
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrowth of epithelial cells on films of enzymatically synthesized poly(gallic acid) crosslinked to carboxymethylcellulose†
Alejandra Romero-Monteroa,
Alberto Tecantea,
Roeb García-Arrazolaa,
Carmina Montiela,
Luis J. del Valleb,
Jordi Puiggalíb and
Miquel Gimeno *a
*a
aFacultad de Química, Departamento de Alimentos y Biotecnología, Universidad Nacional Autónoma de México, Ciudad Universitaria, Ciudad de México, Mexico. E-mail: mgimeno@unam.mx
bChemical Engineering Department, Escola d'Enginyeria de Barcelona Est-EEBE, c/Eduard Maristany 10-14, Barcelona, Spain
First published on 22nd March 2017
Abstract
Poly(gallic acid), a novel polymer obtained by enzymatic polymerization of gallic acid, was successfully cross-linked to carboxymethylcellulose using citric acid as a cross-linker. Non-woven sheets were prepared with the resulting copolymer and their in vitro biocompatibility was assessed. The results on the characterizations of the produced films by mechanical tests, water vapour permeability, contact angle, and antioxidant activity by electronic paramagnetic resonance were adequate for skin tissue regeneration. Films were also decomposed under physiological conditions using universal buffers at pH 3, 7 and 10. In vitro experiments with fibroblast-like and epithelial-like cells showed good adhesion and proliferation onto the PGAL-co-CMC sheets. These non-woven sheets can consequently be considered as novel biocompatible and biodegradable films with high-responsiveness for biomedical or tissue engineering applications.
Introduction
The biomaterials composed of polymers from natural sources found applications in tissue engineering owing to their good biocompatibility, superior responses, few side effects and reduction or even suppression of secondary treatments or surgeries.1–5 Additionally, they are also studied in neurology, medical imaging, bionic medicine, biomechanics or bio-nanotechnology.6 Generally, the improvement of the properties of these biomaterials to meet the requirements for specific biomedical application includes adequate mechanical properties, stability in aqueous environments, vapour barrier properties and cell biocompatibility, all these concomitantly with synthetic reproducibility in the absence of toxic processes and reagents.6,7 In this regard, the use of non-toxic carboxylic acids as cross-linking agents between two or more polymer backbones, such as citric acid (CA), proved successful to render tissue engineering materials with superior biocompatibility compared to those derived from other cytotoxic crosslinkers, such as glutaraldehyde or formaldehyde.8 Among the studied polymers matrices, the approaches using the naturally abundant cellulose and its derivatives are encouraging owing to their low costs and availability in addition to good biocompatibility. Specifically, the water-soluble carboxymethyl celluloses (CMC)s have multiple biomedical uses such as in heart, chest and cornea surgeries in addition to orthopaedic and pharmaceutical applications, among others.9,10On the other hand, plant polyphenols have attracted wide interest in the biomedical field especially due to their radical scavenging activities (RSA) and antimicrobial capacities. Free radical species promote interaction within cells leading to proteins, membrane and gene damage and additionally, the oxidative stresses generated by the so-called radical oxygen species (ROS) are involved not only in wound healing processes but also in other several metabolic syndromes and inflammatory processes.11–13 Thus, the preparation of matrices with antioxidant activity to protect cells and tissues from oxidative damage has been achieved by incorporation of oxidant radical scavenger molecules, e.g. water-soluble forms of vitamin B6 (pyridoxine, pyridoxal and pyridoxamine), and small polyphenols such as coumaric and caffeic acids.14,15 Nonetheless, the characteristics in most of the natural polyphenol extracts, specially their low molecular stability against temperature or UV-irradiation, as well as high hydrophobicity or their limited extractions in large quantities restricts their bioavailability and processability; thereby their modifications play an important role to produce synthetic derivatives with adequate characteristics and scale-up feasibility.16 Enzyme-mediated modifications offer environmentally favourable and mild process conditions that resulted in non-toxic products.17,18 Glycosylation or esterification of plant antioxidants proved to be successful for these purposes.19,20 Additionally, the oxidative polymerization of phenols using oxidoreductases has been studied to attain novel macromolecular structures and functionalities.21,22 We have recently reported the laccase from Trametes versicolor-mediated polymerization of the naturally abundant gallic acid (GA) in mild process conditions to attain a novel electrically semi-conducting poly(gallic acid) (PGAL).23 This polymer displays a controlled molecular structure with phenyl–phenyl linkages and number average molecular weight of ca. 7000 Da, which can vary according to synthetic conditions, and importantly, this enzymatically synthesized PGAL presents high water solubility, which allows for innocuous processability. However, the rigidity of PGAL did not allow for film formation by itself. In the present work, PGAL is crosslinked to CMC using CA as the cross-linking agent by a non-toxic methodology.24,25 This novel material (PGAL-co-CMC) displays adequate physicochemical characteristics for tissue engineering. The antioxidant power of PGAL–CMC films was demonstrated by solid-state electronic paramagnetic resonance (EPR) as well as the antimicrobial activity for both PGAL and films by the study of the inhibition of common pathogen bacterial strains in hospitals. In order to assess its potential as biomaterial for skin tissue regeneration, the viability of epithelial cells were studied on the produced PGAL-co-CMC films.
Results and discussion
Synthesis and characterization of the produced PGAL-co-CMC films
A possible molecular structure of the target molecule is depicted in Fig. 1 as the product of the reaction scheme. Noteworthy, both polymers have more than one hydroxyl moieties for esterification and CMC presents random methylation and carboxylation in its repeat units. Therefore, the product molecular structures might be several. The production of PGAL (Fig. 1) was confirmed by the absence of signals in 1H-NMR spectra. | ||
| Fig. 1 Schematic representation of the cross-linking reaction between PGAL and CMC with CA as cross-linking agent to a possible molecular structure in product. | ||
Additionally, it was confirmed by the broad FT-IR bands between 3000 and 3400 cm−1 assigned to O–H stretching and the absence of characteristic C–O–C vibration band, as reported elsewhere23 (see ESI data file 1 and 2† for 1H-NMR and FTIR spectra for PGAL, respectively). SEC analyses for PGAL against PEG standards displayed 6000 g mol−1 and the concentration of saturation (Cs) in water for the synthesized molecule was determined as 500 mg mL−1, which is worth to remark from the standpoint of innocuous processability.26
The FT-IR spectrum for the PGAL-co-CMC (entry 3 in Table 1), shown in Fig. 2, is compared with those for PGAL, CA and CMC, where expected bands for native polymers are observed. The bands assigned to carbonyl moieties in AC are also present, 1700–1725 cm−1, which sustains the cross-linking.
| Entry | PGAL (%) | Gly (%) | σa (MPa) | Young's modulus (MPa) | σ (puncture assay, MPa) | Thickness (m) × 105 | Cross-linking degreeb (mol mL−1) × 103 |
|---|---|---|---|---|---|---|---|
| a σ true stress is related to Hencky strain in tensile and puncture test.b Cross-linking degree was calculated from Flory–Huggins polymer–solvent interaction equation. | |||||||
| 1 | 5 | 2 | 3.92 ± 0.33 | 23.06 ± 3.56 | 42.34 ± 2.24 | 1.2 ± 0.1 | 9.3 |
| 2 | 5 | 3 | 7.78 ± 0.74 | 14.40 ± 0.75 | 44.12 ± 2.26 | 1.3 ± 0.3 | 9.3 |
| 3 | 5 | 4 | 14.23 ± 0.35 | 11.32 ± 0.83 | 47.44 ± 1.46 | 1.2 ± 0.2 | 9.8 |
| 4 | 10 | 2 | 7.86 ± 0.54 | 21.67 ± 1.25 | 43.40 ± 2.89 | 1.1 ± 0.2 | 10.8 |
| 5 | 10 | 3 | 7.51 ± 0.58 | 10.50 ± 0.40 | 47.68 ± 0.82 | 1.3 ± 0.2 | 10.1 |
| 6 | 10 | 4 | 28.40 ± 0.36 | 9.46 ± 0.76 | 65.85 ± 3.70 | 1.2 ± 0.3 | 11.1 |
| 7 | 15 | 2 | 12.80 ± 0.69 | 19.22 ± 1.46 | 45.44 ± 1.40 | 1.4 ± 0.1 | 19.1 |
| 8 | 15 | 3 | 12.22 ± 0.31 | 9.82 ± 0.60 | 63.24 ± 1.63 | 1.3 ± 0.3 | 19.2 |
| 9 | 15 | 4 | 31.02 ± 2.89 | 5.85 ± 0.28 | 75.83 ± 0.25 | 1.4 ± 0.2 | 17.8 |
| 10 | 0 | 2 | 8.99 ± 0.79 | 6.44 ± 0.61 | 39.54 ± 2.35 | 1.2 ± 0.1 | 1.3 |
| 11 | 0 | 3 | 14.73 ± 1.11 | 4.88 ± 0.41 | 41.15 ± 1.34 | 1.2 ± 0.2 | 1.1 |
| 12 | 0 | 4 | 17.65 ± 0.99 | 3.91 ± 0.26 | 43.80 ± 2.49 | 1.3 ± 0.1 | 1.5 |
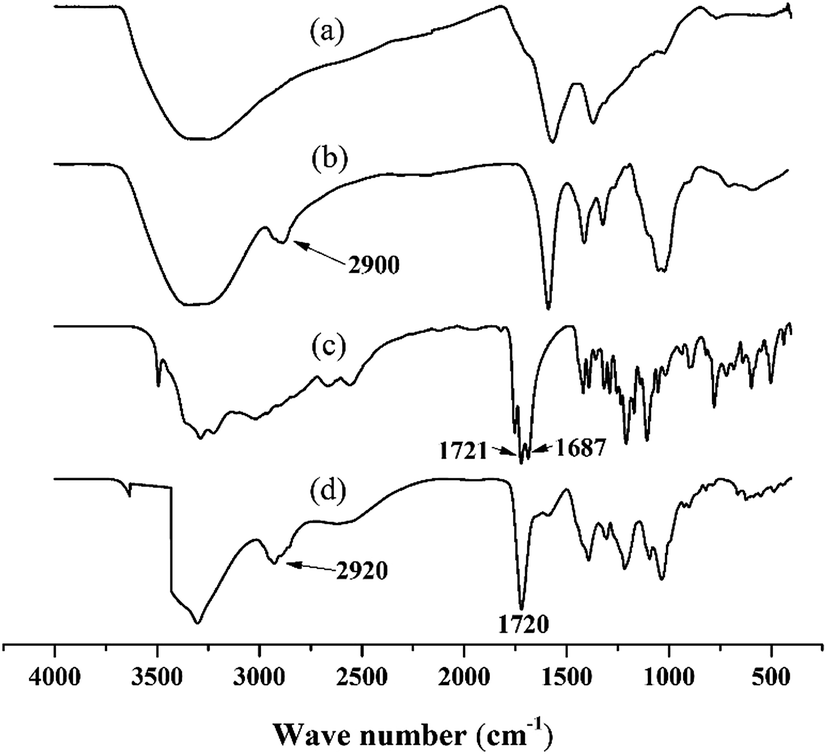 | ||
| Fig. 2 Comparative FT-IR spectra for PGAL (a); CMC (b); AC (c); and PGAL-co-CMC (d). | ||
Solid-state EPR in films was implemented to detect the signal of unpaired electrons in the “hyperfine structure” owing to the interaction of the magnetic momentum of any unpaired electron and the vicinal nucleus with spin number different to zero (Fig. 3). This method allowed non-bonding electrons to be detected qualitatively through the magnetic excitation of the phenolic –OH moieties. The obtained results were compared to solid state EPR for PGAL measured under the same conditions. The analyses display an intense signal for PGAL, which decays for PGAL-co-CMC films. This feature can be reasonably attributed to the decrease of free –OH moieties because of the cross-linking, as shown in Fig. 3A and B, respectively.
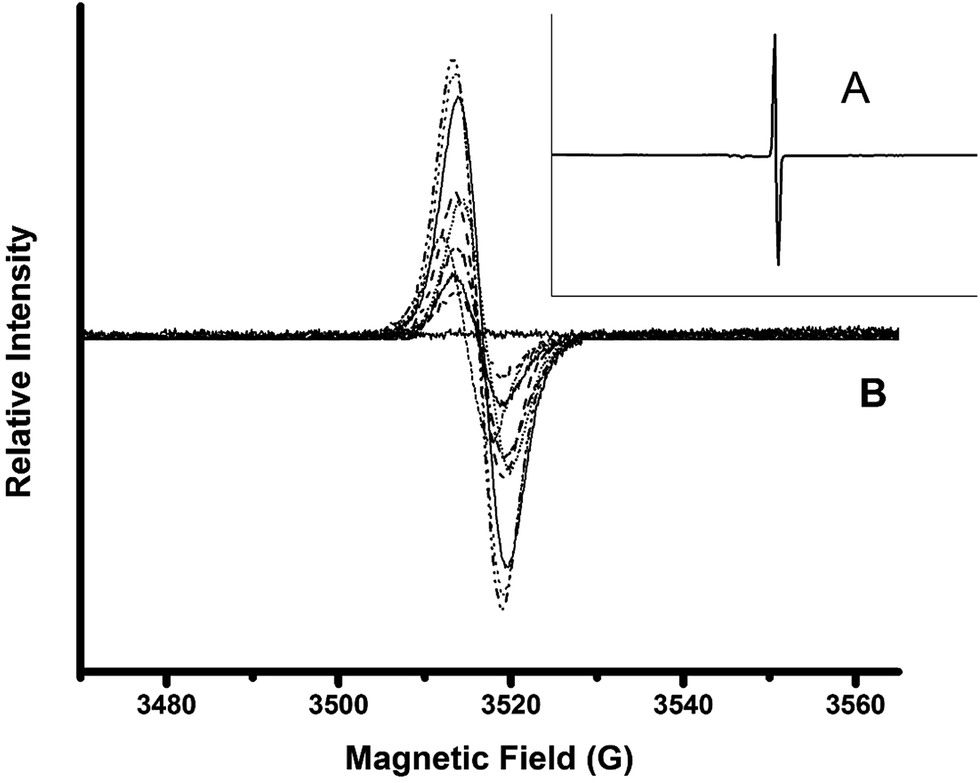 | ||
| Fig. 3 Solid state EPR spectra for PGAL (A); and for the PGAL-co-CMC films (B). Decrease in PGAL feed decrease paramagnetic field intensities. Control measurement without sample displays a flat continuous line. | ||
The comparison between EPR spectra of films points out that the signal intensities are related to the PGAL feed. The decrease in PGAL feed resulted in the decrease of the paramagnetic field intensities, most intense bands in Fig. 3B correspond to entries 7–9, intermediate to 4–6 and lowest intensity bands to 1–3 in Table 1. Additionally, the intensity decreases as the Gly content increases which might be ascribed to the interaction of the plasticizer with the –OH moieties in the phenolic and CMC repeat units.
Mechanical properties and degree of cross-linking of PGAL-co-CMC films
The data of maximum tensile strength at break and puncture as well as Young's modulus for each of the formulations used in the tensile test are shown in Table 1. Data clearly indicated that PGAL and Gly concentrations had a significant influence on the properties of the final product. These results might suggest that besides of the cross-linking, the semi crystalline CMC allows generating ion-dipole and hydrogen bonding interactions between its chains. However, keeping in mind that this polyphenol has exposed hydroxyl and carboxyl moieties in each phenolic repeat unit, the dipole–dipole interactions might be favoured, thereby improving the tensile strength. The density of cross-linking of the polymer matrices in deionized water has been studied as an indirect method to estimate the degree of cross-linking as reported by Xing et al.27 The results shown in Table 1 substantiate the effect of PGAL in the cross-linking which can be ascribed to the pendant hydroxyl and carboxyl moieties at the polyphenol backbone. Contrarily, control experiments without PGAL addition, entries 10–12 in Table 1, displayed poor bonding among CMC chains. This was also evidenced when comparing the data of equilibrium swelling ratio (Qm) and percentage of swelling among samples (see ESI data file 4† for Qm and percentage of swelling for entries 1–12 in Table 1 and a graphical representation of the swelling percentage for each entry in Table 1).Additionally, the strength at break is also affected by the amount of plasticizer. The experimental evidence suggests that the three hygroscopic compounds with high hydrogen-bonding moieties might be advantageous in dermal regeneration studies, where skin hydration is a crucial factor. The fracture force, true stress, is influenced together with the percentage of PGAL and by the amount of Gly, although the highest contribution of the latter is 4 wt% to enhance the flexibility. Therefore, the increase in the amount of the plasticizer could decrease the material strength by limiting the relatively weak intermolecular interactions between chains.28 However, this is not observed in PGAL-co-CMC, which might be ascribed to the cross-linking.
Regarding the Young's modulus, all formulations can be classified as soft materials according to ASTM standards. The data in Table 1 indicate that the presence of PGAL and plasticizer tend to increase the elastic modulus. Non-cross-linked polymers have a greater free volume and consequently more freedom of movement. This condition could explain the increase in flexibility. Also, the plasticizer can contribute to molecular mobility and therefore increase the flexibility and elongation at break. Control experiments without PGAL allowed the separate effect of the polyphenol and plasticizer to be observed. Therefore, it can be concluded that the increase in Gly concentration results in more tolerance to stretching, i.e. more capacity to withstand strain. This plasticizing effect can be interpreted as an enhanced flexibility (the measured elongation for each entry in Table 1 is shown in ESI data file 5† to exemplify both effects). On the other hand, the increase in PGAL concentration resulted in higher tensile strength, regardless of Gly concentration. This means films with higher resistance to fracture. Noteworthy, Young's modulus is less sensitive to changes in polyphenol and plasticizer concentration but indicates the intrinsic elastic character or individual films, below their elastic limit.
The true stress to puncture for all formulations displays a remarkable influence of PGAL and Gly initial concentrations. However, puncture and tensile strength are not comparable and must be considered for each specific application of the films. For tissue engineering, specifically in skin regeneration, the elastic characteristics of our films are superior to those of the human skin, which Young's modulus varies between 4.2 × 105 Pa to 8.5 × 105 Pa for young and old human skin, respectively.29 Therefore, although the forces in human skin and that in our mechanical analyses are not comparable, it can be concluded that the material presents good qualities for application in tissue engineering.
Thermal characterization
TGA analyses (ESI data file 2†) demonstrated the thermal stability of the crosslinked polymers since an appreciable degradation was only observed when the temperature was higher than 200 °C. All samples showed an initial weight loss that can be associated with the adsorbed water (i.e. note the peak at 100 °C in the DTGA curve). This loss was always close to 10% and indicates a higher affinity of PGAL-co-CMC polymers with water than PGAL (a loss of only to 5% was determined in this case) which might be ascribed to the ratio of hydroxyl groups in the repeat units. Decomposition of PGAL was characterized by a complex mechanism that followed at least two degradation steps with DTGA peaks at 230 and 320 °C and rendered a very high char yield (49%) due to the high aromatic content of PGAL. Crosslinked polymers showed also an intermediate degradation process associated with CMC that rendered an absolute and distinctive DTGA peak at 265 °C. Char yield decreased for example to 12% for PGAL-co-CMC entry 9 and to 16% for entry 1, values that clearly contrasted with that attained with the aromatic PGAL component alone. Decrease on the char yield was logically observed when samples incorporated higher amounts of the volatile Gly component and had a lower content of PGAL.DSC heating runs showed that samples were amorphous, displaying only a clear glass transition at temperatures near 0 °C when the PGAL content was low (e.g. 5 wt%) (see ESI data file 2† for TGA and DSC traces). This temperature slightly increased with the PGAL content and was also influenced by the incorporation of the plasticizer Gly component. The highest value was logically attained for the sample having maximum PGAL (i.e. 15 wt%) and minimum Gly (i.e. 2 vol%) content, being the observed Tg significantly lower than the one observed for the PGAL homopolymer (i.e. 42 °C respect to 88 °C). The increase of the Gly content to 4 vol% decreased the glass transition temperature to 25 °C. In this way, flexible PGAL–CMC samples could be easily prepared if an appropriate amount of Gly was incorporated. Despite having a crosslinked structure, PGAL-co-CMC samples were more flexible than PGAL due to their lower aromatic content and the incorporation of a small ratio of plasticizer molecules.
WVP and contact angle analyses on PGAL-co-CMC films
Two PGAL-co-CMC samples, entry 1 and 2 in Table 1 were selected for the WVP analyses based on ASTM E-96 standard calculations and the correction method proposed by Gennadios in 1994,30 as shown in Table 2. The increase of Gly increases the WVP in films owing to the lowered intramolecular forces, which in turn increases the free space and therefore allow water molecules diffusion through the macromolecular structure.28However, the results obtained differ from those reported for linear polysaccharides and are in the order of magnitude of low-density polyethylene and waxes at the same temperature and humidity. This result suggests that cross-linking with PGAL restricts the diffusion of water molecules throughout the film. Noteworthy, films swelled and showed no weight gain while placed in the humidity-controlled chamber (50% RH).
Fig. 4 shows the contact angle behaviour of PGAL–CMC films. The increase of PGAL tends to increase the contact angle, which might be ascribed to the hygroscopic characteristics of PGAL. Samples with the lower amount of PGAL (i.e. 5%) showed similar contact angles to the CMC film (16.5°). However, the contact angle decreases by the increase in plasticizer, and this was most noticeable when the concentration of PGAL was high (i.e., 10% and 15%).
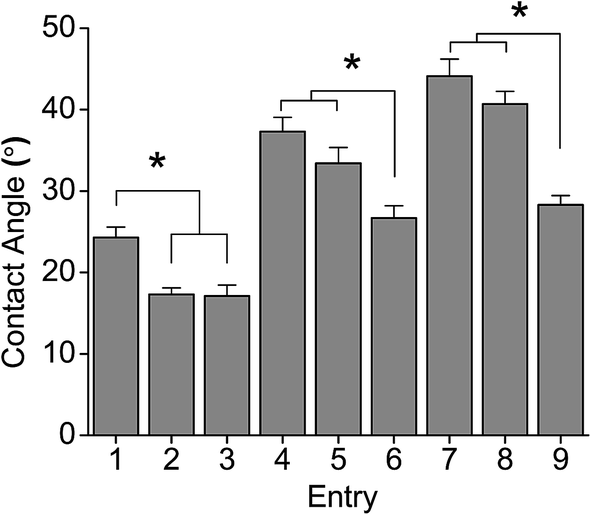 | ||
| Fig. 4 Contact angle measurements on PGAL-co-CMC film sample entries in Table 1. *p < 0.05 determined by ANOVA of means of multiple comparisons by Tukey test. | ||
This behaviour points out that the plasticizer plays an important role in the hydrophilic characteristics of the films (see ESI data file 3† for contact angle to glycerol and PGAL relationships).
Hydrolytic degradation
The in vitro hydrolytic degradation of PGAL-co-CMC films was conducted at the physiological temperature (37 °C) and pHs of 3, 7 and 10 that were provided by the universal buffer consisting of citrate-phosphate-borate/HCl solutions.31,32 The degradation was studied in film samples of entries 7, 8 and 9 in Table 1 owing to the highest content of PGAL thereby to assess its effect compared to that for CMC, as reported elsewhere,33 and the results are shown in Fig. 5. In all cases, the samples showed a very fast weight loss at the beginning of exposure (i.e. 42–72% after only three days) and a subsequent moderate weight loss between 7 and 15 days when a plateau was reached. Results pointed out the first process that involved the dissolution of the excess of CA and the added Gly plasticizer as well as the degradation and rapid dissolution of molecules with a low degree of cross-linking. The differences observed between samples with different Gly content after three days of exposure can be justified considering the dissolution of the plasticizer and the increase in the matrix hydrophilicity that facilitates the diffusion of water molecules and degradation products through the films. Note that the weight loss at three days of exposure clearly increased with the Gly content (e.g. 42, 52 and 62% for entries 7, 8 and 9, respectively, for pH 3). After the indicated three days of exposure, the degradation behaviour was still dependent on the initial Gly feed, suggesting that some plasticizer molecules remained in the films due to the strong hydrogen bonding interactions between CMC and PGAL backbones. Thus, the weight loss increased for the pH 3 medium from 42 to 60%, 52 to 79% and 62 to 95% for 2, 3 and 4 vol% Gly feed, respectively.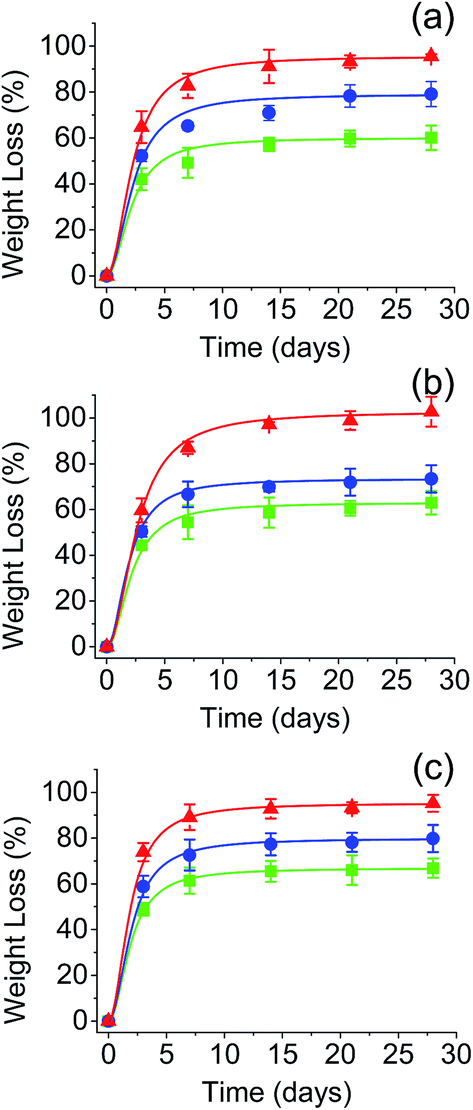 | ||
Fig. 5 Effect of pH in the hydrolytic degradation at 37 °C of the PGAL-co-CMC films. (a) pH 3, (b) pH 7, and (c) pH 10. Symbols (■), (●) and (▲) corresponds to samples with PGAL/Gly vol% ratios of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (entry 7), 15:3 (entry 8) and 15:4 (entry 9), respectively. :2 (entry 7), 15:3 (entry 8) and 15:4 (entry 9), respectively. | ||
It is worth noting that the pH of the medium had a slight influence on the degradation behaviour despite the presence of ionic COO− groups in PGAL and CMC. Logically, ionization should influence on the molecular solubility and molecular conformation. In fact, it is well known that the conformation of CMC depends on the pH of the medium forming dense aggregates in solution at low pHs; i.e. below its pKa,33 while extended chain conformations are favoured in the presence of carboxylate ions. Data displayed in Fig. 5 also show that the main difference between samples degraded at different pHs corresponds to the initial dissolution, which is highest at pH 10.
Specifically, weight losses increased up to 48, 59 and 72% for entries 7, 8 and 9, which were samples loaded with 2, 3 and 4 vol% of Gly, respectively. Contrarily, weight losses attained after 15 days were comparable when the pH was varied. For example, 60, 63 and 67% were determined for entry 7 at pHs 3, 7 and 10. Noteworthy, the weight loss of these samples in the period between 3 and 15 days was rather constant (i.e. 18, 20 and 19% for pHs 3, 7 and 10, respectively). In summary, hydrolysis of ester groups was not significantly changed by pH to effectively reduce cross-linking and render soluble fragments.
On the other hand, Fig. 6 shows different images taken during the process of dissolution and hydrolytic degradation of the films.
 | ||
| Fig. 6 SEM micrographs of PGAL-co-CMC film entry 9 in Table 1. Initial sample (a) and after 3 days (b and c), 7 days (d and e) and 14 days (f) of exposure to universal buffer pH 7 at 37 °C. | ||
Films prepared by solvent evaporation had a smooth and homogeneous surface as depicted in Fig. 6a. At the beginning of exposure to the hydrolytic medium (i.e. three days), the polymer matrix was slightly swelled as revealed by the ampoules formed on its surface and that can be clearly observed in the low magnification images (Fig. 6b).
In addition, small surface erosions are observed at high magnification (Fig. 6c). These became progressively deep and giving rise to holes and cracks, while at low magnification, a terrace structure derived from the hatching of the previously indicated ampoules/bubbles can be detected. Finally, the remaining material shows again a smooth surface with some reminiscences of the terrace structure (Fig. 6f).
Effect of the PGAL-co-CMC films in the bacterial growth
Fig. 7a shows the formation of bacterial growth inhibition halos for samples with different composition and different bacterial strains. This qualitative observation demonstrates the antibacterial activity against both Gram-positive (e.g. Staphylococcus aureus and Enterococcus faecalis) and Gram-negative bacteria (e.g., Escherichia coli and Pseudomonas aeruginosa). The qualitative assay showed scarce differences among the PGAL-co-CMC film samples with different PGAL content and more logically between samples with different Gly content.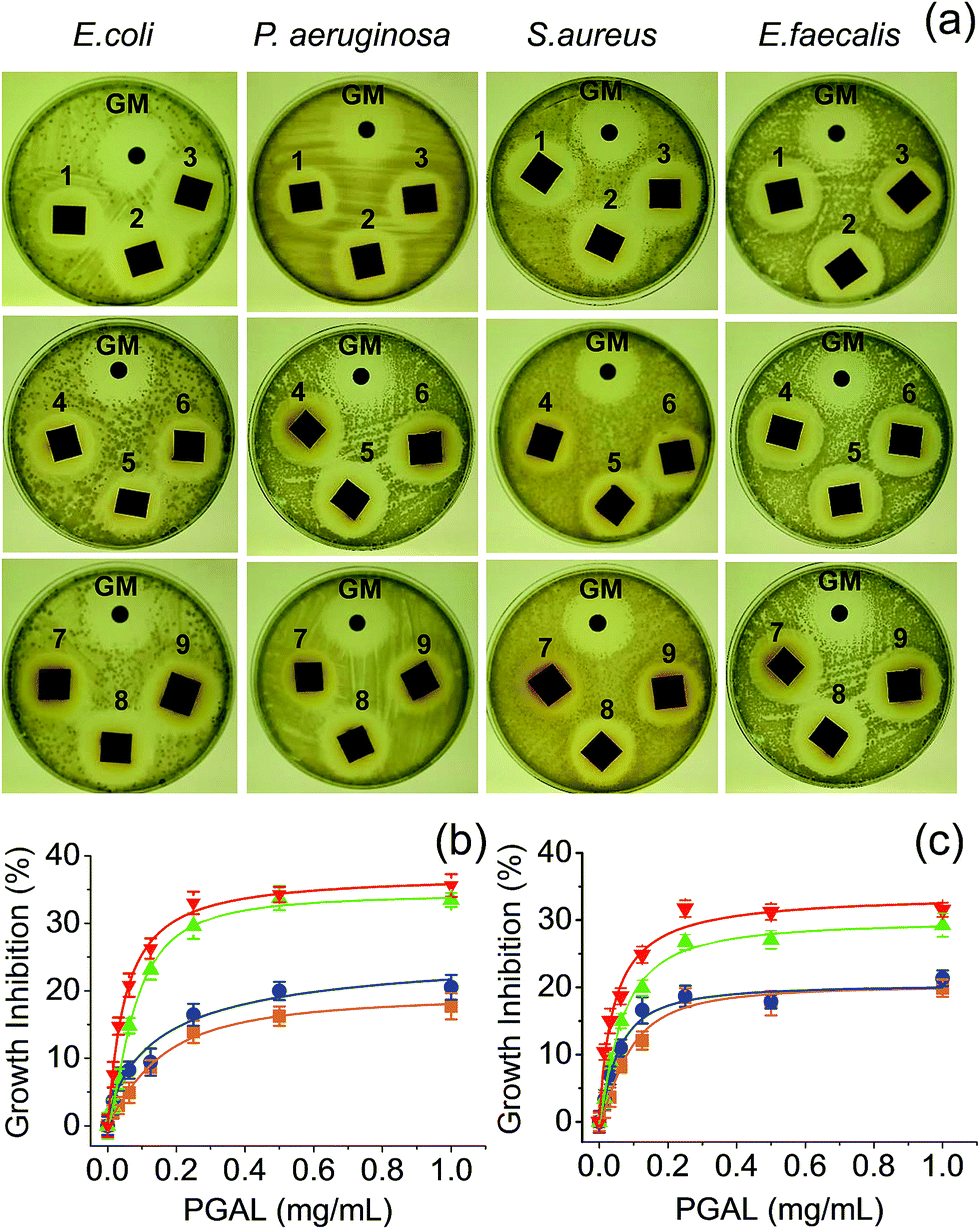 | ||
| Fig. 7 Inhibition of the bacterial growth. (a) Halos of inhibition produced by the PGAL-co-CMC films. The samples named 1–9 corresponded to the entry in the Table 1, and GM is the gentamicin control disk. (b and c) Growth inhibition by PGAL after 24 and 48 h of bacterial culture (b and c, respectively). Symbols E. coli (■), P. aeruginosa (●), E. faecalis (▲) and S. aureus (▼). | ||
Quantitative measurements indicate that the antibacterial effect is limited because the entire bacterial population was not eliminated, even at the highest assayed concentration of PGAL (1 mg mL−1). Results also showed two features concerning the antibacterial action of PGAL; first, the activity is only bacteriostatic because it reduces the population by 35 and 20% at 24 h of culture (Fig. 7b) and around 30 and 15% at 48 h (Fig. 7c) for Gram-positive and Gram-negative bacteria, respectively. Second, the antibacterial activity has greater effectiveness against the tested Gram-positive strains. The decrease in antibacterial activity as the culture time progresses is consistent with the decrease of the bioactive ingredient. Finally, the diffusion on the agar plate could consider a concentration smaller than 1 mg mL−1, and this would explain the similarity of the sizes of the inhibition halos for all samples as shown in Fig. 7a.
Cytotoxicity of PGAL and cell viability on PGAL-co-CMC films
Fig. 8a displays the cytotoxicity experimental curves for PGAL, which are characterized by a typical sigmoidal form. Curves allow determining in the part of the linear slope, the cytotoxic dose (LD50) that corresponds to the region of linear dependence between cell viability percentage and PGAL concentration. This dose varied in the 0.2–0.6 mg mL−1 range for the different tested cell lines except for MDCK epithelial-like cells that required a remarkable higher dose (i.e. 1.1 mg mL−1). Interestingly, these cells are characterized by their monolayer configuration and consequently to a capability to form a protective cell barrier that justifies the relative higher dose needed to produce a cytotoxic effect. Indeed, a usual dose is found when this cell barrier is lost as it is for example the case of the SIAT cells derived from MDCK that had an LD50 value of 0.4 mg mL−1. It is also interesting to compare the values obtained from epithelial lines derived from prostate epithelium. Thus, the PNT2 line derived from a normal tissue shows a higher LD50 (0.3 mg mL−1) than the PC-3 prostate tumor line (0.2 mg mL−1). Finally, the Vero line, which is the one that forms the most reduced cell barrier shows the lowest LD50 (0.2 mg mL−1). On the other hand, a slightly higher LD50 (i.e. 0.6 mg mL−1) was determined for fibroblast type cells such as Cos-1.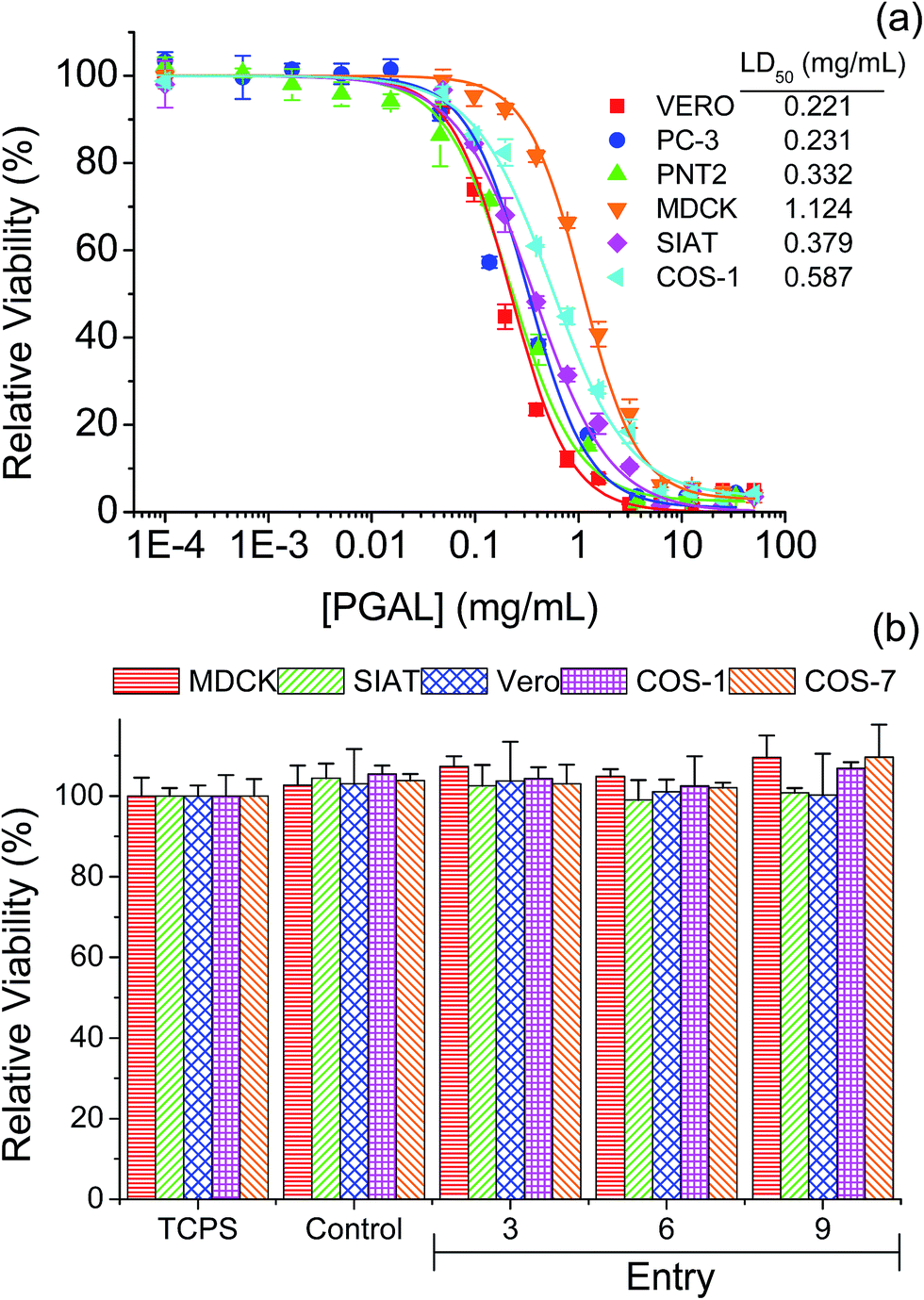 | ||
| Fig. 8 Cytotoxicity curves of PGAL (a). Cell viability on films of PGAL-co-CMC (50 mg mL−1), cells was evaluated at 96 h of culture (b). | ||
The viability of the seeded cells on different PGAL-co-CMC (50 mg mL−1) films is shown in Fig. 8b. The test was performed considering PGAL contents of 5, 10 and 15 wt% with the maximum load of the plasticizer, entries 3, 6 and 9 in Table 1, respectively, to assess the effect of the PGAL contents. In all cases, films do not show cytotoxic effects on cells seeded on their surface and led to viability entirely comparable with that determined for the surface of the culture plate (TCPS) and also to the CMC matrix used as a control. Thus, it is noteworthy that a high viability after 96 h of culture has been detected for all tested cell line (e.g. epithelial-like MDCK, SIAT and Vero cells and fibroblasts-like Cos-1 and Cos-7 cells).
The lack of toxicity of the studied samples implied also that the weight loss detected during exposition of the studied samples in the hydrolytic degradation media at pH 7 and 37 °C mainly corresponds to soluble materials different than PGAL. Note that the weight loss is around 50% for the indicated period and the LD50 occurs at a concentration of 0.2–0.6 mg mL−1. Two points were considered to establish this cultivation as a control point. First, the kinetics of hydrolytic degradation at pH 7 and 37 °C, corresponding to conditions in which about 50% of the weight loss of the material can occur and second, the LD50 of PGAL occurs at a concentration of 0.2–0.6 mg mL−1. Thus in theory, for a sample of approximately 50 mg cultured in 1 mL of medium, the maximum release of PGAL would correspond to a concentration of 1.25, 2.5 and 3.75 mg mL−1 for samples having 5, 10 and 15 wt% of PGAL, respectively. These values clearly exceeded the concentration corresponding to LD50, demonstrating that most of PGAL was retained in the cross-linked film and the safety of the films as substrates for cell adhesion and growth. The morphology of the cultured cells on the surface of the films is shown in Fig. 9. As an example, the monolayer arrangement of epithelial-type Vero cells is shown on the surface of CMC (control, Fig. 9a) and on surfaces of the studied PGAL-co-CMC films (Fig. 9b–d). Other epithelial cell lines such as MDCK and SIAT were also characterized by the formation of monolayers on the surfaces of PGAL-co-CMC films. Finally, the distribution of more isolated cells on this surface is shown for fibroblast cells such as Cos-1 and Cos-7.
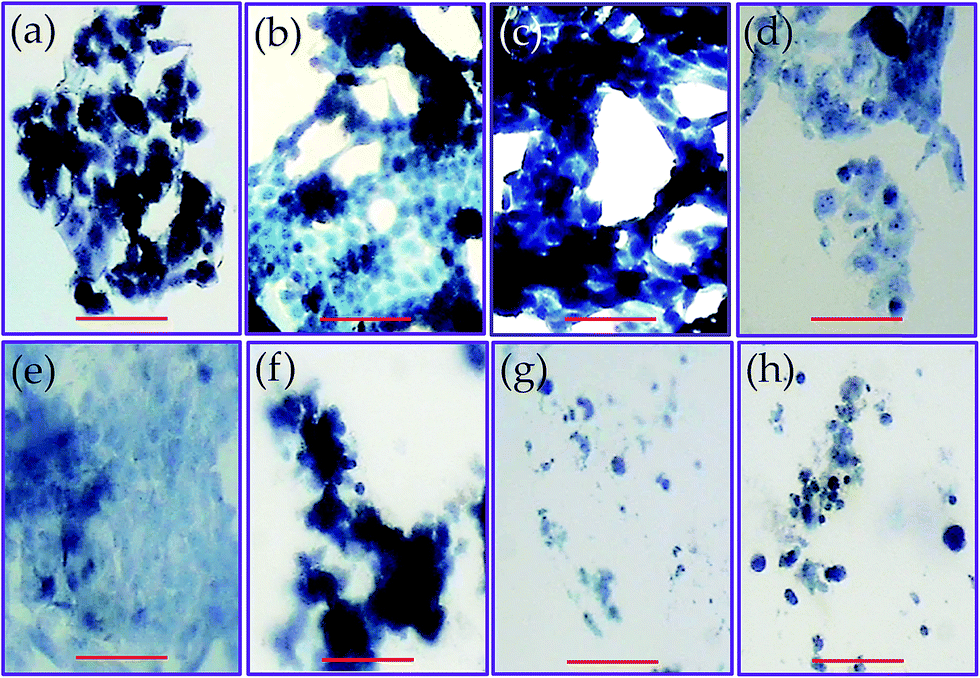 | ||
| Fig. 9 Images of cell proliferation on films of PGAL-co-CMC. Vero cells (epithelial-like cells) on the CMC film or control (a), and films of entry 3 (b), entry 6 (c), and entry 9 (d). Epithelial-like cells on (entry 9) films: MDCK cells (e), and SIAT cells (f). Fibroblast-like cells on the films (entry 9) films: Cos-7 cells (g), and Cos-1 cells (h). Cells were stained with trypan-blue and observed by optical microscopy (scale bar = 20 μm). | ||
Conclusions
The use of citric acid as a cross-linking agent made possible to obtain a novel copolymer of PGAL and CMC. The experimental evidence demonstrates that the PGAL-co-CMC films are potential candidate biomaterials for skin tissue regeneration studies. Skin cells were successfully grown on films of this material. The films were non-toxic and presented antioxidant and antimicrobial properties, which are desired features in dermatological and chirurgical fields. Further work toward the application of these materials in skin tissue regeneration is in progress.Experimental
Materials
LTV (lyophilized protein stored at −20 °C) was purchased to Fluka (USA). Specific enzymatic activity was determined by the 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) method; 50 μL of an aqueous enzyme solution (10 mg mL−1) was added to 2.9 mL of ABTS solution (9.1 mM) in phosphate buffer (pH 5, 100 mM). Solutions were measured spectrophotometrically in a Thermo Scientific Genesys 10S UV-vis (USA) at 405 nm (25 °C). Enzymatic activity unit (U) is defined as the amount of protein required to oxidize 1.0 μmol of ABTS per minute at 25 °C and pH = 5 (E405 ABTS = 36.8 M−1 cm−1). The LTV used in this work presented a specific enzymatic activity of 2.35 U g−1. GA, ABTS, glycerol (Gly), DPX mountant for histology and 2,2-diphenyl-1-picrylhydrazyl (DPPH) were supplied by Sigma-Aldrich (USA). CMC (sodium salt) was supplied by Química Hércules SA de CV (Mexico). CA, calcium chloride, monobasic sodium phosphate and sodium hydroxide were acquired from JT Baker Mexico. Ethyl alcohol (technical grade, >99%) was supplied by Química Barsa SA de CV (Mexico).Enzymatic synthesis of PGAL
PGAL was produced following the procedure described by López et al. in 2013 with some modifications.23 In a typical experiment, GA (8.5 g) was dissolved in 250 mL of acetate buffer (0.25 M, pH = 5) with the addition of 20 mL of NaOH (2 M) in a 500 mL three-necked round-bottom amber flask equipped with a magnetic stirrer at 25 °C. Constant oxygen concentration was kept by an Elite 800 air pump (Hagen Inc, Canada) and monitored by an Apliens ez-DO sensor (Applikon, USA). Then, LTV was added (26.5 U), and the reaction was stirred allowed to stir at 25 °C for 24 h. Reaction contents were precipitated dropwise into 2 L of cold ethanol (5 °C) under stirring, vacuum filtered (0.45 μm) and dried in Stable temp Cole Palmer (USA) vacuum oven connected to a Vacuubrand PC3 RZ 2.5 system at 10 × 10−4 mbar at 50 °C for 2 h to give PGAL as powder in 96% yield.Cross-linking of PGAL with CMC using CA
In a typical experiment; CMC (1 g) was dissolved in 50 mL of water at 80 °C under stirring in a 100 mL round-bottom flask. Then, experiments were conducted with different amounts of PGAL; 5, 10 and 15 wt% with respect to CMC. Then, 6.5 × 10−3 mol (1.25 g) of CA and 30 mg of monobasic sodium phosphate were added to the stirred solution and allowed to react for 2 h at 80 °C. Gly was added in different proportions of 2, 3 and 4 vol% (wet basis) and stirred at 80 °C for further 15 min. Reaction contents were poured into polyethylene Petri dishes (25 mL per sample) and dried at room temperature until complete evaporation of the solvent. Films were removed from the Petri dishes and neutralized to remove the excess of CA before cell viability studies.Chemical characterizations
1H-NMR spectra were recorded in a Varian MR-400 spectrometer in D2O. FT-IR spectra of PGAL and PGAL-co-CMC films were acquired in a Perkin-Elmer ATR-FTIR Spectrum 400 instrument. Size exclusion chromatography (SEC) measurements were conducted in an Agilent 1210 series (USA) liquid chromatography appliance equipped with an RID detector and a Ultrahydrogel-500 (7.8 × 300 mm, Waters, USA) column eluted with deionized water containing LiCl (0.1 M) mobile phase at 0.8 mL min−1 and 30 °C. Standard narrow molecular weight polyethylene glycol (PEG) was used for calibration for PGAL. Pullulan standards were used for CMC. Samples were dissolved in the mobile phase (1 mg mL−1) and filtered (0.45 μm) before injection in the chromatographer. Solid state EPRs for PGAL and PGAL–CMC films were conducted in a Bruker EPR Elexsys E500 spectroscope with PGAL (100 mg) or sections of PGAL-co-CMC films placed in a quartz tube a room temperature in the 80–4080 G range of magnetic field and 9.8 × 109 Hz microwave frequency.Cross-linking density
Samples were allowed to swell in deionized water at room temperature and the weight change was monitored until swelling equilibrium volumes (Vs) after 48 h by displacement of pure toluene and measured in a pycnometer (Brand, Germany) using eqn (1).27
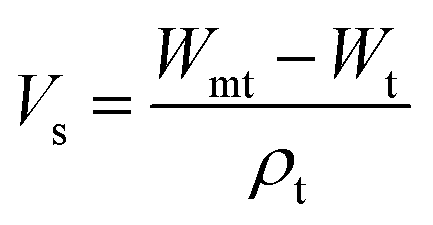 | (1) |
Then, equilibrium swelling ratio (Qm) was determined by eqn (2).
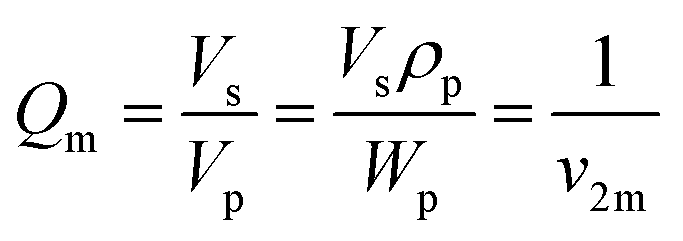 | (2) |
The Flory–Huggins swelling equation for ionic polymer was adapted for isotropic equilibrium swelling of an ionic or amorphous crosslinked polymer hydrogel, thus adjusting for crosslinked polymer in solution with eqn (3).
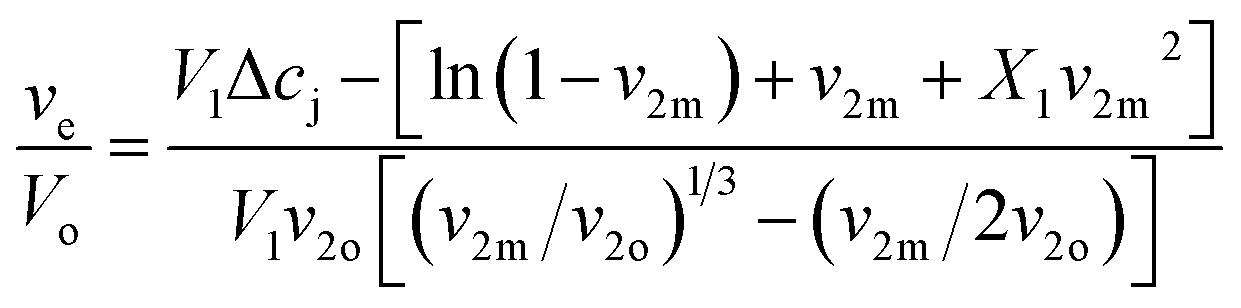 | (3) |
Mechanical analyses of PGAL–CMC films
All mechanical analyses were conducted and processed as specified in the American Society for Testing Materials Methods (ASTM).34 Films were preconditioned before analyses by placing them in a humidity-controlled chamber according to ASTM D618 method (50 ± 5% RH and 23 ± 2 °C) for 24 h. Then, the thickness of films was measured with a Cole-Parmer 08204-80 micrometer at five different and random points (4 at the perimeter and one in the centre). Films were subjected to tensile, and puncture tests using a SINTECH 1/S (MTS, USA) with a load cell of 100 N. Tensile test was performed at room temperature following the ASTM D882-97 standard. In a typical experiment; films were cut into samples of 1 cm wide by 8 cm long and clamped in the jaws to a length of 5 cm. Samples were assessed at an extension rate of 150 mm min−1 to record the material's resistance to breaking. All analyses were conducted by quintuplicate for each formulation. Fracture force to puncture was determined according to the method described by Gontard et al. in 2003,28 which specifies an adjustment for polysaccharide-base matrices with the use of Gly as the plasticizer. In a typical experiment; the preconditioned films were cut into circles of 10 cm in diameter, attached to a metal ring and subjected to the force of a puncture in an area of 1.3 cm diameter at a descent rate of 50 mm min−1. Samples were analysed by quintuplicate for each formulation. Results reflect the average of 5 replicates (n = 5 for each experiment). Control experiments were identically carried out without the addition of PGAL.Water vapour permeability and contact angle measurements
Water vapour permeability (WVP) was determined gravimetrically following the ASTM E96-00 standard, upon the transmission coefficient of water vapour by monitoring the weight increase in time relative to moisture passing through the film. Films were cut and placed in pre-conditioned cells which were filled with anhydrous calcium chloride (previously dried at 200 °C to ensure 0% RH) leaving an air gap of 0.5 cm on its surface. Films were attached to an acrylic ring using four screws placed symmetrically and a layer of silicone to maintain air tightness. After placing the film, the initial weight was recorded and placed in a humidity-controlled chamber (50% RH and 22 °C). The humidity difference between the inside of the cell and the chamber provided the driving force for the vapour to pass through the film toward the desiccant. Cells were removed every 24 h to record the weight gain in an analytical balance (Ohaus Model GA200, Germany) with a precision of 0.0001 g for three months. Analyses were done by quintuplicate for each formulation and data were analysed according to the ASTM standard and using a correction for hydrophilic materials (e.g. polysaccharides).30 Contact angles were measured at room temperature with sessile drops using an OCA-15 plus Contact Angle Microscope (Dataphysics, USA) and SCA20 software. Contact angle values of the right and left sides of distilled water drops were measured and averaged. Measurements were performed 5 s after the drop (1 μL) was deposited on the sample surface. All data were an average of ten measurements on different surface locations.Hydrolytic degradation of films
Polymer film sections (1 × 1 cm2) were employed for the different degradation studies. In vitro hydrolytic degradation assays were carried in universal buffers at pH 3, 7 and 10 at 37 °C and 70 °C.32 Samples were kept under orbital shaking in bottles filled with 5 mL of the degradation medium and sodium azide (0.03 wt%) to prevent microbial growth for selected exposure times. Then, samples were thoroughly rinsed with distilled water, dried to constant weight under vacuum and stored over P4O10 before analysis. Degradation studies were performed in triplicate and the given data corresponded to the average values. Weight retention (Wr) of the specimens was determined by the percentage ratio of weight after degradation (Wd) to initial weight before degradation (W0) according to eqn (4).
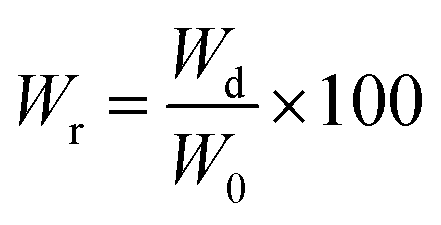 | (4) |
Thermal characterizations
Calorimetric data were recorded by differential scanning calorimetry (DSC) using a TA Instrument Q100 series equipped with a refrigerated cooling system (RCS) operating from −90 to 550 °C. Experiments were performed under a flow of dry nitrogen with a sample of ≈5 mg. The instrument was calibrated for temperature and heat of fusion using an indium standard. Heating and cooling runs were carried out at a rate of 20 °C min−1 and 10 °C min−1, respectively. Thermal degradation was studied at a heating rate of 20 °C min−1 in a Q50 Thermogravimetric Analyzer (TGA) of TA Instruments with ranged test temperatures from 0 to 600 °C.Antimicrobial tests
E. coli, P. aeruginosa, S. aureus and E. faecalis bacteria strains (CECT, Valencia, Spain) were selected to evaluate the antimicrobial effect of PGAL and PGAL-co-CMC films. Strains were previously grown aerobically to exponential phase in broth culture (5 g L−1 beef extract, 5 g L−1 NaCl, 10 g L−1 peptone, and pH 7.2). Growth experiments were performed in sterile Eppendorf microtubes. 1 mL of different concentrations of PGAL prepared in culture medium was added into each microtube. Then, 0.1 mL of broth culture containing 103 CFU was seeded into the microtubes. Cultures were incubated at 37 °C and agitated at 80 rpm. Aliquots of 50 μL were taken at predetermined time intervals for absorbance measurement at 650 nm in a microplate reader (Biochrom Ltd., UK). Turbidity was directly related to bacterial growth. All assays were conducted in six replicates and the values averaged. Regarding the qualitative method, the culture medium was prepared with LB agar and sterilized at 121 °C for 30 min in an autoclave. Plates were filled with 15 mL of medium and kept at rest to solidify the medium at room temperature. Agar diffusion plate were seeded with 104 UFC mL−1 of each bacterium separately and using a nylon swab, then the 1 × 1 cm2 samples of PGAL-co-CMC films were placed onto agar surface. A commercial gentamicin (GM) disc was used as a positive control. Inhibition halos images were taken after incubation of the plates for 24 h at 37 °C.In vitro cytotoxicity and cell viabilities
Vero, PC-3, PNT2, MDCK, SIAT, Cos-7 and Cos-1 cells (ATCC, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 2 mM of L-glutamine at 37 °C in a humidified atmosphere with 5% CO2 and 95% air. Culture medium was changed every two days and, for subculture, cell monolayers were rinsed with phosphate buffered saline (PBS) and detached by incubation with trypsin EDTA (0.25%) at 37 °C for 2–5 min. Cell concentration was determined by counting the number of cells with a Neubauer chamber and employing 4% trypan blue as a dye vital. Detached cells with viability ≥ 95% were used for cell culture assays. To determine the cytotoxic concentration (LD50) of PGAL, the viability and cell death (mortality) were determined by different concentrations of PGAL prepared by dilution in culture medium. PGAL cytotoxicity was determined from 200 μL aliquots containing 104 cells that were seeded in each well in a 96-wells culture plate and cultured for 24 h to allow cell attachment to the plate. Afterward, the medium was aspired and new samples were prepared in half dilution by adding fresh culture medium. Cellular viability was evaluated after another 24 h of incubation and determined by the MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium; 5 mg mL−1) and then, the relative viability was calculated for each sample as the percentage quotient between the viability of the sample and the control (medium without PGAL). Cytotoxicity was evaluated using five replicates and the results were averaged and graphically represented. For the proliferation assays on the PGAL-co-CMC films, the procedure was as follows; samples of 1 × 1 cm2 of the films were sterilized by exposure to UV light for 15 min. Samples were then adhered using a small drop of silicone (Silbione® MED ADH 4300 TRV, Bluestar Silicones France SAS, Lyon, France) in the wells of a 24-well culture dish. Then, 1 mL of culture medium was added and the samples were maintained for 24 h under culture conditions to ensure adequate rehydration of the films. Medium was aspirated and the samples washed two times with sterile PBS. Cells were seeded by depositing an aliquot of 100 μL containing 2 × 104 cells on the surface of the samples (50 mg mL−1). The cultures were maintained for 96 h to allow cell growth, and the media were renewed every two days. Finally, cell viability was determined by the MTT assay. Each sample was evaluated using five replicates and the results were averaged and graphically represented. To obtain representative images from proliferation assays, the samples were fixed overnight with formaldehyde 2.5% in PBS at 4 °C, and then washed five times with PBS. Samples were stained with 4% trypan-blue and the excess of dye was removed by aqueous washings. Samples were placed on a slide and mounted with a drop of DPX, then covered with a cover slip and dried overnight prior to analyses in the optical microscope.Statistical analysis
Values were averaged and shown in tables and graphs, together with their respective standard deviations. Statistical analysis was performed by ANOVA test to compare the means of all groups, and then Tukey's test was applied to determine a statistically significant difference between two groups. The test confidence level was set at 95% (p < 0.05) (OriginPro software, Microcal Corp., USA).Acknowledgements
We thank CONACyT for scholarship (ARM) and funding by project SEP-BASICA 165757. We also like to thank DGAPA PAPIIT for funding project 200115.Notes and references
- R. J. Soto, J. R. Hall, M. D. Brown, J. B. Taylor and M. H. Schoenfisch, Anal. Chem., 2017, 89, 276–299 CrossRef CAS PubMed.
- K. Miki, A. Kimura, T. Inoue, H. Matsuoka, H. Harada, M. Hiraoka and K. Ohe, Bull. Chem. Soc. Jpn., 2015, 88, 792–803 CrossRef CAS.
- Y. Ramota, M. Haim-Zadab, A. J. Dombb and A. Nyska, Adv. Drug Delivery Rev., 2016, 107, 153–162 CrossRef PubMed.
- K. Ariga, K. Minami, M. Ebara and J. Nakanishi, Polym. J., 2016, 48, 371–389 CrossRef CAS.
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 518–521 CrossRef.
- Handbook of Bioplastics and Biocomposites Engineering Applications, ed. S. Pilla, John Wiley & Sons, 2011, vol. 81 Search PubMed.
- A. Espadín, N. Vázquez, A. Tecante, L. T. Dios, M. Gimeno, C. Velasquillo and K. Shirai, J. Appl. Polym. Sci., 2014, 131, 40252 CrossRef.
- V. Coma, I. Sebti, P. Pardon, F. H. Pichavant and A. Deschamps, Carbohydr. Polym., 2003, 51, 265–271 CrossRef CAS.
- M. P. Adinugraha and D. W. Marseno, Carbohydr. Polym., 2005, 62, 164–169 CrossRef CAS.
- H. Kang, R. Liu and Y. Huang, Polymer, 2015, 70, A1–A16 CrossRef CAS.
- P. G. Pietta, J. Nat. Prod., 2000, 63, 1035–1042 CrossRef CAS.
- O. S. Nimmi and P. George, Int. J. Pharm. Pharm. Sci., 2012, 4, 505–510 CAS.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouysegu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed.
- E. Llorens, L. J. del Valle, A. Díaz, M. T. Casas and J. Puiggalí, Macromol. Res., 2013, 21, 775–787 CrossRef CAS.
- E. Llorens, L. J. del Valle and J. Puiggalí, Macromol. Res., 2014, 22, 388–396 CrossRef CAS.
- Y. Zheng, W. B. Song and L. J. Xuan, Org. Biomol. Chem., 2015, 13, 10834–10843 CAS.
- T. Oguchi, S. I. Tawaki, H. Uyama and K. Kobayashi, Bull. Chem. Soc. Jpn., 2000, 73, 1389–1396 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- M. Jacobsson, J. Malmberg and U. Ellervik, Carbohydr. Res., 2006, 341, 1266–1281 CrossRef CAS PubMed.
- L. Xu, T. Qi, L. Xu, L. Lu and M. Xiao, J. Carbohydr. Chem., 2016, 35, 1–23 CrossRef.
- H. Uyama, H. Kurioka, I. Kaneko and S. Kobayashi, Chem. Lett., 1994, 3, 423–426 CrossRef.
- S. I. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- J. López, J. M. Hernández-Alcántara, P. Roquero, C. Montiel, K. Shirai, M. Gimeno and E. Bárzana, J. Mol. Catal. B: Enzym., 2013, 97, 100–105 CrossRef.
- D. Alonso, M. Gimeno, R. Olayo, H. Vázquez-Torres, J. D. Sepúlveda-Sánchez and K. Shirai, Carbohydr. Polym., 2009, 77, 536–543 CrossRef CAS.
- M. A. Hernández-Valdepeña, J. Pedraza-Chaverri, I. Gracia-Mora, R. Hernández-Castro, F. Sánchez-Bartez, J. Nieto-Sotelo, C. Montiel, K. Shirai and M. Gimeno, Food Chem., 2016, 199, 485–491 CrossRef PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- Q. Xing, K. Yates, C. Vogt, Z. Qian, M. C. Frost and F. Zhao, Sci. Rep., 2014, 4(4706), 1–10 Search PubMed.
- N. Gontard, S. Guilbert and J. L. Cuq, J. Food Sci., 1993, 58, 206–211 CrossRef CAS; ASTM, Standard Test Method for Tensile Properties of Thin Plastic Sheeting, American Society for Testing and Materials, 2002 Search PubMed.
- P. G. Agache, C. Monneur, J. L. Leveque and J. De Rigal, Arch. Dermatol. Res., 1980, 269, 221–232 CrossRef CAS PubMed.
- A. Gennadios, C. L. Weller and C. H. Gooding, J. Food Eng., 1994, 21, 395–409 CrossRef.
- T. Theorell and E. Stenhagen, Biochem. Z., 1939, 299, 416–419 Search PubMed.
- Y. Márquez, J. C. Martínez, P. Turon, L. Franco and J. Puiggalí, Fibers, 2015, 3, 348–372 CrossRef.
- I. Dogsa, M. Tomsic, J. Orehek, E. Benigar, A. Jamnik and D. Stopar, Carbohydr. Polym., 2014, 111, 492–504 CrossRef CAS PubMed.
- American Society for Testing and Materials, Standard test Method for Tensile Properties of Thin Plastic Sheeting, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00883j |
| This journal is © The Royal Society of Chemistry 2017 |