
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual crystal structure and chirality of uridine 5′-monophosphate coordination polymer†
Leilei Gua,
Qi-ming Qiua,
Pei Zhoua,
Liang Haoa,
Minghua Liu b and
Hui Li*a
b and
Hui Li*a
aKey Laboratory of Clusters Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: lihui@bit.edu.cn; Fax: +86-10-68914780-804; Tel: +86-10-68912667
bNational Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: liumh@iccas.ac.cn
First published on 11th April 2017
Abstract
Two types of Cu(II)–UMP-L coordination polymers, {[Cu2(UMP)2(bpda)2(H2O)3]·10H2O}n (1) and {[Cu2(UMP)2(bpe)2(H2O)2]·7H2O}n (2) (UMP = uridine 5′-monophosphate, bpda = 1,4-bis(4-pyridyl)-2,3-diaza-1,3-butadiene, bpe = 1,2-bis(4-pyridyl)ethylene), were designed and synthesized for researching the effect of auxiliary ligand. X-ray single-crystal diffraction analysis revealed that complex 1 is the first example of a 1D parallel double-bridged coordination polymer in a nucleotide–metal coordination complex, while complex 2 is a 2D grid coordination polymer. Based on their crystal structure analysis, their chirality, including molecular chirality, supramolecular helical chirality and auxiliary ligand's extended axial chirality (EAC), was studied by aqueous solution and solid state circular dichroism (CD) spectroscopy. The length and conjugated auxiliary ligand had an influence on the EAC.
Introduction
Metal–nucleotide coordination complexes are important both in chemistry and biology domains.1 These bio-coordination polymers are appealing as templates to mediate the synthesis of inorganic nanoparticles (NPs),2 accumulating heavy metal and rare earth ions in aqueous solution,3 drug delivery,4 regulating enzyme function in cell5 and switching hierarchical levels in biological systems.6 The complexes of adenosine, cytidine, guanosine and thymidine, uridine phosphates are the five most important nucleotide complexes found in living organisms, in which the metal-binding modes for various nucleotides are different.7–9 For the purine base nucleotides, N7 and O donor of the base motif actively coordinate with metal ions.10–12 In contrast, uridine and its analogues are the least active nucleosides, since the N3 of uridine (Scheme 1) can be a potential donor only when it is deprotonated at pH > 9, and usually carbonyl of uridine is not active.11 The coordinating functional group of UMP to metal ions is actually a phosphate group.13 The coordinating ability of nucleotides changes in the following order: GMP > IMP > AMP > CMP = UMP.14 Therefore, the structurally elucidated UMP coordination complexes are very rare.15,16 Recently, we reported the ternary assembly of a UMP coordination complex, {Ni(UMP)(4,4′-bipy) (H2O)3]·H2O}n, and proposed the extended axial chirality (EAC) for the first time.16 The concept relates the axial chirality in organic chemistry to coordination polymers, which describes a chiral conformation of coordination polymer generated by bridge ligands with a limited rotation of aryl–aryl single bonds through coordination interaction and intermolecular interaction with nucleotides, and the symmetry is broken after the extension. Due to the chiral inducer surrounding the axis of auxiliary ligand clockwise, the absolute axial chiral configuration of the auxiliary ligand is P, otherwise is M configuration. In previous study, several types of bipyridyl-based bridge ligands were used to successfully tune the chirality and structure of the complexes.16–18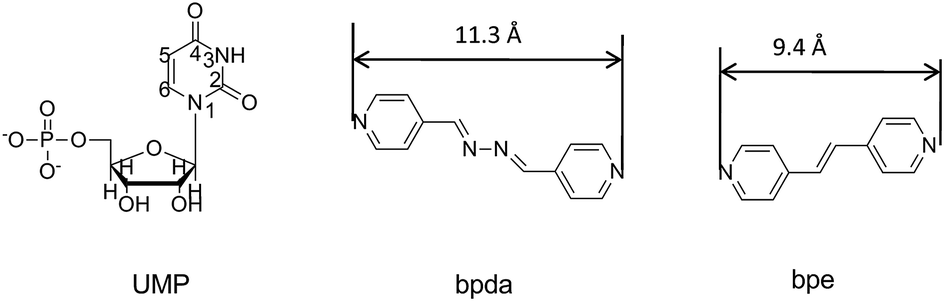 | ||
| Scheme 1 Molecular structures of UMP, bpda and bpe. | ||
In this study, 1,2-bis(4-pyridyl)ethylene (bpe) and 1,4-bis(4-pyridyl)-2,3-diaza-1,3-butadiene (bpda) were chosen to study the length and conjugation of the auxiliary ligand in the structure and chirality of the ternary assembly (Scheme 1). The linker of the two bridge ligands was different, in which bpda with a longer spacer –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC– is a stronger Lewis base (pKb = 8.81) than that of bpe with –CC– (pKb = 9.32).19,20 Their electronic structures were also different and resulted in different π-stacking interactions.21,22 Herein, 1D double-bridged {[Cu2(UMP)2(bpda)2(H2O)3]·10H2O}n (1) and 2D sheet {[Cu2(UMP)2(bpe)2(H2O)2·7H2O]}n (2) were obtained and characterized by X-ray single crystal diffraction. To the best of our knowledge, complex 1 is the first 1D coordinated polymer with double bpda bridges. The chirality was studied based on single crystal structure analysis as well as solution and solid state circular dichroism (CD) spectroscopy.
N–NC– is a stronger Lewis base (pKb = 8.81) than that of bpe with –CC– (pKb = 9.32).19,20 Their electronic structures were also different and resulted in different π-stacking interactions.21,22 Herein, 1D double-bridged {[Cu2(UMP)2(bpda)2(H2O)3]·10H2O}n (1) and 2D sheet {[Cu2(UMP)2(bpe)2(H2O)2·7H2O]}n (2) were obtained and characterized by X-ray single crystal diffraction. To the best of our knowledge, complex 1 is the first 1D coordinated polymer with double bpda bridges. The chirality was studied based on single crystal structure analysis as well as solution and solid state circular dichroism (CD) spectroscopy.
Experimental
Materials and methods
With the exception of the ligands, bpda was prepared according to a literature procedure,19 and all reagents and solvents for the synthesis and analysis were commercially available and used as received without further purification. FT-IR spectra were recorded on a Nicolet-360 FT-IR spectrometer with KBr pellets in the 4000–400 cm−1 region. Elemental analyses (C, H, N) were recorded on a EA3000 elemental analyzer at the Beijing Institute of Technology. Thermo-gravimetric analyses (TGA) were performed on a DTG-60H thermal analyzer from room temperature to 800 °C with a heating rate of 10 °C min−1 in a flowing 50 mL min−1 nitrogen atmosphere. UV-vis spectra were obtained on a TU-1950 spectrophotometer. Powder X-ray diffraction (PXRD) studies of samples were carried out using a Japan Rigaku D/max γA X-ray diffractometer measured with graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å). CD measurements were performed at 25 °C under a constant nitrogen flow on a JASCO J-810 spectropolarimeter. For solid-state (CD) measurement of UMP ligand and complexes 1 and 2, the ratio of crystalline sample and KCl powder was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200, and the mixture was pressed into a disk by a literature method.23
:200, and the mixture was pressed into a disk by a literature method.23
Single crystal X-ray diffraction
X-ray single-crystal diffraction data for complexes 1 and 2 were collected on a Bruker AXS CCD area detector with graphite monochromatic molybdenum Kα (λ = 0.71073 Å) radiation. Unit-cell parameters were determined from automatic centering and were refined by the least-squares method. The structure was solved by direct methods and refined using full-matrix least-square techniques on F2 with the SHELXTL program.24 All non-hydrogen atomic positions were located by a full-matrix least-squares method and refined anisotropically. Hydrogen atoms in water were placed in their geometrically generated positions and other hydrogen atoms bonded with carbon or nitrogen were located from different Fourier maps and refined isotropically. The crystallographic data and refinement parameters are presented in Table 1, and H-bonding and selected bond lengths and angles are summarized in Tables S2–S5 in the ESI.† CCDC-1487536 (1), 1509043 (2) contain the supplementary crystallographic data for this study.| Complex | 1 | 2 |
|---|---|---|
| Formula | C42H68Cu2N12O31P2 | C42H58Cu2N8O27P2 |
| Mr | 1426.10 | 1295.98 |
| Temperature (K) | 296(2) | 153(2) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21 | C2 |
| a (Å) | 10.364(3) | 30.261(6) |
| b (Å) | 20.041(6) | 9.6957(19) |
| c (Å) | 15.393(4) | 21.350(4) |
| α (°) | 90 | 90 |
| β (°) | 98.080(7) | 120.61(3) |
| γ (°) | 90 | 90 |
| V (Å3) | 3165.2(14) | 5391(2) |
| Z | 2 | 4 |
| F(000) | 1480 | 2680 |
| Reflections collected | 27649 |
24399 |
| Independent reflections | 11171 |
13881 |
| Goodness-of-fit on F2 | 1.046 | 1.045 |
| Rint | 0.0909 | 0.0357 |
| R1 [I > 2σ(I)] | 0.0710 | 0.0732 |
| wR2 [I > 2σ(I)] | 0.1672 | 0.1934 |
| R1 (all data) | 0.1104 | 0.0838 |
| wR2 (all data) | 0.1815 | 0.2043 |
| Flack parameter | 0.038(11) | 0.053(7) |
Results and discussion
Auxiliary ligand and Cu–UMP binding
From previous studies, the coordination ratio of the auxiliary ligand going from binary to ternary in the solution could be studied by UV titration.25–28 In order to study the interactions of bpda/bpe with Cu–UMP in an aqueous medium, UV titrations were carried out by adding an auxiliary ligand to the solution of Cu–UMP. UV absorbance was measured as a function of concentration of bpda and bpe (Fig. S2 and Table S1†). With increased auxiliary ligand content the apparent intensity increased at 250–300 nm, and broadened the main absorption at 265 nm; this was due to the strong intermolecular interaction for the strong π–π* transition. A blue shift at 200–225 nm occurred, which was related to the n–π* transitions of the complex. The isosbestic point around 223 nm indicated that a new complex was generated in the solution. Moreover, after the ratio of Cligand/CCu–UMP = 0.5, the main absorption was maintained at 280/293 nm, which illustrated ternary formations at an equimolar concentration of bpda/bpe and Cu–UMP (Fig. 1). After each addition of bpda/bpe, the difference Δλ(λmax − λiso) was obtained as a function of Cligand/(CCu–UMP + Cligand). The plots show that the complex was 1:1 adduct; this further confirms that the ternary assembly was a 1:1:1 assembly in the solution (Fig. S3†).
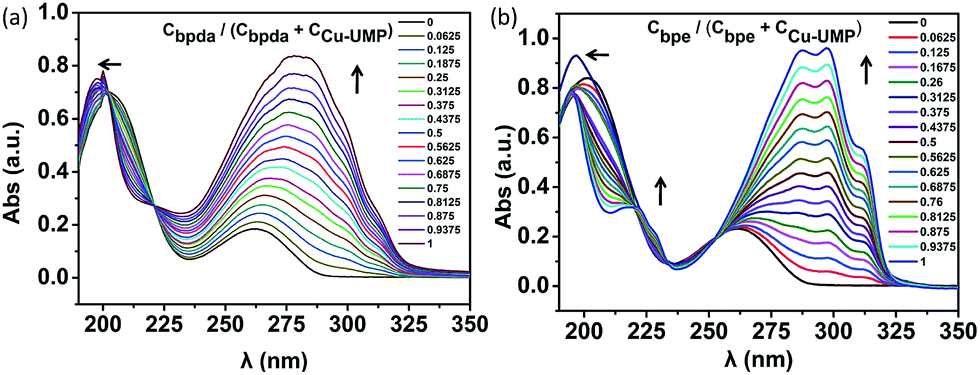 | ||
| Fig. 1 Electronic spectra of Cu–UMP (1:1) upon addition of (a) bpda (b) bpe. The total concentration of Cu–UMP and bpda/bpe is held fixed (3.0 × 10−5 M) and varying the ratio of the components (Cbpda/CCu–UMP and Cbpe/CCu–UMP). | ||
Structure description
However, the coordination spheres of Cu1 and Cu2 are different, in which the remaining coordination position of Cu1 was occupied by a water molecule, while Cu2 was occupied by a terminal UMP ligand (Fig. 2a). Bond distances for Cu–O (1.942(6) Å) and Cu–N (2.035(7) Å) are close to the mean value found for square pyramidal Cu(II) complexes (1.970(2) Å, 2.025(3) Å).16 The building block for a 1D coordination polymer is a dinuclear Cu(II) unit linked by UMP (Cu⋯Cu: 3.940(2) Å), in which each Cu(II) is bridged by a bpda ligand to extend the 1D polymer, which exhibits an unusual double-bridge 1D structure (Fig. 2b). The distance between the two planar bpda ligands within the chain is 3.756(9) Å with a π–π stacking interaction. The width of the 1D belt-type coordination polymer is 14.350(13) Å (Fig. S4†). The orientation of the UMP nucleobases of UMP is parallel with the bpda ligand induced by the intra-chain π–π interactions between the nucleobase and bpda (3.808(9) Å and 3.579(9) Å). This type of UMP orientation relative to the 1D chain leads to the Cu⋯Cu distance of the neighboring belt being 6.779(2) Å and prohibits the bridging function of PO4 group of UMP along the 2D direction. As in our previous study, the similar orientation of nucleobases also leads to a 1D coordination polymer in the CMP–bpe–Co complex and not a 2D coordination polymer.30 The key interactions between the 1D chains are the base-pair H-bonding (N10–H10A⋯O17, 2.023 Å, 2.876 Å, 170.94°; N12–H12⋯O8, 1.935 Å, 2.782 Å, 168.33°). Moreover, there are inter-chain π–π stacking interactions produced by the nucleobases with the bpda ligand (3.609(1) Å and 3.914(9) Å) (Fig. 2c and S5†). In addition, H-bonding formed by the coordinated oxygen atom of water with the phosphate group (O18–H18C⋯O3, 1.701 Å, 2.538 Å, 167.63°) is another force to assemble the 1D chains into a 2D sheet (Fig. 2c and S5†). The 2D sheets can be further organized into a 3D supramolecular framework through H-bonding (O6–H6⋯O12, 1.891 Å, 2.687 Å, 163.24°; O14–H14A⋯O20, 1.727 Å, 2.545 Å, 174.96°) between hydroxyl group of pentose and oxygen of phosphate group as well as hydroxyl group of pentose and coordinated water (Fig. S5†). There is helical H-bonding formed by the hydroxyl group of pentose with the oxygen of phosphate group (O6–H6⋯O12, 1.891 Å, 2.687 Å, 163.24°) (Fig. 2d and S7†), which is a P helix with a screw pitch of 20.0(1) Å.
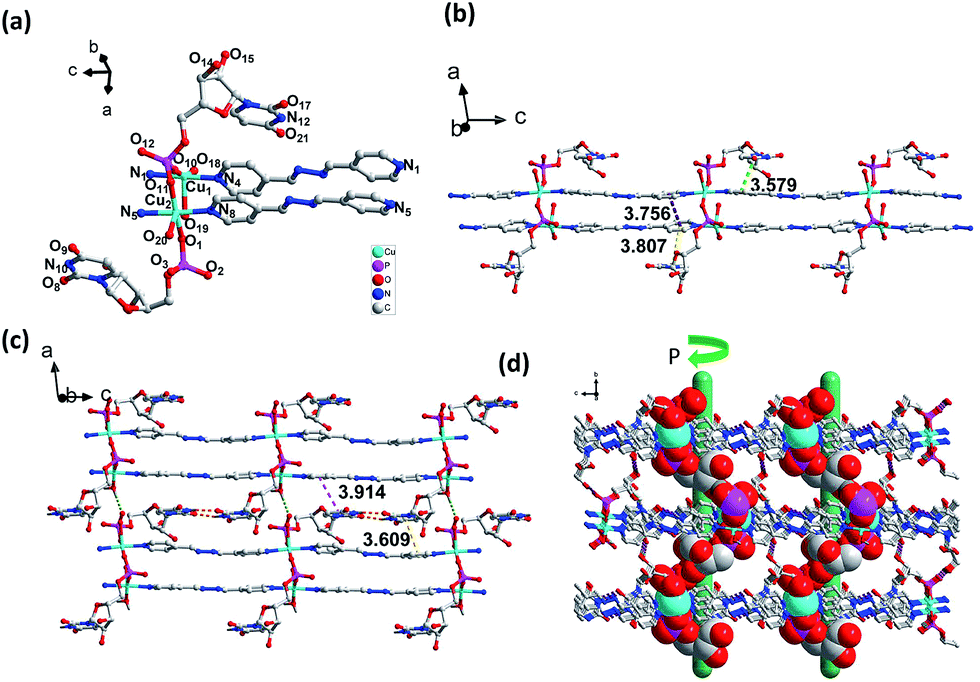 | ||
| Fig. 2 (a) The asymmetric unit of complex 1. The solvent molecules and hydrogen atoms are not shown for clarity. (b) 1D chain of complex 1 viewed down from the b axis. (c) 2D layer based on inter-chain H-bonding (N10–H10A⋯O17, 2.023 Å, 2.876 Å, 170.94°; N12–H12⋯O8, 1.935 Å, 2.782 Å, 168.33°) and π–π interactions. (d) 3D supramolecular framework based on H-bonding (O6–H6⋯O12, 1.891 Å, 2.687 Å, 163.24°; O14–H14A⋯O20, 1.727 Å, 2.545 Å, 174.96°) viewed down from the a-axis and P helix formed by H-bonding (O6–H6⋯O12, 1.891 Å, 2.687 Å, 163.24°). | ||
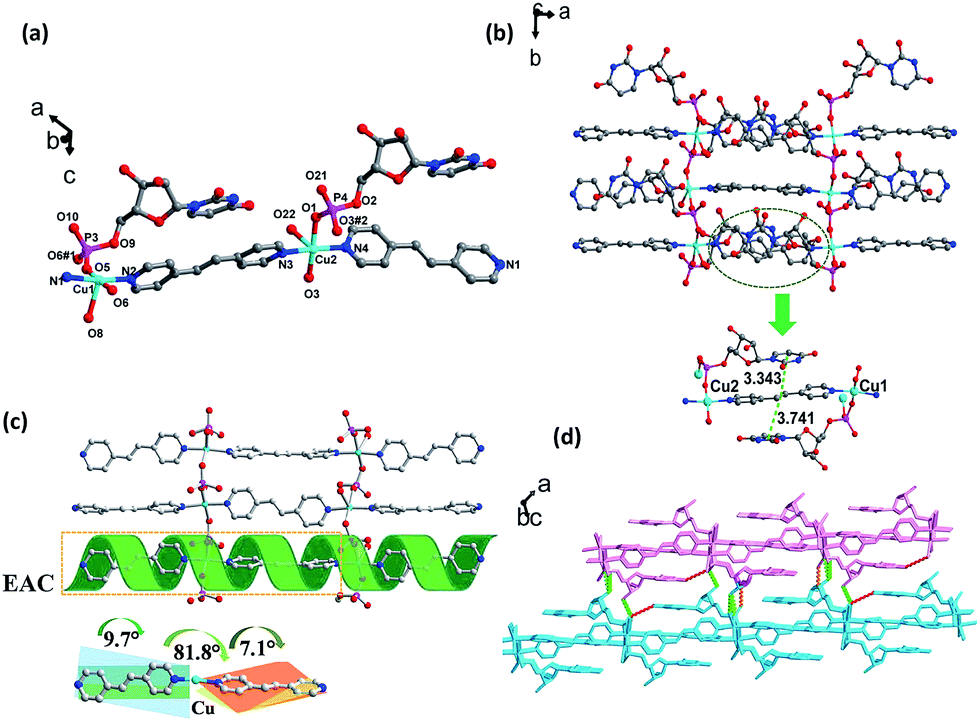 | ||
| Fig. 3 (a) The asymmetric unit of complex 2. The solvent molecules and hydrogen atoms are not shown for clarity. (b) 2D coordination network of complex 2 viewed down from the c axis and the intra-layer π–π stacking interactions of complex 2. (c) 2D coordination network of complex 2 viewed down from c axis, the dihedral between bpe and the presentation of EAC, and a cartoon used to express the EAC visually. (d) The inter-layer H-bonding (O16–H16⋯O6, 1.871 Å, 2.698 Å, 168.04°; O8–H8⋯O16, 2.525 Å, 3.274 Å, 152.27°; O17–H17A⋯O10, 2.044 Å, 2.878 Å, 166.40°). | ||
Circular dichroism (CD) spectroscopy
According to the structural analysis, supramolecular helical chirality and the EAC appeared in complexes 1 and 2. In order to identify the new chirality, the aqueous solution and solid-state CD spectra were recorded.17,30 All the samples were phase pure, which was verified by single crystal by X-ray powder diffraction (XRPD) measurements (Fig. S13†).The aqueous solution CD spectra (Fig. 4a) for UMP had two typical broad peaks centered near 250 and 280 nm. Specifically, the stronger and wider negative envelope near 247 nm corresponds to the intermolecular π–π* and n–π* transitions. The positive absorption near 285 nm for UMP relates to the intermolecular π–π* interaction between nucleotide bases, which indicates that the UMP is a D-ribonucleotide.31 Compared to the ligand, there was a new negative absorption at 300 nm in 1 and 2. Because the coordination of Cu(II) with UMP prevents the mutarotation of the ligand, making ligands keep the β-anomers in their complexes.16,17
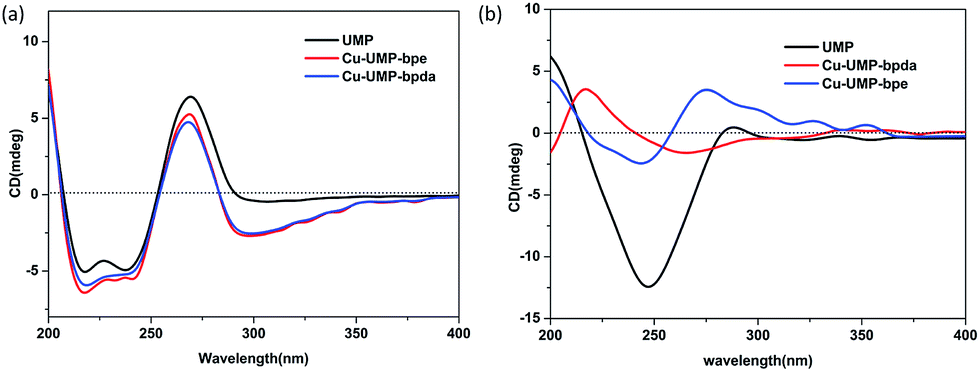 | ||
| Fig. 4 (a) The CD spectra of the solution of UMP and its complexes. The spectrum was obtained by measuring a 0.25 mM solution in a 1 mm cell. (b) The solid-state CD spectra of the UMP ligand and its complexes 1 and 2 (KCl:[sample] = 200:1). | ||
The solid-CD (Fig. 4b) for complex 1 was significantly different compared with UMP. A positive and new peak at 227 nm indicated that an additional chiral source was present, which was a result of the supramolecular helix changing the symmetry of electronic transition of UMP.16,17,32 The 227 nm CD signal transition is the excitation coupling of the π–π* transitions of the intra- and intermolecular uridine chromophores. On comparison, it was found that the solid-CD for complex 2 was similar to UMP, except for the weak positive acromions above 300 nm, and this can be attributed to the EAC of bpe, which was caused by the π–π* transitions between bpe and pyrimidine of UMP; this can be confirmed by structure research.33
Conclusion
Two nucleotide coordination complexes Cu–UMP–bpda/bpe were synthesized and their different architectures were studied by X-ray single crystal diffraction analysis. A 1D double-bridged structure was obtained that was totally different from the previous structure of bpda and nucleotide complexes. The important aim of the crystallography study of Cu–UMP–bpda/bpe is establishing a structural fundament to comprehensively understand the relative functional properties, such as chirality. Furthermore, the extended axial chirality (EAC) and supramolecular chirality were investigated in detail based on their crystal structure and aqueous solution and solid-state CD spectral analysis. The torsion of bpe leads to EAC and the 1D parallel double-bridged structure of bpda limits the rotation of aromatic rings, and hence no EAC appeared for 1. Moreover, the large torsion of UMP leads to a supramolecular helix by H-bonding in 2. Our present study provides a profound understanding of the relationship between structure and chirality, and promises to provide another effective method for researching nucleotide self-assembly through changing the length of the auxiliary ligand.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (no. 21271026, 21471017).Notes and references
- B. Roy, A. Depaix, C. Périgaud and S. Peyrottes, Chem. Rev., 2016, 116, 7854 CrossRef CAS PubMed.
- M. Yamada and K. Abe, Polym. J., 2014, 46, 366 CrossRef CAS.
- L. Berti and G. A. Burley, Nat. Nanotechnol., 2008, 3, 81 CrossRef CAS PubMed.
- V. Allain, C. Bourgaux and P. Couvreur, Nucleic Acids Res., 2012, 40, 1891 CrossRef CAS PubMed.
- K. Sureka, P. H. Choi, M. Precit, M. Delince, D. A. Pensinger, T. N. Huynh, A. R. Jurado, Y. A. Goo, M. Sadilek and A. T. Iavarone, et al., Cell, 2014, 158, 1389 CrossRef CAS PubMed.
- V. A. Tverdislov, Biophysics, 2013, 58, 128 CrossRef CAS.
- K. Aoki and K. Murayama, Met. Ions Life Sci., 2012, 10, 43 CAS.
- R. K. O. Sigel and H. Sigel, Acc. Chem. Res., 2010, 43(974), 9 Search PubMed.
- P. Zhou, R. Shi, J. F. Yao, C. F. Sheng and H. Li, Coord. Chem. Rev., 2015, 292, 107 CrossRef CAS.
- C.-Y. Zheng and H. Li, Inorg. Chem. Commun., 2013, 34, 30 CrossRef CAS.
- H. Sigel and R. Griesser, Chem. Soc. Rev., 2005, 34, 875 RSC.
- M. Benedetti, G. Tamasi, R. Cini, L. G. Marzilli and G. Natile, Chem.–Eur. J., 2007, 13, 3131 CrossRef CAS PubMed.
- D. Maruyama, Y. Nishitani, T. Nonaka, A. Kita, T. A. Fukami, T. Mio, H. Yamada-Okabe, T. Yamada-Okabe and K. J. Miki, Biol. Chem., 2007, 282, 17221 CrossRef CAS PubMed.
- L. A. Herrero and A. J. Terron, Biol. Aspects Inorg. Chem., 2000, 5, 269 CrossRef CAS.
- M. Kato, A. K. Sah, T. Tanase and M. Mikuriya, Eur. J. Inorg. Chem., 2006, 2504 CrossRef CAS.
- P. Zhou, J. F. Yao, C. F. Sheng and H. Li, CrystEngComm, 2013, 15, 8430 RSC.
- P. Zhou and H. Li, Dalton Trans., 2011, 40, 4834 RSC.
- N. Chen, M. X. Li, P. Yang, X. He, M. Shao and S. R. Zhu, Cryst. Growth Des., 2013, 13, 2650 CAS.
- M. Cattaneo, F. Fagalde and N. E. Katz, Inorg. Chem., 2006, 45, 6884 CrossRef CAS PubMed.
- W. S. Zou, Q. J. Shen, Y. Wang and W. J. Jin, Chem. Res. Chin. Univ., 2008, 24, 712 CAS.
- A. J. Calahorro, E. S. Sebastián, A. Salinas-Castillo, J. M. Seco, C. Mendicute-Fierro, B. Fernández and A. Rodríguez-Diéguez, CrystEngComm, 2015, 17, 3659 RSC.
- D. M. Ciurtin, Y. B. Dong, M. D. Smith, T. Barclay and H. C. Zur Loye, Inorg. Chem., 2001, 40, 2825 CrossRef CAS PubMed.
- J. B. Hou, G. Tang, J. N. Guo, Y. Liu, H. Zhang and Y. F. Zhao, Tetrahedron: Asymmetry, 2009, 20, 1301 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- Q.-X. Zhou, W.-H. Lei, Y.-J. Hou, Y.-J. Chen, C. Li, B.-W. Zhang and X.-S. Wang, Dalton Trans., 2013, 42, 2786 RSC.
- S. Brahma, S. K. A. Ikbal, S. Dey and S. P. Rath, Chem. Commun., 2012, 48, 4070 RSC.
- W. Liu, H. Zhu, B. Zheng, S. Cheng, Y. Fu, W. Li, T. C. Lau and H. Liang, Nucleic Acids Res., 2012, 40, 4229 CrossRef CAS PubMed.
- P. Suvanvapee, J. Boonmak, F. Klongdee, C. Pakawatchai, B. Moubaraki, K. S. Murray and S. Youngme, Cryst. Growth Des., 2015, 15, 3804 CAS.
- A. Meyer, G. Schnakenburg, R. Glaum and O. Schiemann, Inorg. Chem., 2015, 54, 8456 CrossRef CAS PubMed.
- P. Zhou, C. Wang, Q. M. Qiu, J. F. Yao, C. F. Sheng and H. Li, Dalton Trans., 2015, 44, 17810 RSC.
- A. I. S. Holm, E. S. Worma, T. Chakraborty, B. R. Babu, J. Wengel, S. V. Hoffmann and S. B. Nielsen, J. Photochem. Photobiol., A, 2007, 187, 293 CrossRef CAS.
- G. Pescitelli, L. D. Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211 RSC.
- S. M. Quarrie, A. Blanc, N. J. Mosey, C. M. Crudden, R. P. Lemieux and M. P. Thompson, J. Am. Chem. Soc., 2008, 130, 14099 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structure information, spectra (IR, UV-vis and CD), ESI-MS, XRD and crystallographic data (CIF). CCDC 1487536 and 1509043. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00851a |
| This journal is © The Royal Society of Chemistry 2017 |