DOI:
10.1039/C7RA06103J
(Paper)
RSC Adv., 2017,
7, 39349-39358
Preparation and characterization of nanoparticle reinforced alginate fibers with high porosity for potential wound dressing application†
Received
2nd June 2017
, Accepted 10th July 2017
First published on 11th August 2017
Abstract
A novel fiber dressing was fabricated by blending nano-silica or hydroxyapatite with alginate via microfluidic spinning, which demonstrated delayed degradation, greater mechanical performance and superior bioactivity due to the reinforcing alginate fibers. The prepared fibrous dressings exhibited higher surface area and porosity, which was conducive to cell growth. The addition of nano-silica and hydroxyapatite particles in the alginates improved the mechanical properties and biomineralization on the surface of the fibers. Besides, the degradation time scale and swelling behavior of the nanoparticle reinforced alginate fibers were delayed compared to the original alginate fibers, indicating a longer duration in contact with the wound. The prompt proliferation of fibroblast and keratinocyte cells suggested no significant cytotoxicity of the alginate containing silica and hydroxyapatite. Taken together, porous alginate fibers reinforced by nano-silica and/or hydroxyapatite showed high potential for wound dressing applications.
1 Introduction
Designable biomaterials play the central roles in the fields of regenerative medicine and tissue engineering, as they can direct the cellular behavior and repair capacity of the host.1–5 Among them, artificial wound dressings have some specific requirements: appropriate gelation properties, minimal immunogenicity and the ability to facilitate cell growth and differentiation.6–8 Compared with traditional dressings, novel wound dressings prepared from a tissue engineering strategy have eliminated most inflammation risks and thereby accelerated the wound healing process.9,10 As is well known, the raw materials to fabricate wound dressings are synthetic polymers, natural polymers and their mixtures.11 Although synthetic polymers are intensively used in the current market, some synthetic polymers are not considered as good candidates because of their lack of biodegradability. It was also difficult to clean the wound, as the wound surface is covered by the undegradable wound dressing, which may easily induce secondary trauma and inflammation.12,13 Therefore, considerable research has been devoted to using natural polymers for the fabrication of wound dressings.14
Alginate is a typical natural polymer that has been extensively studied, owning to its high biocompatibility,15 low toxicity, relatively low cost and the safe mild gelation condition with divalent cations such as Ca2+ and Ba2+,16 which are preferred as candidates for biomedical applications,17,18 as diverse as drug delivery,19,20 tissue scaffold,21,22 wound dressing,23 et al. These highly porous scaffolds are more conducive to cell growth and proliferation, nutrient supplement and new tissue regeneration.24 A few studies report the use of alginate to fabricate wound dressings for treatment of diabetic feet,25–28 which demonstrated excellent therapeutic effect. However, poor mechanical performance and the uncontrolled degradation of alginate hamper the practical application of these scaffolds.21 To solve this problem, further research on the fabrication of composite dressings show that, with a combination of other materials, the mechanical limitations could be avoided.21,29–33
Recent progress in bone repair have shown that the addition of nano-silica and nano hydroxyapatite particle can increase mechanical performance and facilitated cell proliferation and differentiation.34 Hydroxyapatite is the mostly investigated additive for the fabrication of scaffolds, due to its close resemblance to bone tissue inorganic phase,35–37 while silica is found to promote biomineralization deposition on the surface of scaffolds, which is beneficial to bone regeneration.38 Additionally, it can also increase the surface area for the superior interface with the polymer matrix and provide preferable mechanical performance.
Inspired by these studies, herein we reported the fabrication of alginate-based wound dressings by the addition of nanoscale silica and hydroxyapatite. Through microfluidic spinning, alginate fibers having inorganic particles were acquired. The characterization for prepared fibers confirmed that the incorporation of nanoparticles increased surface area and pore volume. Fibers containing nano particles exhibited superior mechanical performance and lower degradation, when compared to pure alginate fibers. In addition, the incorporation of nano-silica into alginates was beneficial to the mineral deposited on the surface of surface, which delayed the degradation period. Cell culture results confirmed that the blended fibers were conducive to cell growth. These results meant that the concept of adding bioactive nanoparticles in alginate fibers may hold great potential for healing and other biomedical applications.
2 Materials and methods
2.1 Fabrication of biocomposite porous fibers
Three distinct core fluids were applied: sodium alginate solution (3% (w/v)) was prepared by dissolving sodium alginate powder (Sigma-Aldrich, St. Louis, MO, USA) in deionized water (DW): 0.10 mg of nano-silicon dioxide (SiO2) powder (Sinopharm Chemical Reagent Co., Ltd. Shanghai, China) added into 1 ml sodium alginate solution as the one core flow and the other one was 0.10 mg of hydroxyapatite powder in 1 ml of 3% alginate solution, the last one was above protocol being followed in which the addition of inorganic particles were excluded. The sheath flow contains 3% (w/v) calcium chloride (CaCl2) in ethanol (Ling Feng Chemical Reagent Co., Ltd. Shanghai, China), which was utilized in inducing the instant gelation of alginate solution. To further solidify fibers, 8% (w/v) CaCl2 was dissolved in ethanol as the coagulation bath. The composite porous fibrous dressings were fabricated by a self-developed microfluidic spinning system.39 Three types of fibrous dressing were marked as: SA (alginate fibrous dressings), SA–HAP (alginate-hydroxyapatite composite fibrous dressing), SA–SiO2 (alginate–silica composite fibrous dressings) and listed in the Table 1.
Table 1 Core-sheath ingredient of three kinds of fiber
| |
SA |
SA–HAP |
SA–Si |
| Core flow |
Alginate |
Alginate–HAP |
Alginate–SiO2 |
| Sheath flow |
CaCl2–EA |
CaCl2–EA |
CaCl2–EA |
2.2 Swelling behavior of alginate fibers
The swelling behavior of various fibers were measured based on the solvent uptake by immersing a certain quantity of dry fibers in PBS at room temperature. After a fixed time intervals, the samples were taken out and blotted onto the filter paper to remove excessive water on fiber surface. The weight change of fibers was transformed to swelling ratio using the formula:
| Swelling ratio (%) = [(Ws − Wi)/Wi] × 100% |
where Wi is the initial starting weight of fiber, and Ws denotes the weight of fiber reached swelling equilibrium. All experiments were performed in triplicate.
2.3 Degradation behavior of alginate fibers
The degree of degradation was characterized by immersing a certain quantity of fibers in PBS at room temperature. After a fixed time intervals, the alginate fibers were taken out and dried in an oven at 60 °C, then weighed under ambient conditions. The drying step was continued until a constant weight was achieved. The degradation degree of fibers was represented by the mass loss and then calculated with the formula:
| Mass loss (%) = [(W0 − W1)/W0] × 100% |
where W0 is the initial starting weight of fiber, and W1 denotes the weight of the partially degradative fiber. All experiments were performed in triplicate.
2.4 Inductively coupled plasma-atomic emission detector (ICP-AED)
The samples were prepared and measured according to the detection standards of JY/T 015-1996 (ref. 40) using inductive coupling plasma atomic emission spectroscopy. The element content of calcium in three groups of fibers were determined using ICP-AED (Leeman Prodigy, America). Measurements were performed in triplicate.
2.5 In vitro biomineralization studies
The in vitro biomineralization of the prepared fibrous dressings were investigated by incubating the dressings in PBS for 15 days. Then the fibers were taken out and washed with deionized water to remove the extra minerals. In order to investigate the deposition on the surface of fiber, the specimens were subjected to SEM (HITACHI S-4800, Japan) analysis and XRD characterization.
2.6 The studies of surface area and pore size distribution
The Brunauer–Emmett–Teller (BET) surface area and BJH pore size distribution of the prepared alginate fibers were detected by N2 adsorption–desorption isotherms using a MICROMERITICS ASAP2020 surface analyzer at 77 K. All samples were degassed at 393 K for 3 h before the measurement.
2.7 Mechanical characterization
The mechanical properties of various fibers were evaluated and compared by tensile tests using a Tensile Tester (XQ-1C, Shanghai, China). The measurement was carried out at room temperature, using a gauge length of 10 mm and a stretching speed of 20 mm min−1. The results were characterized as a breaking stress and an elongation ration at rupture. All experiments were performed with a minimum of six samples.
2.8 Fourier transform infrared spectroscopy
In order to investigate the difference in structure of distinct fibers before and after incubation with 15 days in the PBS solution. FT-IR spectrum was performed via a universal ATR diamond accessory of Nicolet 6700 (Thermo Fisher, USA). Transmission and ATR spectra were recorded with spectral region between 500 and 4000 cm−1.
2.9 X-ray diffraction studies
To make a comparison of crystallinity of three types of fibers before and after incubated in the PBS medium for 15 days, XRD analysis was performed using a D/max-2500PC-ray diffractometer (Rigaku, Japan) equipped with a Cu kα source.41–43 The one-dimensional X-ray diffraction patterns with the intensity curves, and a function of 2θ, were obtained from integrating the two-dimensional scattering patterns of various fibers. The angle of diffraction was varied from 10° to 55° to identify the difference in the crystal structure of various fibers during the process of degradation.
2.10 Cytotoxicity
2.10.1 Proliferation assay. In order to evaluate the cytotoxicity of various dressings, the proliferation and migration assay were carried out based on the HaCaT cells (human keratinocyte cell line) and human dermal fibroblasts, respectively. 50 mg of various alginate fibers were immersed in 50 ml DMEM and incubated at 37 °C for 5 days, respectively. Thus, 1 mg ml−1 alginates extracted medium were obtained and filtered with a needle filter. HaCaT cells (human keratinocyte cell line from ACTT) and human dermal fibroblasts (a gift from Dr Mark A. Carlson, University of Nebraska Medical Center, NE, USA)44 were seeded in 96-well plates at a density of 5000 cells per well and incubated in 5% CO2 at 37 °C for 12 h. After the attachment of cells, the medium were replaced by the extracted medium in which they were incubated with SA, SA–Si and SA–HAP, cells cultured with DMEM served as the blank groups. At the fixed intervals, 10 μl of 5 mg ml−1 MTT solution were added and cultured for another 3 h, respectively. Then, the culture medium were removed and 100 μl DMSO were added in the plates. Finally, the absorbance was measured at 490 nm.45 The relative growth rate of cells was represented by RGR and calculated with the formula
| RGR (%) = (the absorbance of sample/the absorbance of negative control) × 100% |
2.10.2 Migration assay. Human dermal fibroblast were seeded in 3 cm culture dishes at a density of 1 × 106 cells per well and incubated in 5% CO2 at 37 °C for 12 h, respectively. After the cells grew to 90% confluence, an artificial scratched wound were created with 20 μl-micropipette tips. The cell culture medium were replaced by SA, SA–HAP and SA–Si extracted medium and cultured for 12, 24 and 48 h, respectively. At each fixed intervals, the artificial scratched wounds were observed by a phase contrast microscope. For the HaCaT cells (human keratinocytes cell line), the above protocol were followed and the culturing time was set as 24, 48 and 72 h respectively. Both cells were cultured with DMEM served as blank control group.
2.11 Statistical analysis
All the measured data were expressed as mean ± standard deviation (SD). The data were analyzed by the Student's t test, and differences were considered significant at P < 0.05.
3 Results and discussions
3.1 Swelling behavior of fibers
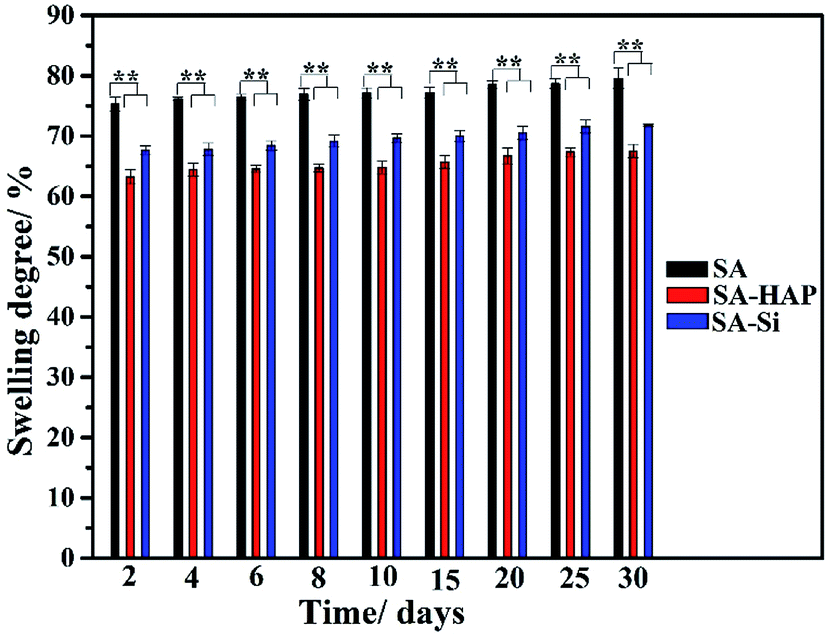
As exhibited in Fig. 1, SA experienced the maximum swelling degree for all the incubation times and SA–Si displayed larger swelling ratio when compared to SA–HAP. The weight increment of SA was 79.56 ± 1.71% at day 30, whilst that of SA–HAP and SA–Si was 67.53% ± 1.10% and 71.78% ± 0.58%, respectively. In addition, the swelling degree among three types of fibers increased slightly with the increasing of immersing times. Moreover, it could be apparently observed that specimens containing nanoparticles showed lower swelling degree, suggesting that the incorporation of nanoparticles obstructed the permeation of PBS, and thus weakened the swelling process. Furthermore, the deposition of hydroxyapatite on the surface of SA–Si specimen hindered the swelling behavior. Our previous study found that the degradation of alginate fibers initiated from the swelling process.39
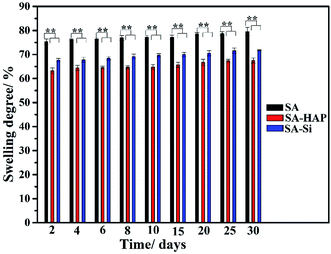 |
| | Fig. 1 Swelling behavior of three types of fibers. These tests (n = 3) were repeated three times. **p < 0.05. | |
3.2 Degradation behavior of fibers and the morphology characterization of fibers
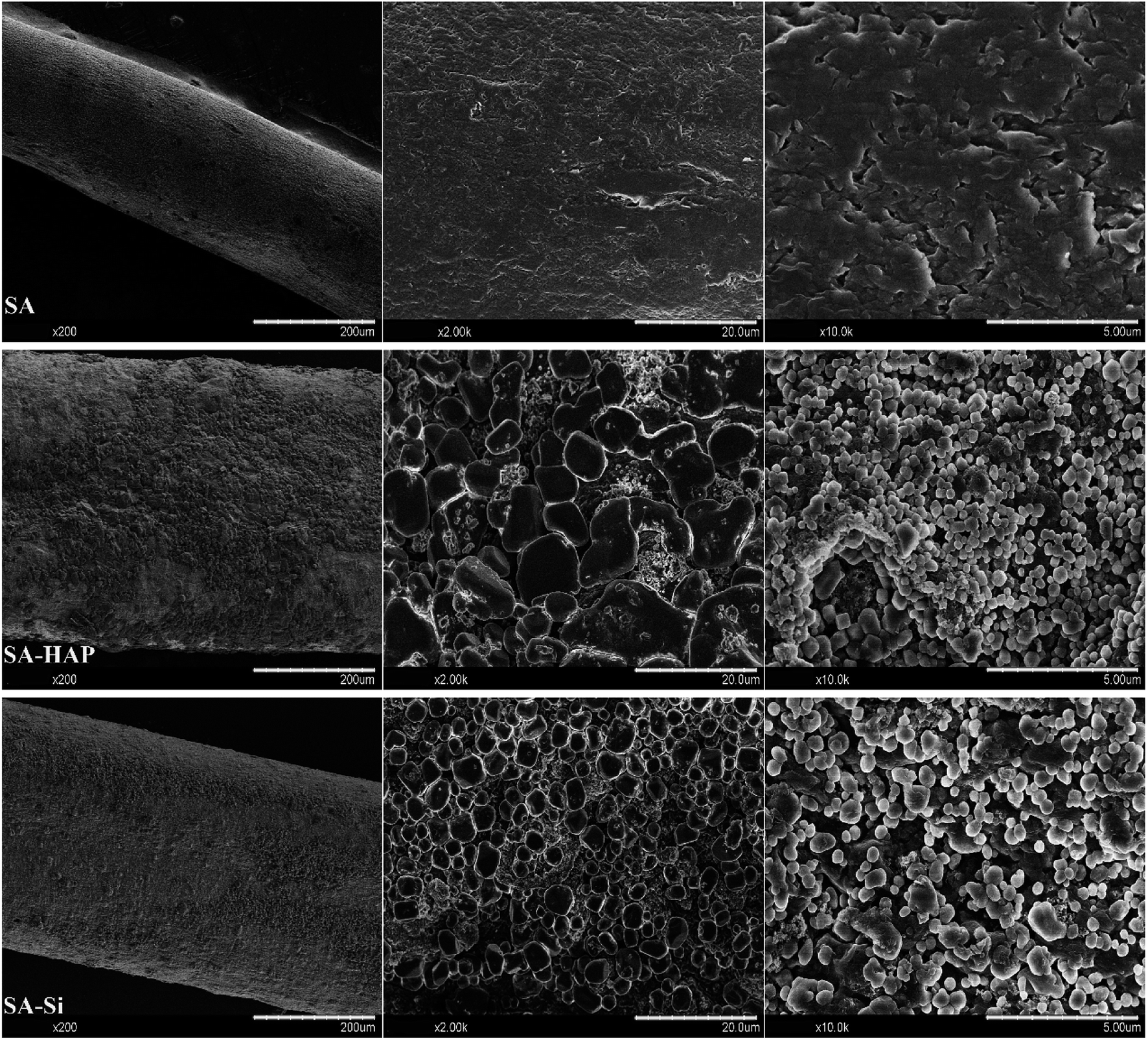
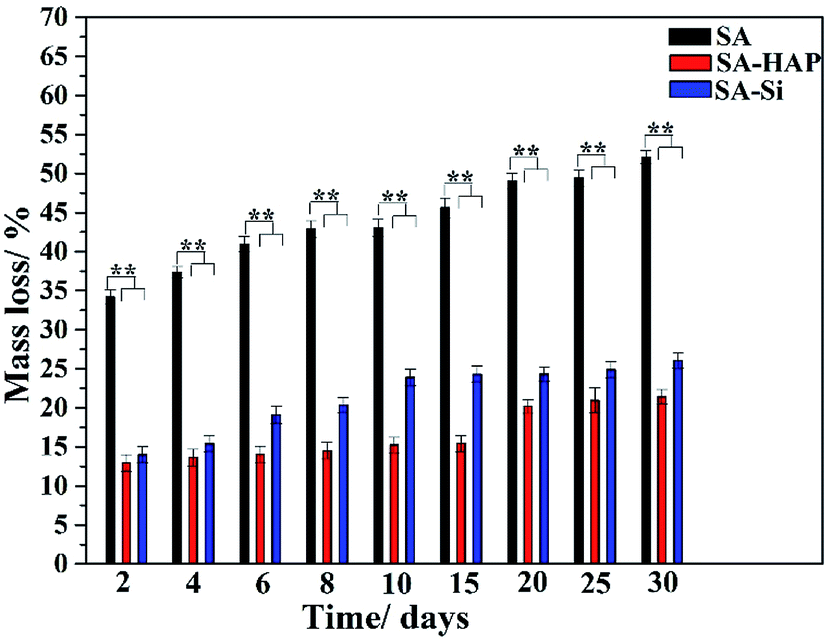
As well known, the crucial parameter to be considered in the biomedical materials was the controlled degradation timescale of fibrous dressings as it offered enough time for tissue ingrowth and skin reparation, which was essential to tissue engineering regenerative. The degradation behavior of the prepared fibers incubated in the PBS solution were investigated as displayed in Fig. 3. It could be clearly seen that SA indicated the maximum mass loss for all the incubation times (Fig. 3), whilst SA–HAP demonstrated the lower degradation rate compared to SA–Si. Besides, it was found that all the specimens displayed an increase in the weight loss with the increasing incubation times, which was in accordance with the swelling profile. Ca2+ ions of the egg-box structure in the alginates molecular chain exchanged with Na+ ions of the PBS solution when SA immersed in the PBS medium, leading to the degradation process of fibers. However, the degradation of fibers embedded nanoparticle were significantly delayed, according to the degradation profile of SA–HAP and SA–Si. As demonstrated in Fig. 2, it could be seen that SA–HAP and SA–Si showed some additive covered on the surface, whereas SA displayed smooth surface. Under the larger magnification, SA displayed tiny crack, which was triggered inevitably during the preparation process. SA–HAP and SA–Si composite reinforced fibers exhibited lots of small and round crystal-like homogeneous particles attached to the fibrous surface. It demonstrated that the addition of inorganic nano-silica or/and hydroxyapatite particles into the alginate coated evenly on the surface of fibers. The morphology characterization illustrated that the embedded inorganic nano-hydroxyapatite and silica covered evenly on the alginate-based fiber surface. Therefore, the delayed degradation of SA–HAP and SA–Si was attributed to the low solubility deposition on the surface on fibers, which weakened the ion-exchange interaction. In order to verify the assumption, the quantification of Ca and Na content in the fibers were investigated as follow. Moreover, the insoluble deposition impeded the permeation of PBS medium, resulting in the delayed degradation timescale, which also had an impact on the mechanical performance. It was essential to conduct in vitro biomineralization study, which was applied to investigate the insoluble deposition on the surface of specimens.
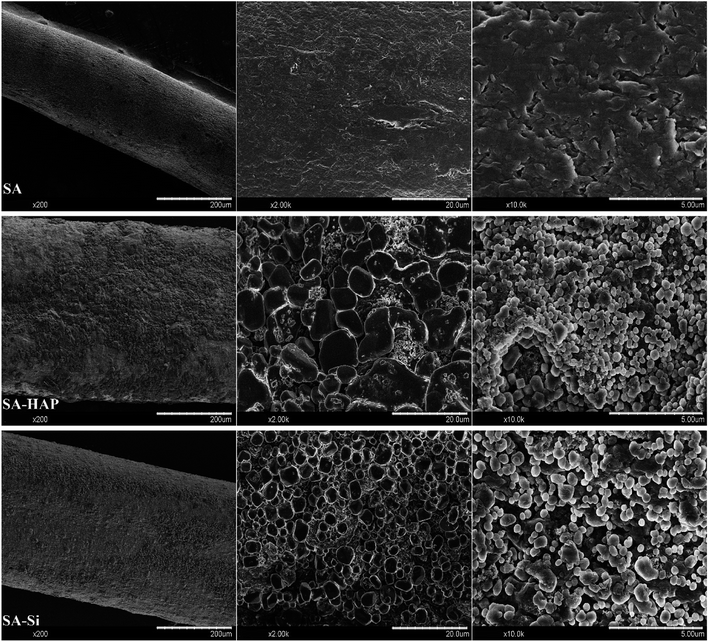 |
| | Fig. 2 The morphological characterization of alginate-based fiber dressings before immersed in PBS. | |
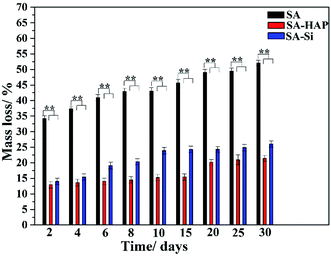 |
| | Fig. 3 Degradation profile of three kinds of fibers. These tests (n = 3) were repeated three times. **p < 0.05. | |
3.3 ICP-AED
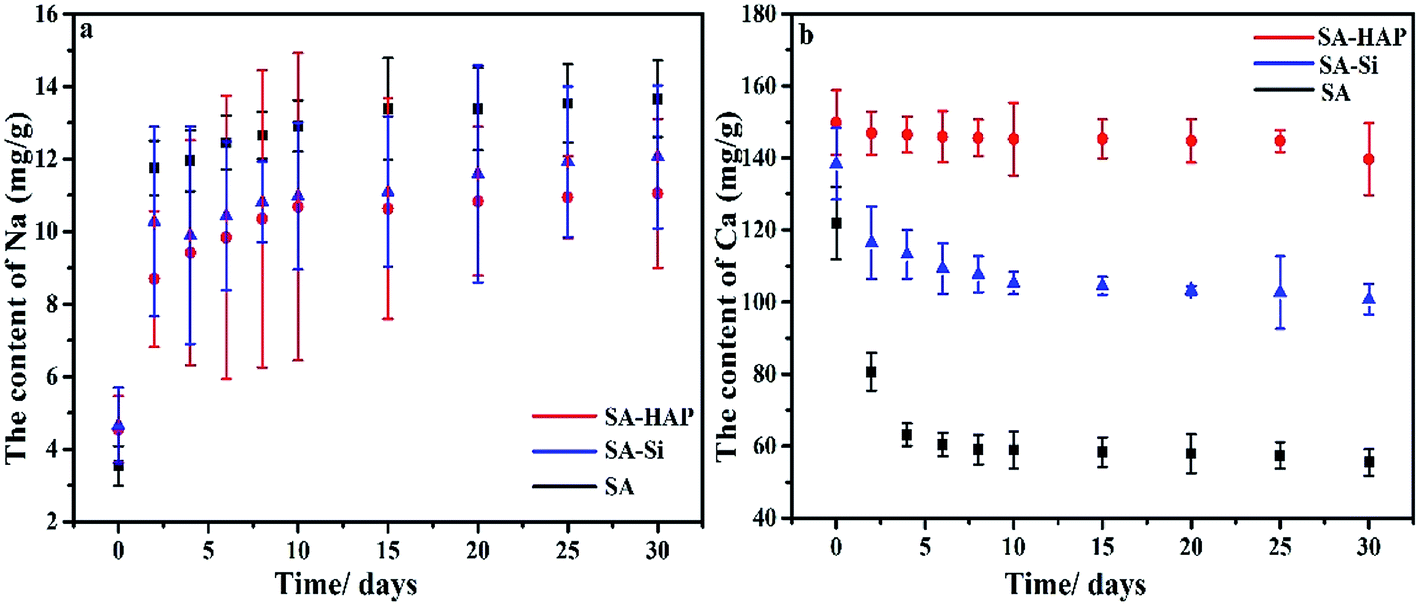
To further prove the inclusion of nanoparticle silicon dioxide and hydroxyapatite obstructed the ion-exchange between Ca2+ of the alginates and Na+ of the PBS medium, we detected the Ca and Na content in the fibers after different incubation time. It was concluded that the degradation degree was positive to the Na content in fibers, but negative to the Ca content. As exhibited in Fig. 4, SA exhibited the maximum Na content and minimum Ca at all degradation time, suggesting that SA group experienced the highest degradation. On the contrary, SA–HAP group had the lowest degradation with the least Na content but the largest Ca content. Since the amount of Ca and Na agreed with the of nanoparticle incorporation, we speculated that the addition of these particles could hinder the ion-exchange interaction, and subsequently elongated the degradation.
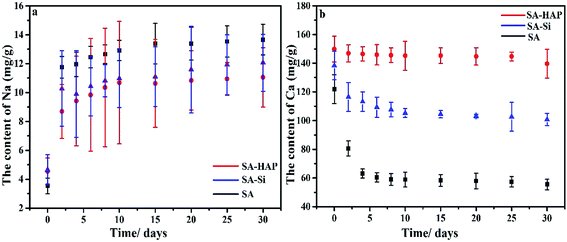 |
| | Fig. 4 Na (a) and Ca (b) content in the fibers with different core flow. | |
3.4 In vitro biomineralization studies
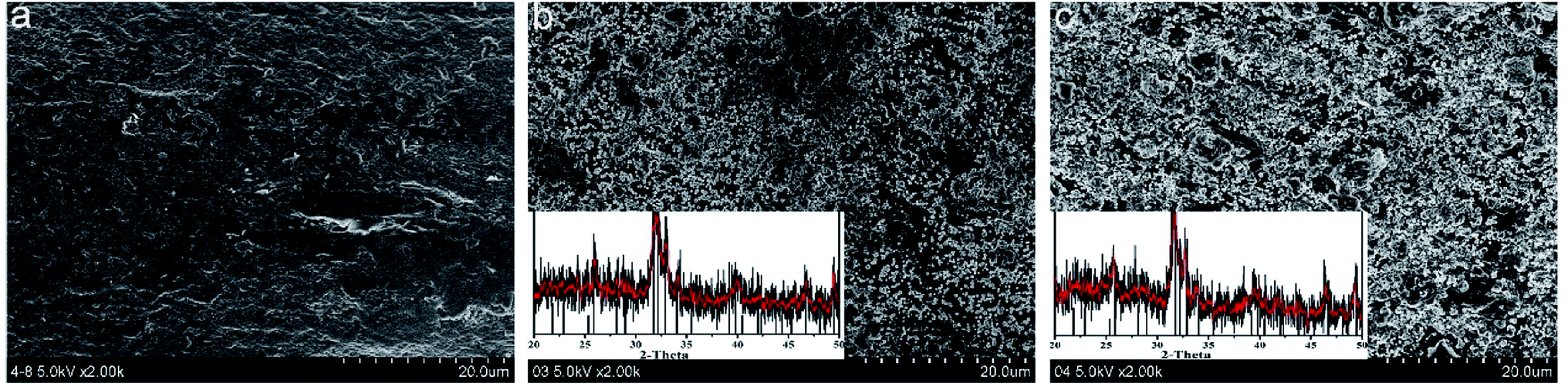
In vitro biomineralization of various fibers were investigated by immersing the fibers in PBS. Surface morphologies and mineral deposition incubated in the PBS solution for 15 days were observed by SEM (Fig. 5). The surface of SA–HAP and SA–Si were covered with numerous round crystals, indicating the mineral deposition on the fibers. The incorporation of nano-silica in SA–Si was supposed to initiate the formation of the hydroxyapatite layer on the surface of fiber, which was ascribed to Si–O units bind with Ca2+ ions in the CaCl2 solution. After which, the positive charged calcium silicate would interact with the negative charged phosphate ions in the PBS solution, resulting in the precipitation of calcium phosphate into the hydroxyapatite layer. In order to identify the amount of deposition, the biomineralization of fibers were also subjected to the XRD for demonstrating the phase of crystal. The inset XRD patterns after incubation of 15 days proved the characteristic peak of hydroxyapatite. This peak agreed well with standard card of JCPDS # 09-0432 and strongly verified the deposition of hydroxyapatite on fiber surface.
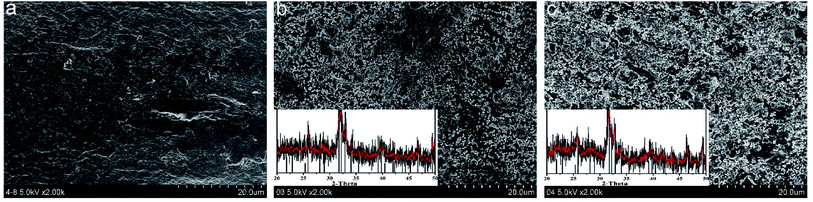 |
| | Fig. 5 SEM images of the fibrous dressings (a) SA, (b) SA–HAP and (c) SA–Si after immersed in the PBS solution for 15 days, inset shown the mineral deposition of crystal and XRD patterns. | |
3.5 Surface area and pore size
The specific surface areas of three types of alginate fibrous wound dressings were determined by BET method. The corresponding surface area, pore size and total pore volume of various fibers were tabulated in Table 2. It could be seen that the minimum surface area and total pore volume of SA were 0.2177 m2 g−1 and 0.000043 cm3 g−1, respectively whereas fibers containing nano silica and hydroxyapatite particle showed higher value in both them. The obtained BET surface area and total pore volume indicated that the inclusion of nano particle in the alginate fibers could improve the surface area, which was believed to be beneficial for cell growth.
Table 2 The parameters obtained from N2 absorption–desorption isotherms
| |
Surface area (m2 g−1) |
Pore size (nm) |
Total pore volume (cm3 g−1) |
| SA |
0.2177 |
0.7885 |
0.000043 |
| SA–HAP |
0.2520 |
6.50126 |
0.00041 |
| SA–Si |
0.4544 |
8.0504 |
0.000915 |
3.6 Mechanical properties
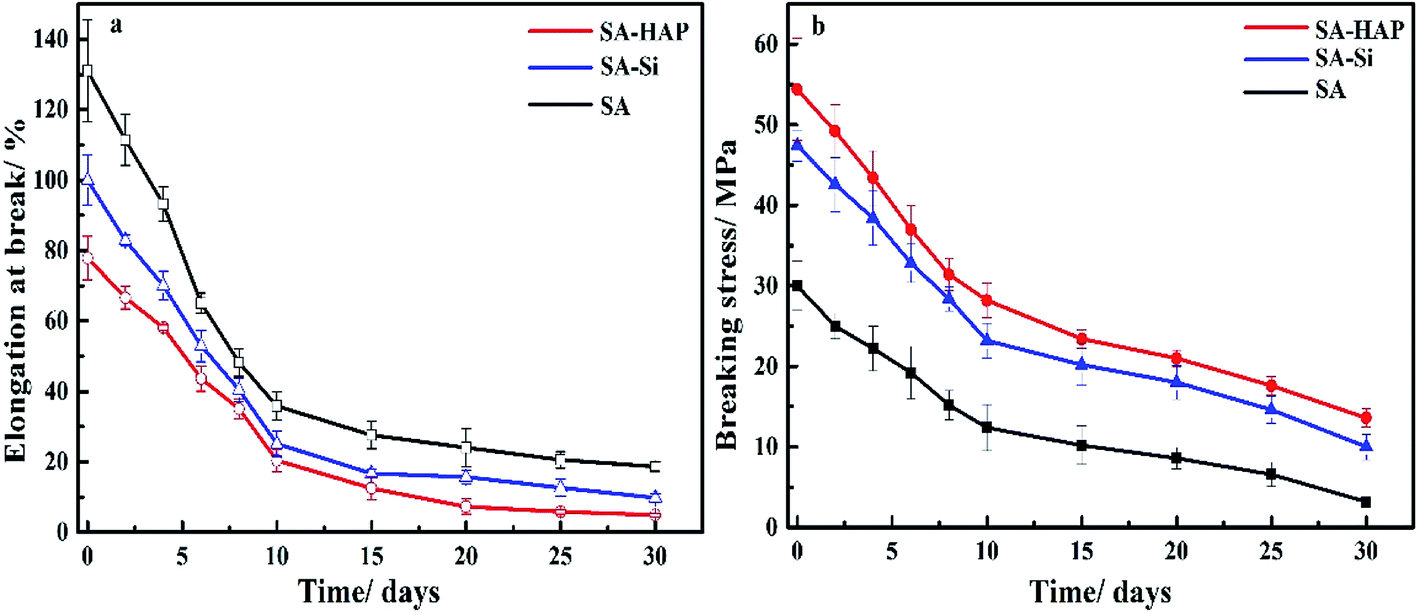
Fibrous materials for wound healing must provide enough mechanical support for in vitro application. Therefore, the elongation at rupture and breaking stress were measured to characterize the effect of degradation on mechanical performance over time. A descending trend was observed from the mechanical curves in Fig. 6, which was ascribed to the degradation. The breaking stress of SA–HAP ranged from 54.4 to 13.6 MPa throughout the incubation time and that of SA–Si ranged from 47.4 to 10 MPa. Nevertheless, the elongation at rupture of three types of samples showed opposite results to breaking stress. This can be explained by the breaking stress of alginate fibers relied upon the crosslinking between Ca2+ and carbonyl group (–COOH) of alginates, the more crosslinking the better breaking stress. On the contrary, more crosslinking raised the stiffness of fibers and resulted in smaller elongation at rupture. Therefore, it can be concluded that fibers with higher Ca content exhibited preferable breaking stress whilst poor stiffness. Quantitative results of Ca content in fibers also indicated that SA–HAP had more Ca content than other groups, which further proved that the incorporation of silica dioxide and hydroxyapatite in the alginate wound dressings could improve the mechanical performance.
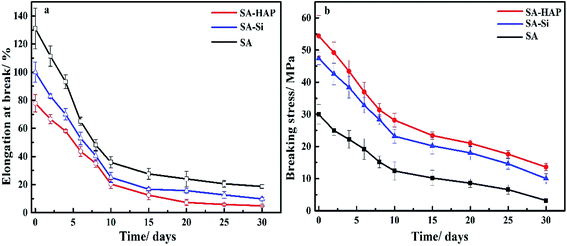 |
| | Fig. 6 Elongation at rupture (a) and breaking stress of various fibers (b). | |
3.7 Fourier transform infrared spectroscopy and X-ray diffraction
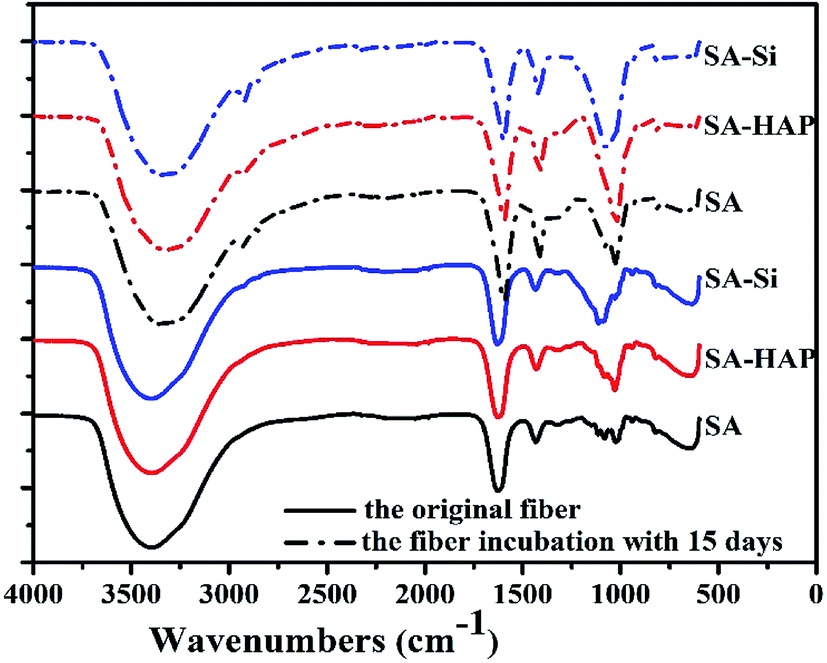
FTIR analysis were conducted to evaluate the change of fiber composition before and after incubated in PBS for 15 days. As shown in Fig. 7, similar absorption bands were found in various fibers and no additional peaks or noticeable shifts were observed. The peak at 3417 cm−1, 1627 cm−1 and 1438 cm−1 were assigned to vibrations of the hydroxyl groups (OH), the asymmetric and symmetric –COO stretching vibrations, respectively.
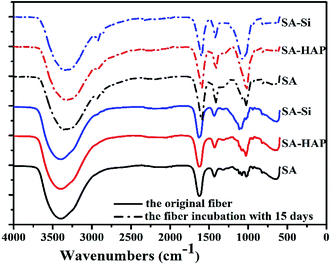 |
| | Fig. 7 FTIR spectrum of various fibers before and after soaking in the PBS media for 15 days. | |
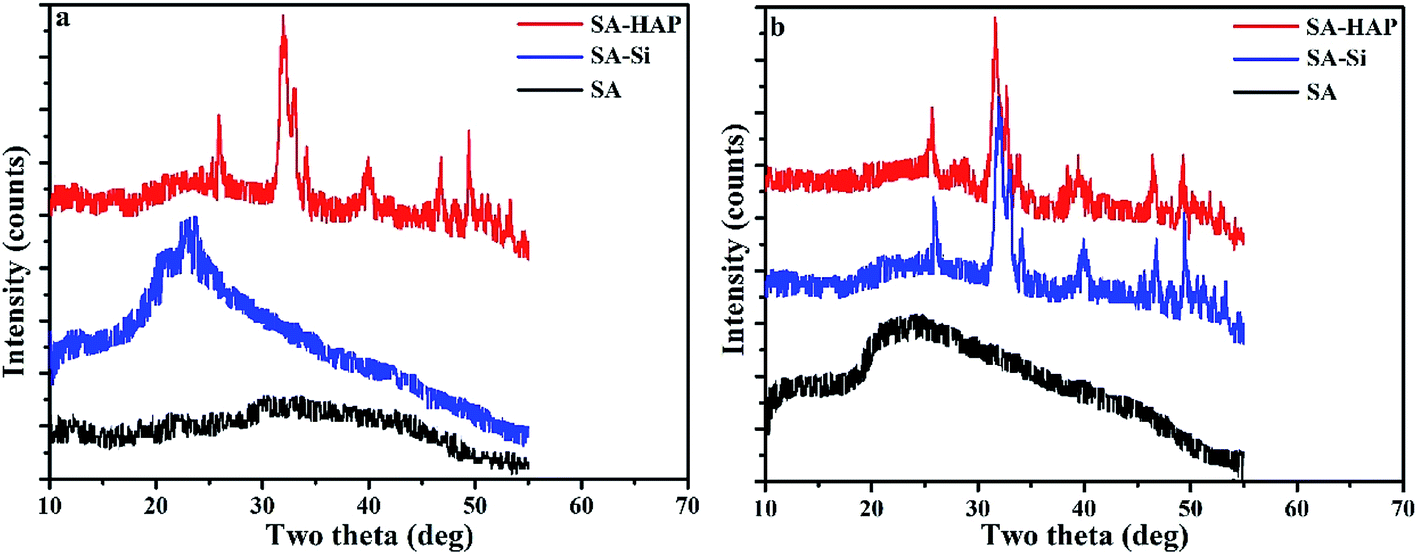
X-ray diffraction patterns of fibers were also conducted and included in Fig. 8. The crystallinity of SA, SA–Si and SA–HAP without immersing in the PBS were 9.02%, 21.08% and 36.43% respectively. And the crystallinity of SA, SA–Si and SA–HAP after incubated in the PBS at 15 days were 18.01%, 35.77% and 37.05% respectively. For pure alginates, it could be clearly seen that fiber after degradation exhibited stronger peak intensity, suggesting a drop in the fibrous amorphous area during the process of degradation. A sharp difference of XRD patterns before and after in PBS for 15 days was noticed in SA–Si group. The X-ray patterns of SA–Si degraded at 15 days was similar to that of SA–HAP. Characteristic peak of hydroxyapatite was found in SA–Si, confirming the deposition of hydroxyapatite on the surface of SA–Si during degradation.
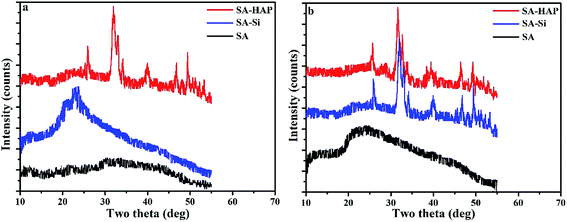 |
| | Fig. 8 The XRD patterns of fibers before (a) and after (b) incubated in the PBS at 15 days. | |
3.8 Cytotoxicity
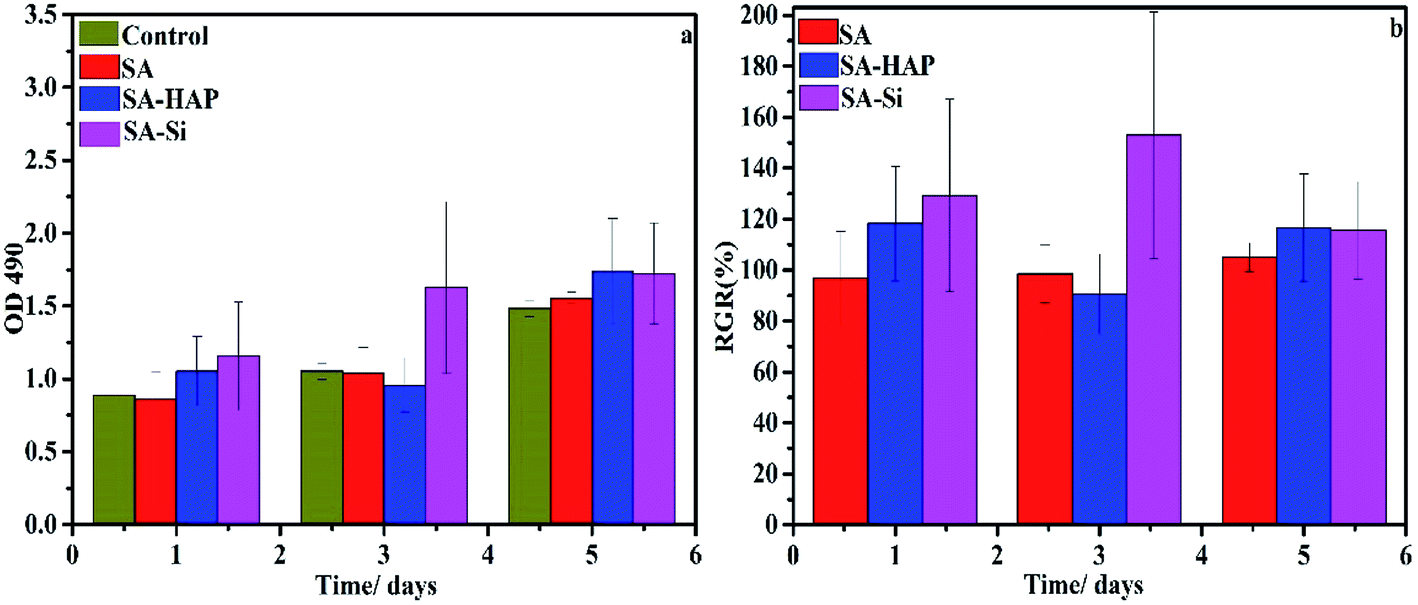
3.8.1 Proliferation assay. To ascertain any negative biological influence of the prepared fibers, proliferation of dermal fibroblast and keratinocytes were examined based on the MTT assay (Fig. 9 and 10). It could be concluded that SA, SA–HAP and SA–Si had no effect of both fibroblast and keratinocyte cells. Increase of cell proliferation on SA–HAP and SA–Si were faster than that of the control group. The relative growth rates of fibroblasts in SA, SA–HAP and SA–Si were 96.96%, 118.33% and 129.35% at 1 day, respectively, while the relative growth rates of keratinocytes were 78.88%, 83.66% and 152.35%, respectively. All these rates were higher than 75%, suggesting that all the fibrous wound dressings had no significant cytotoxicity according to the cytotoxicity grading criteria.45
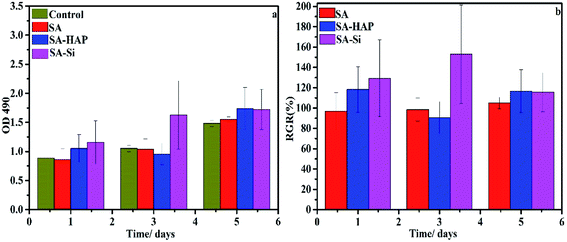 |
| | Fig. 9 The effect of fibrous wound dressings on skin cell proliferation. The proliferation (a) and the relative growth rate (RGR/%) (b) of fibroblasts treated with SA, SA–HAP and SA–Si fibrous wound dressings extracted medium for 1, 3 and 5 days. The DMEM medium served as a control. | |
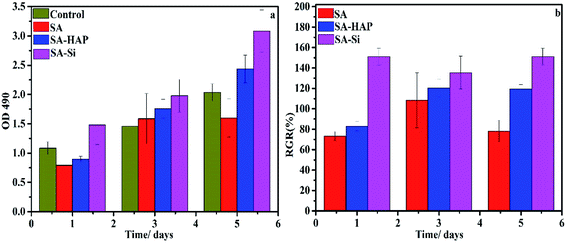 |
| | Fig. 10 The effect of fibrous wound dressings on skin cell proliferation. The proliferation (a) and the relative growth rate (RGR/%) (b) of keratinocytes treated with SA, SA–HAP and SA–Si fibrous wound dressings extracted medium for 1, 3 and 5 days. The DMEM medium served as a control. | |
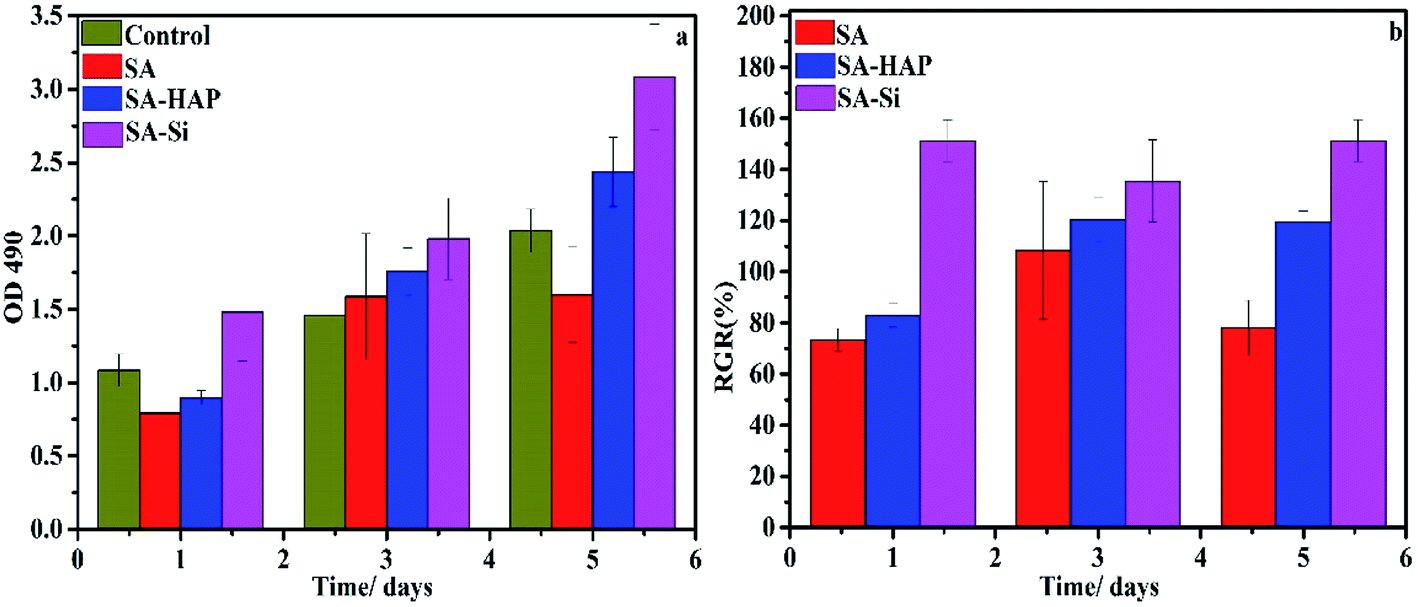
3.8.2 Migration assay. The in vitro scratch wound healing assay was further used to characterize cell migration, which was essential factor for wound healing. The artificial wound healing assays were demonstrated by preparing and culturing keratinocytes and dermal fibroblast, with the artificial cell-scratched regions as displayed in Fig. 11 and 12. The artificial wound were treated with SA, SA–HAP and SA–Si extracted medium and DMEM medium to determine the influence on the cell migration. Apparently, the wound gaps showed a decreasing tendency with an increase in time scales. For fibroblasts, it took 24 h for the cells to cover 90% of the artificial wound area. This speed was equal to that of the control groups. The keratinocytes needed 72 h to cover 90% of the wound indicating that the treatment by SA, SA–HAP and SA–Si extracted medium took almost same time to heal scratch wound. As such time is in accordance to the control group, the treatment had no negative impact on the migration of dermal cells.
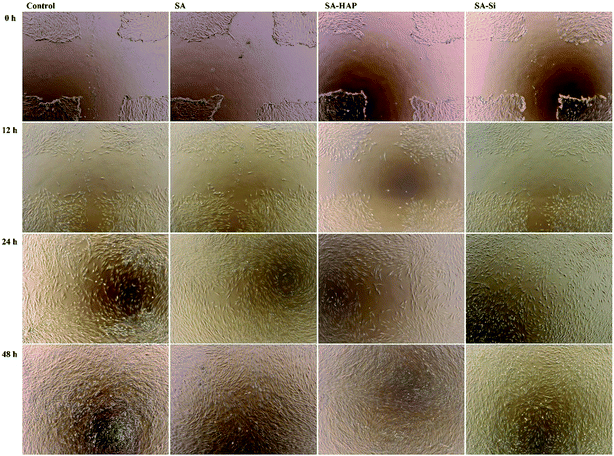 |
| | Fig. 11 The effect of fibrous wound dressings on skin cell migration. Fibroblast migration in the scratched fibroblast free area after treated with SA, SA–HAP and SA–Si extracted medium incubated with 1, 3 and 5 days. The DMEM medium serves as a control. | |
 |
| | Fig. 12 The effect of fibrous wound dressings on skin cell migration. Keratinocyte migration in the scratched keratinocyte free area after treated with SA, SA–HAP and SA–Si extracted medium incubated with 1, 3 and 5 days. The DMEM medium serves as a control. | |
4 Conclusions
In summary, porous wound dressings were prepared by blending alginate with HAP and SiO2. Compared with pure alginate dressings, the addition of inorganic nanoparticles significantly improved the mechanical performances and delayed the degradation time, while posed no obvious impact on the proliferation and migration of dermal fibroblast and keratinocytes. These results suggested that the concept of adding bioactive nanoparticles in alginate fibers may hold great potential for healing and other biomedical applications.
Acknowledgements
This work was supported by “Chen Guang” Project from Shanghai Municipal Education Commission and Shanghai Education Development Foundation (No. 14CG34) and the Fundamental Research Funds for the Central Universities (CUSF-DH-D-2017009). In addition, I appreciated that Dr Mark A. Carlson, Dr Shixuan Chen and Dr Jingwei Xie gave me some favour.
References
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- N. A. Peppas and R. Langer, Science, 1994, 263, 1715–1720 CAS.
- J. A. Hubbell, Nat Biotech, 1995, 13, 565–576 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- X. Liu and P. X. Ma, Ann. Biomed. Eng., 2004, 32, 477–486 CrossRef PubMed.
- J. Bonadio, E. Smiley, P. Patil and S. Goldstein, Nat. Med., 1999, 5, 753–759 CrossRef CAS PubMed.
- D. W. Hutmacher, J. Biomater. Sci., Polym. Ed., 2001, 12, 107–124 CrossRef CAS PubMed.
- S. Yang, K. F. Leong, Z. Du and C. K. Chua, Tissue Eng., 2001, 7, 679–689 CrossRef CAS PubMed.
- S. Thomas, J. Wound Care., 2000, 9, 56–60 CrossRef CAS PubMed.
- W.-S. Chang and H.-H. Chen, Food Hydrocolloids, 2016, 53, 75–83 CrossRef CAS.
- I. Yuvarani, S. Senthilkumar, J. Venkatesan, S.-K. Kim, A. A. Al-Kheraif, S. Anil and P. N. Sudha, J. Biomater. Tissue Eng., 2015, 5, 665–672 CrossRef.
- S. Thomas, J. Wound Care., 2000, 9, 115–119 CrossRef CAS PubMed.
- J. Baier Leach, K. A. Bivens, C. W. Patrick Jr and C. E. Schmidt, Biotechnol. Bioeng., 2003, 82, 578–589 CrossRef PubMed.
- S. Kumar, V. Deepak, M. Kumari and P. K. Dutta, Int. J. Biol. Macromol., 2016, 84, 349–353 CrossRef CAS PubMed.
- R. LogithKumar, A. KeshavNarayan, S. Dhivya, A. Chawla, S. Saravanan and N. Selvamurugan, Carbohydr. Polym., 2016, 151, 172–188 CrossRef CAS PubMed.
- S. G. Reddy and A. S. Pandit, Polim.: Cienc. Tecnol., 2013, 23, 13–18 CrossRef CAS.
- R. Ghaffarian, E. P. Herrero, H. Oh, S. R. Raghavan and S. Muro, Adv. Funct. Mater., 2016, 26, 3382–3393 CrossRef CAS PubMed.
- J. Hayashizaki, Proc. 7th Intern. Symp. Ceramics in Medicine, 1994, 1994, vol. 7, pp. 41–46 Search PubMed.
- A. Shukla, V. Mishra, B. S. Bhoop and O. P. Katare, Int. J. Pharm., 2015, 495, 220–233 CrossRef CAS PubMed.
- M. J. Coathup, T. C. Edwards, S. Samizadeh, W.-J. Lo and G. W. Blunn, J. Biomed. Mater. Res., Part B, 2016, 104, 1328–1335 CrossRef CAS PubMed.
- C. Sharma, A. K. Dinda, P. D. Potdar, C.-F. Chou and N. C. Mishra, Mater. Sci. Eng., C, 2016, 64, 416–427 CrossRef CAS PubMed.
- H.-L. Kim, G.-Y. Jung, J.-H. Yoon, J.-S. Han, Y.-J. Park, D.-G. Kim, M. Zhang and D.-J. Kim, Mater. Sci. Eng., C, 2015, 54, 20–25 CrossRef CAS PubMed.
- S. Thomas, J. Wound Care., 2000, 9, 163–166 CrossRef CAS PubMed.
- J. Venkatesan, I. Bhatnagar, P. Manivasagan, K.-H. Kang and S.-K. Kim, Int. J. Biol. Macromol., 2015, 72, 269–281 CrossRef CAS PubMed.
- M. Fischer, F. Gebhard, T. Hammer, C. Zurek, G. Meurer, C. Marquardt and D. Hoefer, J. Biomater. Appl., 2017, 31, 1267–1276 CrossRef PubMed.
- F. L. Game, J. Apelqvist, C. Attinger, A. Hartemann, R. J. Hinchliffe, M. Londahl, P. E. Price and W. J. Jeffcoate, Diabetes/Metab. Res. Rev., 2016, 32(suppl. 1), 154–168 CrossRef PubMed.
- L. J. Borda, F. E. Macquhae and R. S. Kirsner, Curr. Dermatol. Rep., 2016, 5, 287–297 CrossRef.
- M. Saco, N. Howe, R. Nathoo and B. Cherpelis, Dermatol. Online J., 2016, 22, 27617934 Search PubMed.
- T.-y. Chen, H.-c. Huang, J.-l. Cao, Y.-j. Xin, W.-f. Luo and N.-j. Ao, RSC Adv., 2016, 6, 35577–35588 RSC.
- C. Sharma, A. K. Dinda and N. C. Mishra, J. Biomater. Tissue Eng., 2012, 2, 133–142 CrossRef CAS.
- S. Gautam, A. K. Dinda and N. C. Mishra, Mater. Sci. Eng., C, 2013, 33, 1228–1235 CrossRef CAS PubMed.
- S. Gautam, C.-F. Chou, A. K. Dinda, P. D. Potdar and N. C. Mishra, Mater. Sci. Eng., C, 2014, 34, 402–409 CrossRef CAS PubMed.
- C. Sharma, A. K. Dinda and N. C. Mishra, J. Appl. Polym. Sci., 2013, 127, 3228–3241 CrossRef CAS.
- L. Kong, Y. Gao, G. Lu, Y. Gong, N. Zhao and X. Zhang, Eur. Polym. J., 2006, 42, 3171–3179 CrossRef CAS.
- W. Nie, C. Peng, X. Zhou, L. Chen, W. Wang, Y. Zhang, P. X. Ma and C. He, Carbon, 2017, 116, 325–337 CrossRef CAS.
- S. K. Gupta, A. K. Dinda, P. D. Potdar and N. C. Mishra, BioMed Res. Int., 2013, 2013, 23841083, DOI:10.1155/2013/651945.
- C. Isikli, V. Hasirci and N. Hasirci, J. Tissue Eng. Regener. Med., 2012, 6, 135–143 CrossRef CAS PubMed.
- J. A. Sowjanya, J. Singh, T. Mohita, S. Sarvanan, A. Moorthi, N. Srinivasan and N. Selvamurugan, Colloids Surf., B, 2013, 109, 294–300 CrossRef CAS PubMed.
- X. Zhang, C. Huang and X. Jin, J. Appl. Polym. Sci., 2017, 134 DOI:10.1002/app.44396.
- S.-F. Lim, Y.-M. Zheng, S.-W. Zou and J. P. Chen, Environ. Sci. Technol., 2008, 42, 2551–2556 CrossRef CAS PubMed.
- Z. Jiang, H. Azim, R. A. Gross, M. L. Focarete and M. Scandola, Biomacromolecules, 2007, 8, 2262–2269 CrossRef CAS PubMed.
- A. Babaei and A. Arefazar, J. Appl. Polym. Sci., 2015, 132 DOI:10.1002/app.41969.
- M. H. Rahaman and H. Tsuji, J. Appl. Polym. Sci., 2013, 129, 2502–2517 CrossRef CAS.
- S. Chen, L. Ge, A. Mueller, M. A. Carlson, M. J. Teusink, F. D. Shuler and J. Xie, Nanomedicine, 2017, 13, 1435–1445 CrossRef CAS PubMed.
- K.-H. Sun, Z. Liu, C. Liu, T. Yu, T. Shang, C. Huang, M. Zhou, C. Liu, F. Ran, Y. Li, Y. Shi and L. Pan, Sci. Rep., 2016, 6, 23931 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06103j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Chen Huang
,
Chen Huang