spontaneous activity appeared on the electrodes. The number of active electrodes depended on the relation of the neural network and electrode densities, in which both the initial plating concentration and the applied surface treatment (see Fig. 2A–D and the corresponding discussion) played an important role. The peak-to-peak amplitude of the signals measured in the range of 200–250 µV was comparable to that of conventional 2D cultures grown directly on the MEA surface.Fig. 5C presents a representative fragment of the extracellular signal picked up by the electrode visible in Fig. 5A, together with its calculated spike count and the calcium activity trace of region (a) selected around the electrode, showing a strong correlation between the recorded intracellular and extracellular signals. However, the calcium activity time traces of different regions of interest plotted in Fig. 5B show a high variation. Even though all the traces are dominated by network-wide bursts towards the beginning and end of the curves with a decaying activity in between, the detailed differences lack any clear trend or correlation between the traces and the positions of the selected regions relative to the electrode. The developed platform provides a unique tool to study the underlying signal propagation for further insights, as well as to reveal the relation between intracellular calcium and extracellular spiking activity.
Even though network-wide bursts are characteristic to mature cultures in vitro and have been studied extensively, the fundamental mechanisms are not fully understood due to their complex parameter dependence.51,91,92 On the one hand, neural network activity is highly influenced by the network density and culture age,93 and bursting patterns were shown to be affected by the ratio of excitatory and inhibitory connections.94,95 On the other hand, astrocytes can influence the network-wide activity both directly and indirectly. Astrocytes exhibit calcium waves spreading over the whole population,96 while they also interact bidirectionally with the neurons, inducing action potentials and synchronizing populations both in vitro and in vivo.97,98 In Fig. 5C, one can follow the buildup of a network-wide burst through the extracellular spiking activity showing two distinctive steps. First only the spiking frequency increases, then with a second boost in spike count spikes with two-, threefold higher amplitudes appear as well. At the same time, Ca2+ traces plotted in Fig. 5B imply subtle differences between the activity patterns of the different regions, see ESI Movie M1† for a dynamic representation. A comprehensive, pixel-based correlation analysis could shed light on more details of the activity propagation within the prepared 3D cultures, but such a systematic study is outside the scope of the present work.
Since a neuron-specific synapsin-based calcium indicator has been used for the experiments and the astrocytes are unable to produce action potentials directly, both the measured calcium and electrical signals should originate exclusively from neuronal cells excluding astrocytes, resulting in synchronized responses similarly to the case presented in Fig. 5C.99 However, some of the recordings were temporally lacking such synchronicity, exemplified in Fig. S9† and supporting Movie M2 of the ESI.† Peaks of the calcium activity corresponded to network-wide bursts also in this case, and spike counts and interspike intervals of the local extracellular activity oscillated with similar periodicity, but there was a pronounced phase shift between the two properties changing over time. This decoupling between the intracellular and extracellular signals is rather strange, however, since the observed periodicity is on the timescale characteristic to the calcium waves induced by the astrocytes, they might play a key role in this phenomenon as well.
Conclusions and outlook
The present work introduced a paper-based system to create 3D cultures of primary neurons with user-defined topology to study their network dynamics. Hippocampal and cortical primary neurons grew and formed healthy cultures on both non-treated and surface treated cellulose substrates with high viability for over 4 weeks. Immunolabeling showed that neurons invaded the substrate and formed complex 3D networks throughout the fiber matrix. The structure of the filter membranes provided enough mechanical stability to transfer the cultures between setups used for incubation and measurement, in an on-demand manner and without changing the neural activity or affecting the substrate itself.The developed platform provides a simple and robust technique for investigating the intimate relationship between topology and function by studying the activity patterns of 3D neural cultures with arbitrary topology in a high-throughput manner, combining network-wide intracellular and local extracellular signals. Network topology can be controlled by patterning the paper substrates to physically confine network development, as well as via cell adhesion using different surface treatments to control sphere formation, and as such, the homogeneity of the network.
Since filter membranes are available with various pore sizes, the penetration depth of the cell bodies can be altered by the specifications of the paper used for the experiments. A fine tuning of such properties would allow to precisely control the interaction between subcultures plated from the opposite sides of the substrate at the levels of both the interconnections of the neurites and the mixing of the cell bodies, providing a unique tool for studying co-cultures and also envisioning the feasibility of paper-based 3D neural scaffolds for applications.
The presented technique is by nature compatible with local chemical and electrical stimulation protocols, both during and after maturation. Therefore, parameters influencing network synchronization, signal propagation, and the developed activity patterns in different culturing conditions, including stimulation protocols, could be systematically studied through a high-throughput screening to reveal basic information processing phenomena in 3D neural cultures with different network topologies. In addition, the role of astrocytes and related phenomena could also be further investigated using the aforementioned co-culturing capabilities via the double-sided plating of cells.
The simplicity and versatility of the platform provides groups with little or no related experience the possibility to expand their research towards 3D neural cultures with arbitrary topology. All components of the presented setup used for the experiments could be easily substituted with similar components according to availability, including but not limited to the MEA setup and laser cutter. In addition, the on-demand availability of the prepared paper-based cultures for measurements makes the technique cheap and much higher throughput than current in vitro methods.
While the present work focused exclusively on cultures of primary neurons, applications using other cell types could also benefit from components of the developed technology. As an example, stem cells are also highly viable in cellulose-related substrates,100,101 and the presented method with surface treatments promoting sphere formation could be an alternative approach to current culturing systems inducing embryoid body formation, with an improved cell support from the substrate and easier handling of the cultures.102,103
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Swiss National Science Foundation and the 3DNeuroN project in the European Union's Seventh Framework Program, Future and Emerging Technologies, Grant Agreement No. 296590. The authors thank Stephen Wheeler (ETHZ, LBB) for his valuable technical support, Dr Tobias Schwarz and ScopeM (ETHZ) for the assistance and facility support with image deconvolution. The authors also acknowledge the Genetically-Encoded Neuronal Indicator and Effector (GENIE) Project and the Janelia Research Campus of the Howard Hughes Medical Institute for distributing the GCaMP6 vectors. Special thanks go to Prof. Dr Jean-Marc Fritschy and especially Giovanna Bosshard from the Institute of Pharmacology and Toxicology – Morphological and Behavioral Neuroscience at the University of Zurich, for kindly providing us with dissected rat hippocampi and cortical tissues, as well as sharing their knowledge about primary neuron cultures.References
- S. Zhang, Nat. Biotechnol., 2004, 22, 151–152 CrossRef CAS PubMed.
- M. L. Shuler and J. J. Hickman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13682–13683 CrossRef CAS PubMed.
- O. Sporns, Nat. Neurosci., 2014, 17, 652–660 CrossRef CAS PubMed.
- M. A. Lancaster, M. Renner, C.-A. Martin, D. Wenzel, L. S. Bicknell, M. E. Hurles, T. Homfray, J. M. Penninger, A. P. Jackson and J. A. Knoblich, Nature, 2013, 501, 373–379 CrossRef CAS PubMed.
- H. Peretz, A. E. Talpalar, R. Vago and D. Baranes, Tissue Eng., 2007, 13, 461–472 CrossRef CAS PubMed.
- M. Joseph Phillips and D. C. Otteson, Invest. Ophthalmol. Visual Sci., 2011, 52, 1439–1449 Search PubMed.
- Y. Lai, K. Cheng and W. Kisaalita, PLoS One, 2012, 7, e45074 CAS.
- P. D. Dalton and J. Mey, Front. Biosci., Landmark Ed., 2009, 14, 769–795 CrossRef CAS.
- K. Nagamine, T. Hirata, K. Okamoto, Y. Abe, H. Kaji and M. Nishizawa, ACS Biomater. Sci. Eng., 2015, 1, 329–334 CrossRef CAS.
- B. R. Simona, L. Hirt, L. Demkó, T. Zambelli, J. Vörös, M. Ehrbar and V. Milleret, Biomater. Sci., 2015, 3, 586–591 RSC.
- J. M. Jang, S.-H.-T. Tran, S. C. Na and N. L. Jeon, ACS Appl. Mater. Interfaces, 2015, 7, 2183–2188 CAS.
- A. Odawara, M. Gotoh and I. Suzuki, RSC Adv., 2013, 3, 23620 RSC.
- H. Onoe, T. Okitsu, A. Itou, M. Kato-Negishi, R. Gojo, D. Kiriya, K. Sato, S. Miura, S. Iwanaga, K. Kuribayashi-Shigetomi, Y. T. Matsunaga, Y. Shimoyama and S. Takeuchi, Nat. Mater., 2013, 12, 584–590 CrossRef CAS PubMed.
- G. Eng, B. W. Lee, H. Parsa, C. D. Chin, J. Schneider, G. Linkov, S. K. Sia and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4551–4556 CrossRef CAS PubMed.
- A. Béduer, T. Braschler, O. Peric, G. E. Fantner, S. Mosser, P. C. Fraering, S. Benchérif, D. J. Mooney and P. Renaud, Adv. Healthcare Mater., 2015, 4, 301–312 CrossRef PubMed.
- J. P. Frampton, M. R. Hynd, J. C. Williams, M. L. Shuler and W. Shain, J. Neural Eng., 2007, 4, 399–409 CrossRef CAS PubMed.
- G. Palazzolo, N. Broguiere, O. Cenciarelli, H. Dermutz and M. Zenobi-Wong, Tissue Eng., Part A, 2015, 21, 2177–2185 CrossRef CAS PubMed.
- Y. T. Matsunaga, Y. Morimoto and S. Takeuchi, Adv. Mater., 2011, 23, H90–H94 CrossRef CAS PubMed.
- M. Kato-Negishi, Y. Morimoto, H. Onoe and S. Takeuchi, Adv. Healthcare Mater., 2013, 2, 1564–1570 CrossRef CAS PubMed.
- J. G. Hardy, Z. Z. Khaing, S. Xin, L. W. Tien, C. E. Ghezzi, D. J. Mouser, R. C. Sukhavasi, R. C. Preda, E. S. Gil, D. L. Kaplan and C. E. Schmidt, J. Biomater. Sci., Polym. Ed., 2015, 26, 1327–1342 CrossRef CAS PubMed.
- M. D. Tang-Schomer, J. D. White, L. W. Tien, L. I. Schmitt, T. M. Valentin, D. J. Graziano, A. M. Hopkins, F. G. Omenetto, P. G. Haydon and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2014, 1–6 Search PubMed.
- S. Pautot, C. Wyart and E. Y. Isacoff, Nat. Methods, 2008, 5, 735–740 CrossRef CAS PubMed.
- M. Frega, M. Tedesco, P. Massobrio, M. Pesce and S. Martinoia, Sci. Rep., 2014, 4, 5489 CrossRef CAS PubMed.
- M. Innala, I. Riebe, V. Kuzmenko, J. Sundberg, P. Gatenholm, E. Hanse and S. Johannesson, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 302–308 CrossRef CAS PubMed.
- D. Muller, J. P. Silva, C. R. Rambo, G. M. O. Barra, F. Dourado and F. M. Gama, J. Biomater. Sci., Polym. Ed., 2013, 24, 1368–1377 CrossRef CAS PubMed.
- N. Petersen and P. Gatenholm, Appl. Microbiol. Biotechnol., 2011, 91, 1277–1286 CrossRef CAS PubMed.
- N. Lin and A. Dufresne, Eur. Polym. J., 2014, 59, 302–325 CrossRef CAS.
- M. Jorfi and E. J. Foster, J. Appl. Polym. Sci., 2015, 132, 41719 CrossRef.
- M. Märtson, J. Viljanto, T. Hurme, P. Saukko and O. Paper, Eur. Surg. Res., 1998, 30, 426–432 CrossRef.
- E. Ekholm, M. Tommila, A. P. Forsback, M. Märtson, J. Holmbom, V. Ääritalo, C. Finnberg, A. Kuusilehto, J. Salonen, A. Yli-Urpo and R. Penttinen, Acta Biomater., 2005, 1, 535–544 CrossRef PubMed.
- H.-J. Park, S. J. Yu, K. Yang, Y. Jin, A.-N. Cho, J. Kim, B. Lee, H. S. Yang, S. G. Im and S.-W. Cho, Biomaterials, 2014, 35, 9811–9823 CrossRef CAS PubMed.
- J. Viljanto, in Wound Healing and Skin Physiology SE - 48, ed. P. Altmeyer, K. Hoffmann, S. el Gammal and J. Hutchinson, Springer, Berlin Heidelberg, 1995, pp. 513–522 Search PubMed.
- W. Czaja, A. Krystynowicz, S. Bielecki and R. M. Brown Jr, Biomaterials, 2006, 27, 145–151 CrossRef CAS PubMed.
- H. Fink, L. Ahrenstedt, A. Bodin, H. Brumer, P. Gatenholm, A. Krettek and B. Risberg, J. Tissue Eng. Regener. Med., 2011, 5, 454–463 CrossRef CAS PubMed.
- R. Gurny, S. P. Simmons, G. S. Banker, R. Meeker and R. D. Myers, Pharm. Acta Helv., 1979, 54, 349–352 CAS.
- Y.-H. Chen, Z.-K. Kuo and C.-M. Cheng, Trends Biotechnol., 2015, 33, 4–9 CrossRef CAS PubMed.
- H. Hoenich, BioResources, 2006, 1, 270–280 Search PubMed.
- H. Yagoda, Ind. Eng. Chem., Anal. Ed., 1937, 9, 79–82 CrossRef CAS.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210–2251 RSC.
- M. S. Akram, R. Daly, F. da Cruz Vasconcellos, A. K. Yetisen, I. Hutchings and E. A. H. Hall, in Lab-on-a-Chip Devices and Micro-Total Analysis Systems, Springer International Publishing, Cham, 2015, pp. 161–195 Search PubMed.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Y. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–18462 CrossRef CAS PubMed.
- R. Derda, S. K. Y. Tang, A. Laromaine, B. Mosadegh, E. Hong, M. Mwangi, A. Mammoto, D. E. Ingber and G. M. Whitesides, PLoS One, 2011, 6, e18940 CAS.
- B. Mosadegh, M. R. Lockett, K. T. Minn, K. A. Simon, K. Gilbert, S. Hillier, D. Newsome, H. Li, A. B. Hall, D. M. Boucher, B. K. Eustace and G. M. Whitesides, Biomaterials, 2015, 52, 262–271 CrossRef CAS PubMed.
- M. C. Sapp, H. J. Fares, A. C. Estrada and K. J. Grande-Allen, Acta Biomater., 2015, 13, 199–206 CrossRef CAS PubMed.
- F. Deiss, W. L. Matochko, N. Govindasamy, E. Y. Lin and R. Derda, Angew. Chem., Int. Ed., 2014, 53, 6374–6377 CrossRef CAS PubMed.
- C. Vulin, F. Evenou, J. M. Di Meglio and P. Hersen, Methods Cell Biol., 2014, 121, 155–169 Search PubMed.
- K. F. Lei and C. H. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 22423–22429 CAS.
- F. Deiss, A. Mazzeo, E. Hong, D. E. Ingber, R. Derda and G. M. Whitesides, Anal. Chem., 2013, 85, 8085–8094 CrossRef CAS PubMed.
- J. Cang and D. A. Feldheim, Annu. Rev. Neurosci., 2013, 36, 51–77 CrossRef CAS PubMed.
- A. F. M. Johnstone, G. W. Gross, D. G. Weiss, O. H.-U. Schroeder, A. Gramowski and T. J. Shafer, Neurotoxicology, 2010, 31, 331–350 CrossRef CAS PubMed.
- D. A. Wagenaar, J. Pine and S. M. Potter, BMC Neurosci., 2006, 7, 11 CrossRef PubMed.
- M. J. Aebersold, H. Dermutz, C. Forró, S. Weydert, G. Thompson-Steckel, J. Vörös and L. Demkó, TrAC, Trends Anal. Chem., 2016, 78, 60–69 CrossRef CAS.
- H. Bartsch, M. Himmerlich, M. Fischer, L. Demkó, J. Hyttinen and A. Schober, J. Ceram. Sci. Technol., 2015, 6, 315–324 Search PubMed.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS PubMed.
- G. J. Brewer, J. R. Torricelli, E. K. Evege and P. J. Price, J. Neurosci. Res., 1993, 35, 567–576 CrossRef CAS PubMed.
- C. Sommer, C. Straehle, U. Kothe and F. a. Hamprecht, Proc. - Int. Symp. Biomed. Imaging, 2011, pp. 230–233 Search PubMed.
- T.-W. Chen, T. J. Wardill, Y. Sun, S. R. Pulver, S. L. Renninger, A. Baohan, E. R. Schreiter, R. a. Kerr, M. B. Orger, V. Jayaraman, L. L. Looger, K. Svoboda and D. S. Kim, Nature, 2013, 499, 295–300 CrossRef CAS PubMed.
- T. Miyamoto, S. Takahashi, H. Ito, H. Inagaki and Y. Noishiki, J. Biomed. Mater. Res., 1989, 23, 125–133 CrossRef CAS PubMed.
- C. Xu, X. Ma, S. Chen, M. Tao, L. Yuan and Y. Jing, Int. J. Mol. Sci., 2014, 15, 10855–10867 CrossRef CAS PubMed.
- F. G. Torres, S. Commeaux and O. P. Troncoso, J. Funct. Biomater., 2012, 3, 864–878 CrossRef CAS PubMed.
- W. Czaja, D. Romanovicz and R. M. Brown, Cellulose, 2004, 11, 403–411 CrossRef CAS.
- E. J. Vandamme, S. De Baets, A. Vanbaelen, K. Joris and P. De Wulf, Polym. Degrad. Stab., 1998, 59, 93–99 CrossRef CAS.
- D. Kralisch, N. Hessler, D. Klemm, R. Erdmann and W. Schmidt, Biotechnol. Bioeng., 2010, 105, 740–747 CAS.
- C.-H. Huang, K. F. Lei and N.-M. Tsang, Lab Chip, 2016, 16, 2911–2920 RSC.
- R. Rahimi, S. S. Htwe, M. Ochoa, A. Donaldson, M. Zieger, R. Sood, A. Tamayol, A. Khademhosseini, A. M. Ghaemmaghami and B. Ziaie, Lab Chip, 2016, 16, 4319–4325 RSC.
- H. Lau and A. C. Hurt, Front. Microbiol., 2016, 7, 1–7 Search PubMed.
- D. B. Purchas and K. Sutherland, in Handbook of Filter Media, Elsevier, 2002, pp. 117–151 Search PubMed.
- A. C. Glavan, R. V. Martinez, A. B. Subramaniam, H. J. Yoon, R. M. D. Nunes, H. Lange, M. M. Thuo and G. M. Whitesides, Adv. Funct. Mater., 2014, 24, 60–70 CrossRef CAS.
- S. Rao, Front. Neuroeng., 2009, 2, 1–14 Search PubMed.
- L. M. Y. Yu, N. D. Leipzig and M. S. Shoichet, Mater. Today, 2008, 11, 36–43 CrossRef CAS.
- A. E. Schaffner, J. L. Barker, D. a. Stenger and J. J. Hickman, J. Neurosci. Methods, 1995, 62, 111–119 CrossRef CAS PubMed.
- R. W. F. Wiertz, E. Marani and W. L. C. Rutten, J. Neural Eng., 2010, 7, 56003 CrossRef CAS PubMed.
- C. N. Flynn, C. P. Byrne and B. J. Meenan, Surf. Coat. Technol., 2013, 233, 108–118 CrossRef CAS.
- Z. Q. Hua, R. Sitaru, F. Denes and R. a. Young, Plasmas Polym., 1997, 2, 199–224 CrossRef CAS.
- K. Kang, M.-H. Kim, M. Park and I. S. Choi, J. Nanosci. Nanotechnol., 2014, 14, 513–521 CrossRef CAS PubMed.
- J. Credou, H. Volland, J. Dano and T. Berthelot, J. Mater. Chem. B, 2013, 1, 3277–3286 RSC.
- S. J. Craig, A. Shu, Y. Xu, F. C. Foong and R. Nordon, Protein Eng., Des. Sel., 2007, 20, 235–241 CrossRef CAS PubMed.
- A. Wierzba, U. Reichl, R. F. B. Turner, R. A. J. Warren and D. G. Kilburn, Biotechnol. Bioeng., 1995, 47, 147–154 CrossRef CAS PubMed.
- D. M. Morgan, J. Clover and J. D. Pearson, J. Cell Sci., 1988, 91(2), 231–238 CAS.
- F. Kong and Y. F. Hu, Anal. Bioanal. Chem., 2012, 403, 7–13 CrossRef CAS PubMed.
- N. Li and A. Folch, Exp. Cell Res., 2005, 311, 307–316 CrossRef CAS PubMed.
- J. S. Goldner, J. M. Bruder, G. Li, D. Gazzola and D. Hoffman-Kim, Biomaterials, 2006, 27, 460–472 CrossRef CAS PubMed.
- A. G. P. R. Figueiredo, A. R. P. Figueiredo, A. Alonso-Varona, S. C. M. Fernandes, T. Palomares, E. Rubio-Azpeitia, A. Barros-Timmons, A. J. D. Silvestre, C. Pascoal Neto and C. S. R. Freire, BioMed Res. Int., 2013, 2013, 1–14 Search PubMed.
- J. M. S. Saarela, S. M. Heikkinen, T. E. J. Fabritius, A. T. Haapala and R. A. Myllylä, Meas. Sci. Technol., 2008, 19, 55710 CrossRef.
- A. K. Ellerbee, S. T. Phillips, A. C. Siegel, K. a. Mirica, A. W. Martinez, P. Striehl, N. Jain, M. Prentiss and G. M. Whitesides, Anal. Chem., 2009, 81, 8447–8452 CrossRef CAS PubMed.
- R. Lu, W. Shi, L. Jiang, J. Qin and B. Lin, Electrophoresis, 2009, 30, 1497–1500 CrossRef PubMed.
- Handbook of Neural Activity Measurement, ed. R. Brette and A. Destexhe, Cambridge University Press, Cambridge, 1st edn, 2012 Search PubMed.
- B. C. Wheeler, Y. Nam and B. C. Wheeler, Crit. Rev. Biomed. Eng., 2011, 39, 45–61 CrossRef.
- N. Herzog, M. Shein-Idelson and Y. Hanein, J. Neural Eng., 2011, 8, 56008 CrossRef PubMed.
- W. L. Shew, T. Bellay and D. Plenz, J. Neurosci. Methods, 2010, 192, 75–82 CrossRef CAS PubMed.
- E. Maeda, H. P. C. Robinson and A. Kawana, J. Neurosci., 1995, 15, 6834–6845 CAS.
- H. Kamioka, E. Maeda, Y. Jimbo, H. P. C. Robinson and A. Kawana, Neurosci. Lett., 1996, 206, 109–112 CrossRef CAS PubMed.
- E. Biffi, G. Regalia, A. Menegon, G. Ferrigno and A. Pedrocchi, PLoS One, 2013, 8, e83899 Search PubMed.
- M. Chiappalone, A. Vato, L. Berdondini, M. Koudelka-Hep and S. Martinoia, Int. J. Neural Syst., 2007, 17, 87–103 CrossRef PubMed.
- O. Weihberger, S. Okujeni, J. E. Mikkonen and U. Egert, J. Neurophysiol., 2013, 109, 1764–1774 CrossRef CAS PubMed.
- T. D. Hassinger, P. B. Guthrie, P. B. Atkinson, M. V Bennett and S. B. Kater, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13268–13273 CrossRef CAS.
- M. Amiri, N. Hosseinmardi, F. Bahrami and M. Janahmadi, J. Comput. Neurosci., 2013, 34, 489–504 CrossRef PubMed.
- A. Volterra and J. Meldolesi, Nat. Rev. Neurosci., 2005, 6, 626–640 CrossRef CAS PubMed.
- S. Kügler, P. Lingor, U. Schöll, S. Zolotukhin and M. Bähr, Virology, 2003, 311, 89–95 CrossRef.
- L. Wang, C. Xu, Y. Zhu, Y. Yu, N. Sun, X. Zhang, K. Feng and J. Qin, Lab Chip, 2015, 15, 4283–4290 RSC.
- R. Pelton, TrAC, Trends Anal. Chem., 2009, 28, 925–942 CrossRef CAS.
- T. G. Hammond and J. M. Hammond, Am. J. Physiol. Ren. Physiol., 2001, 281, F12–F25 CAS.
- H. Kurosawa, J. Biosci. Bioeng., 2007, 103, 389–398 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Movies M1 and M2 showing the simultaneous recording of intra- and extracellular neuronal activity of the prepared paper-based cultures. Additional details in 9 figures and 1 table. See DOI: 10.1039/c7ra00971b |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |