
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low temperature synthesis of water dispersible F-doped TiO2 nanorods with enhanced photocatalytic activity†
Jiaxu Zhaoa,
Wenting Lia,
Xue Li *a and
Xiaokai Zhangb
*a and
Xiaokai Zhangb
aShandong Provincial Key Laboratory of Fluorine Chemistry and Chemical Materials, School of Chemistry and Chemical Engineering, University of Jinan, 336 West Road of Nan Xinzhuang, Jinan 250022, People's Republic of China. E-mail: chm_lix@ujn.edu.cn; lixue0312@yahoo.com
bCollege of Physics and Electronics, Shandong Normal University, 88 Wenhuadong Road, Jinan 250014, Shandong, People's Republic of China
First published on 18th April 2017
Abstract
F-doped TiO2 nanorods (F-TiO2 NRs) were prepared through a simple hydrolysis of titanium chloride (TiCl4) and a refluxing process at 100 °C and atmospheric pressure in the presence of sodium fluoride (NaF). The prepared samples were characterized by transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), UV-vis spectrophotometry and N2 adsorption analysis. It is found that F− ions are incorporated into the lattice of TiO2 and absorbed on the surface of TiO2 NRs. The NaF/TiCl4 molar ratio is found to play an important role in the formation of the anatase F-TiO2 NRs. When the molar ratio of NaF/TiCl4 is 4.0/1.0, the prepared F-TiO2 NRs show the highest photocatalytic activity for the degradation of methylene blue (MB) under UV and visible irradiation, showing that F-TiO2 NRs are efficient photocatalytic degradation catalysts of MB pollutant in wastewater treatment.
1 Introduction
It is well known that titanium oxide (TiO2) is one of the most promising photocatalysts due to its high efficiency, non-toxicity, low cost, chemical inertness and photostability.1–4 The performance of TiO2 photocatalysts is strongly affected by their structure, heterogeneous interface, size and exposed faces in solution.5,6 Many efforts have been made for the generation of high-efficiency TiO2 photocatalysts, and tailoring the particle shape, the size and the crystallinity of TiO2 has been intensively investigated to increase its surface area and the number of active sites for adsorption and photocatalytic degradation of pollutants.3,7–12 TiO2 crystals, particularly nanostructures with various shapes (i.e., nanowire, nanorod, nanobelt, nanotube, nanosphere, and nanosheet) have been realized in the past decades.13 Among the various morphologies, TiO2 nanorods (TiO2 NRs) can offer a higher surface-to-volume ratio compared to nanospheres, which can increase the density of active surface sites for photocatalytic reactions or load transfer in composites.14–16 Moreover, TiO2 NRs enabled a fast charge collection, thus demonstrating improved solar cell efficiencies.17 Therefore, TiO2 NRs are particularly interesting for optoelectronic applications, such as photovoltaic devices and self-powered UV detectors. Superior photoconduction efficiencies and gains have been experimentally demonstrated for individual, single-crystalline TiO2 NRs.18,19Various strategies for the preparation of TiO2 NRs have been developed, such as metal–organic chemical vapour deposition,20 electrochemical anodisation,21 direct oxidation of titanium sheets,22 and solution-phase reactions in hydrolytic23–25 and non-hydrolytic hydrolytic syntheses.16,26–28 For example, oleate-capped anatase TiO2 NRs were obtained by low-temperature trimethylamino-noxide dihydrate catalyzed hydrolysis of titanium tetraisopropoxide in oleic acid as surfactant at a temperature as low as 80 °C.25 A facile hydrothermal method was developed for the first time to grow oriented single-crystalline rutile TiO2 nanorod films on transparent conductive substrates. A light-to-electricity conversion efficiency of 3% could be achieved by using 4 μm-long TiO2 nanorod films as the photoanode in a DSSC.29 Highly crystalline TiO2 NRs have been synthesized by a hydrothermal process in a cetyltrimethylammonium bromide surfactant solution.30 By using a seeded growth technique, the highly uniform TiO2 NRs could also be generated.31
The photoactivity of TiO2 NRs depends on their crystal phase composition, geometric morphology, surface area, thermal treatment.32 Compared to P25–TiO2 catalyst, the activity of TiO2 NRs in some photocatalytic reactions need to be further improved.33 It has been reported that F-doped TiO2 (F-TiO2) is demonstrated an effective approach to promote the photocatalytic activity of TiO2.34 Fluorine has been used as a structure directing agent to generate TiO2 with exposed high-energy {0 0 1} facets with large surface area or as a capping agent and/or dopant in TiO2 to modify their surface and bulk properties, and consequently, to enhance their photocatalytic performance.35–37 It was demonstrated that F-doped TiO2 could not only slow down the radiative recombination process of photo-generated electrons and holes in TiO2 but also facilitate the generation of more ˙OH radicals which are responsible for the oxidation of organic molecules in photocatalytic reaction.38,39 More recent research has found that fluoride doping could tune the morphology of TiO2.6,40 For example, the flower-like TiO2 nanostructures with exposed {0 0 1} facets were synthesized by a low-temperature hydrothermal process from Ti powders in hydrofluoric acid (HF) solution, and they exhibited enhanced photocatalytic degradation of MB under UV light.41–43 Using TiCl4 as precursor, a series of TiO2 hierarchical nanostructures, such as pompon-like and football-like microspheres composed of aligned rutile and anatase nanoparticles, were synthesized by a facile template-free hydrothermal method in the presence of NaF.44 Hierarchical porous F-doped TiO2 microspheres exhibiting high visible light photocatalytic activity have been fabricated by a one-step low-temperature hydrothermal approach without using any templates.45 Fluorine-doped hierarchical porous single-crystal rutile TiO2 NRs have been synthesized through a silica template method, in which F− ions acts as both n-type dopants and capping agents to make the anisotropic growth of the nanorods.46 Usually a hydrothermal method with HF as a morphology controlling agent is exploited to prepare TiO2 nanomaterials. However, the hydrothermal method need high-pressure equipment such as Teflon-lined stain-less steel. Moreover, the HF is corrosive.
Although great progress has been made in TiO2 NRs, the synthesis of TiO2-based nanomaterials at low temperature and atmospheric pressure still remains a challenge. Moreover, the effect of NaF in the formation of the nanostructure of F-TiO2 NRs has not been investigated so far. In this work, a facile synthesis route has been developed for the preparation of anatase F-TiO2 NRs at low temperature by simple hydrolysis of TiCl4 and then a refluxing process at 100 °C and atmospheric pressure in the presence of NaF. In the process of synthesis, no other organic surfactant was used. By simply adjusting the reaction conditions such as the molar ratio of NaF/TiCl4 the morphology of TiO2 nanoparticles can be tailored. Finally, photocatalytic activities of the synthesized F-TiO2 NRs for the degradation of pollutant MB under UV and visible light irradiation were evaluated.
2 Experimental section
2.1 Materials
2.2 Synthesis of TiO2 NRs
Anatase TiO2 NRs was synthesized through a facile and low temperature hydrothermal method at normal pressure. In a typical procedure, TiCl4 aqueous solution was prepared by dissolving 100 μL of TiCl4 into 1.0 mL of water under strong stirring in an ice water bath, after mixing uniformly another 9.0 mL of water was added. The pH value of the TiCl4 aqueous solution was adjusted to 8–9 by NH3·H2O solution (2.5 M) to obtain titanium hydrate precipitates. The precipitates was washed with pure water for several times to remove Cl− completely and then redispersed into 100 mL of water. Peroxotitanium acid solution was obtained by addition of 110.0 μL of H2O2 (30%) solution to the above titanium hydrate dispersion drop by drop under stirring.47 After peroxotitanium acid the solution was held at 50 °C for 2 hours, given amount of NaF (NaF/TiCl4 = 0, 2.0, 4.0 and 6.0, molar ratio) was added and then the solution was refluxed at 100 °C for 2–6 hours in a thermostat water bath. One can see from the photocatalytic activity of the F-TiO2 NRs obtained at different refluxing temperature that 100 °C is the optimum synthesis temperature when the refluxing was carried out at atmospheric pressure (Fig. S1†). After the reaction finished, the products were collected by centrifugal separation and washed with deionized water for 5 times. All the products were dried in a vacuum oven at 50 °C overnight. To remove the F− ions absorbed on the surface of F-TiO2 NRs, the samples were first washed with 0.1 M NaOH aqueous solution, and then with pure water till its pH value was neutral.2.3 Characterization
X-ray Diffraction (XRD) was performed using a D8 FOCUS diffractometer (Bruker-AXS, Germany) with Cu Kα radiation (λ = 1.5418 Å). Transmission electron microscopy (TEM) and high resolution TEM (HRTEM) were performed on a JEM-2100 electron microscope (JEOL Ltd., Japan) operating at 200 kV. The samples were prepared by mounting a drop of the dispersion on a carbon-coated Cu grid and allowing it to dry in air. Nitrogen adsorption/desorption experiments were performed on a Micromeritics ASAP 2020 surface area analyzer after outgassing at 200 °C for 4 h prior to analysis. The standard multipoint Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface area. The pore size distributions of the materials were derived from the adsorption branches of the isotherms on the basis of the Barrett–Joyner–Halenda (BJH) model. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo ESCALAB 250 with Al Kα excitation.2.4 Photocatalytic activity measurements
The photocatalytic activity was measured by decomposing MB dye. In a typical process, 40.0 mL of MB aqueous solution with a concentration of 10.0 mg L−1 and 2.0–10.0 mg of TiO2 NRs were placed in a quartz conical flask and kept in the dark with stirring for 30 min to achieve adsorption equilibrium. Then the conical flask was irradiated with UV light (λ = 254 nm) produced by 2 × 15 W UV lamps (Spectronics Co., USA) or visible light irradiation (λ > 400 nm) produced by a 400 W metal halide lamp (HPI-T Plus 400 W, Philips) with cut off filter. At given intervals, 3 mL of suspension was extracted and then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min to get rid of the catalysts from the supernatant. Then, the solution was analyzed by recording the UV-vis spectrum of MB at the maximum absorbance of 665 nm.
000 rpm for 5 min to get rid of the catalysts from the supernatant. Then, the solution was analyzed by recording the UV-vis spectrum of MB at the maximum absorbance of 665 nm.
3 Results and discussion
3.1 Synthesis and characterization of TiO2 NRs
The anatase TiO2 NRs were synthesized through a hydrolysis TiCl4 at pH 8–9 to form titanium hydrate precipitates, and then the titanium hydrate precipitates were subjected to a peptizing, complexing and refluxing process at 100 °C and atmospheric pressure using NaF as shape control agent.47,48Without addition of NaF in the synthesis process, there are only irregular or elongated TiO2 nanoparticles of about 9.5 nm are obtained (Fig. 1a). The lattice fringes can be observed clearly (Fig. 1b), the selected-area electron diffraction (SAED) pattern in Fig. 1c displayed the diffraction rings corresponding to lattice planes ca. (101), (004), (200) and (211) of anatase crystal structure of TiO2 is in accordance with the XRD pattern of TiO2 nanoparticles (Fig. 3).
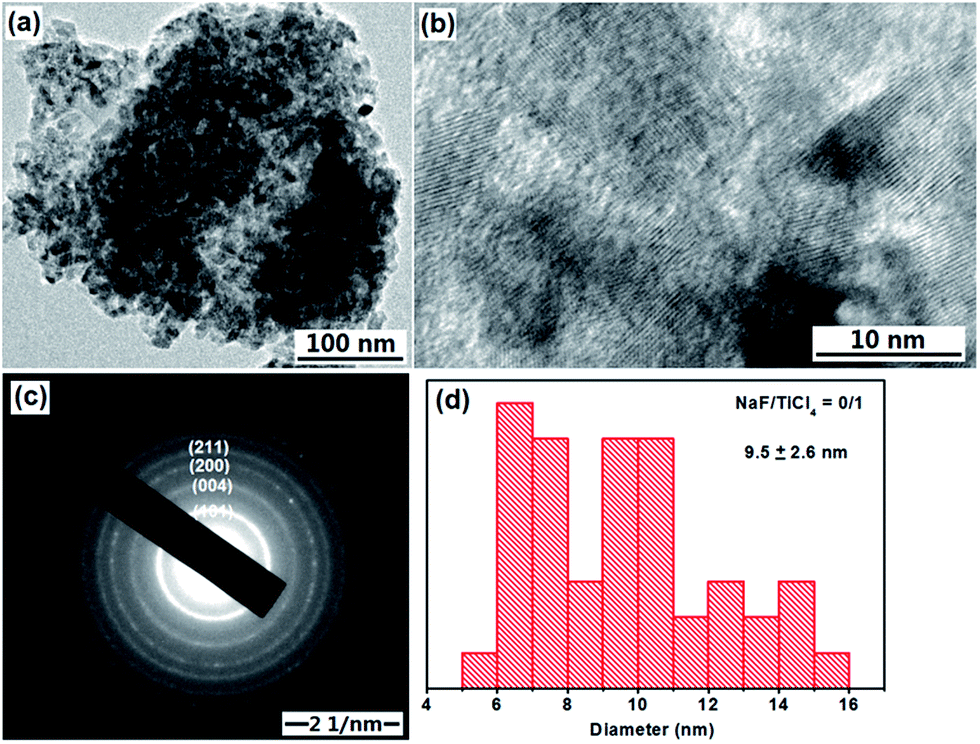 | ||
| Fig. 1 TEM image, HRTEM image, SAED pattern and size distributions of TiO2 nanoparticles obtained through a hydrolysis TiCl4 and a refluxing process at 100 °C for 6 hours. No NaF was added in the synthesis process. | ||
When NaF was added during the refluxing process, a significant change in the morphology of TiO2 nanoparticles was observed. One can see from the TEM images (Fig. 2a and b) that the product consisted of rod-like structures. The average length and diameter determined by TEM image analysis are about 32.7 nm and 9.2 when the molar ratio of NaF/TiCl4 is 4.0/1.0. The lattice fringes of TiO2 NRs were also observed clearly (Fig. 2b). The SAED pattern in Fig. 2c displayed the diffraction rings is similar to that in Fig. 1c. Christy et al.49 and Nian et al.50 prepared TiO2 NRs having diameter 6–10 nm. When the molar ratio of NaF/TiCl4 was changed to 2.0 or 6.0, similar results were obtained. The shape of the TiO2 NRs was maintained, however, the average length and diameter are changed into 26.5 nm and 8.6 for the NaF/TiCl4 molar ratio of 2.0/1.0, 34.7 nm and 8.2 for the NaF/TiCl4 molar ratio of 6.0/1.0 (Fig. S2 and S3†).
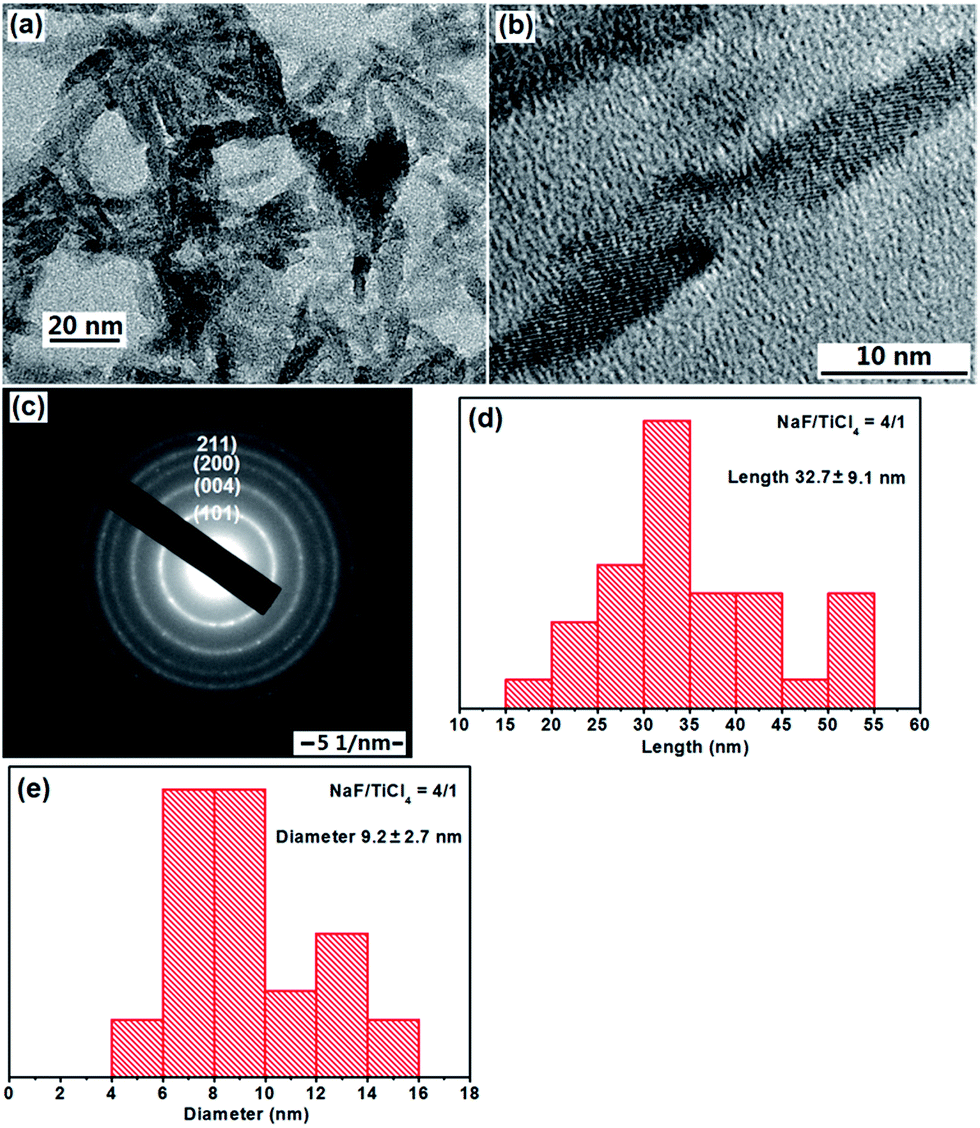 | ||
| Fig. 2 TEM image, HRTEM image, SAED pattern, length and diameter distributions of F-TiO2 NRs obtained through a hydrolysis TiCl4 and a refluxing process at 100 °C for 6 hours. The molar ratio of NaF/TiCl4 was 4.0/1.0. | ||
The crystallographic structure of TiO2 NRs synthesized at different conditions was characterized by powder XRD (Fig. 3). The diffraction peaks could be indexed to anatase-phase TiO2 (JCPDS no. 21-1272), indicating that all the as-prepared products with and without addition of NaF were anatase TiO2. The increased peak intensity and narrow width for the (004) reflex and a comparatively lower diffraction intensity for the (200), (211) reflexes, also indicate the formation of rod-like nanostructures with a preferred growth orientation along the c-axis of the anatase lattice.25 One can also see from Fig. 3 that the crystallinity of the samples increase with the increase of the molar ratio of NaF/TiCl4. Therefore, it is reasonable to conclude that the concentration of NaF plays an important role in the synthesis of the anatase TiO2 NRs.
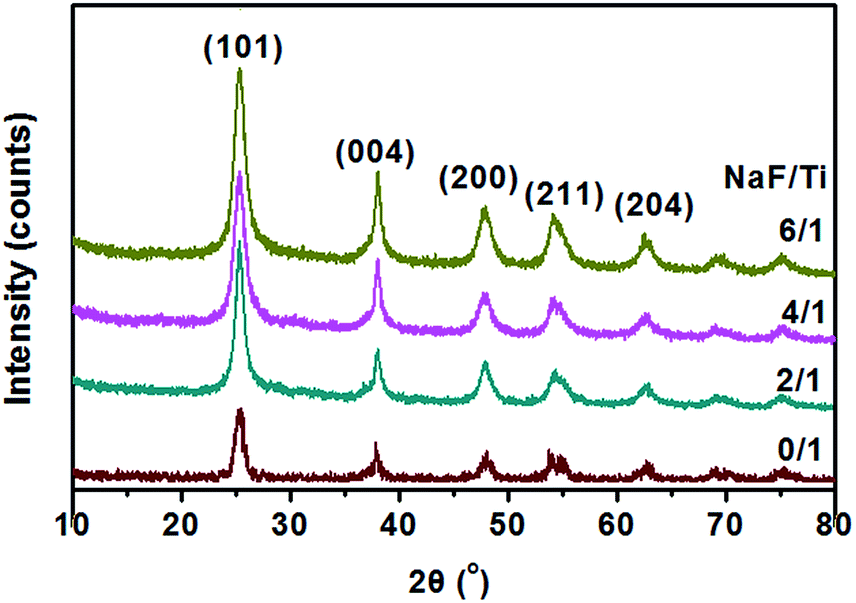 | ||
| Fig. 3 XRD patterns of TiO2 nanoparticles and F-TiO2 NRs obtained with different NaF/TiCl4 molar ratio, all were refluxed at 100 °C for 6 hours. | ||
The surface information of the F-TiO2 NRs prepared with NaF/TiCl4 molar ratio of 4.0/1.0 was detected by XPS. Characteristic peaks of Ti, O, F and C were observed (Fig. 4). The Ti 2p spectrum displays peaks at 458.5 and 464.3 eV corresponding to the 2p3/2 and 2p1/2 spin–orbital components, respectively,51 showing the presence of Ti4+ ions.52 The binding energy of O 1s at 529.8 eV is attributed to the O2− in TiO2. A broadening on the higher bonding energy side at 531.6 eV indicates the presence of another type of oxygen, which was assigned to Ti–OH bonds.53 The binding energy of F 1s core electrons as ∼684.6 eV correspond to the surface ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti–F species formed by ligand exchange between F− and surface hydroxyl groups, which is consistent with the reported results.54–57 Another F 1s core electron peak at 689.8 eV is ascribed to the contribution by surface lattice F (doped F− in the lattice of TiO2).58–61 Therefore, one can conclude that F− ions are incorporated to the lattice of TiO2 and absorbed on the surface of TiO2. From above results, it can be deduced that F atoms play important roles in the formation of rod-like morphology and structure of TiO2. After removal of the F− ions absorbed on the surface by washing with 0.1 M NaOH solution, the binding energy of F 1s at 684.3 eV is attributed to Ti–F species (Fig. 4e). The peak at 689.8 eV becomes very weak, indicating the low concentration of the F− in the lattice of TiO2. The chemical states of elemental Ti and O are not changed (Fig. S4†).
Ti–F species formed by ligand exchange between F− and surface hydroxyl groups, which is consistent with the reported results.54–57 Another F 1s core electron peak at 689.8 eV is ascribed to the contribution by surface lattice F (doped F− in the lattice of TiO2).58–61 Therefore, one can conclude that F− ions are incorporated to the lattice of TiO2 and absorbed on the surface of TiO2. From above results, it can be deduced that F atoms play important roles in the formation of rod-like morphology and structure of TiO2. After removal of the F− ions absorbed on the surface by washing with 0.1 M NaOH solution, the binding energy of F 1s at 684.3 eV is attributed to Ti–F species (Fig. 4e). The peak at 689.8 eV becomes very weak, indicating the low concentration of the F− in the lattice of TiO2. The chemical states of elemental Ti and O are not changed (Fig. S4†).
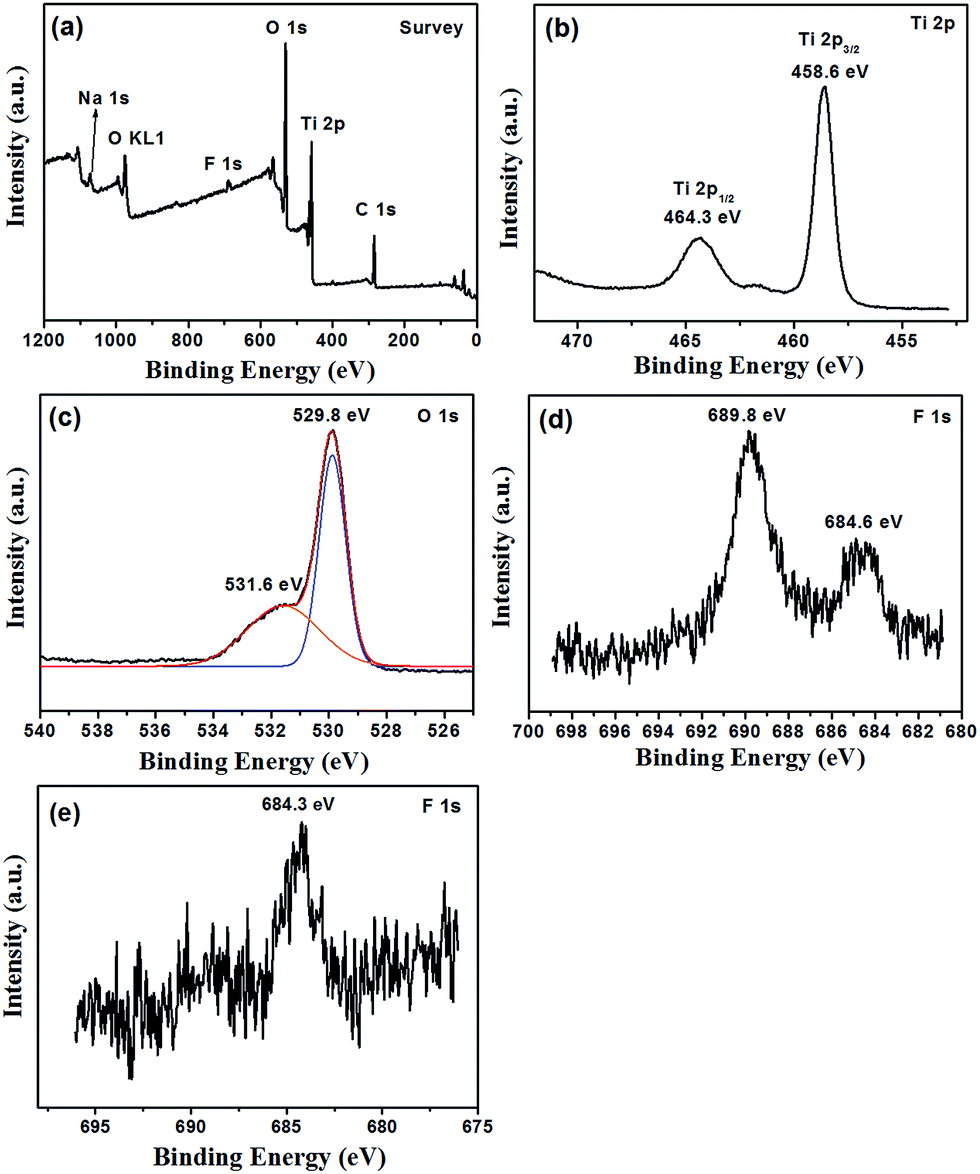 | ||
| Fig. 4 XPS spectra of F-TiO2 NRs synthesized at 100 °C for 6 hours, the molar ratio of NaF/TiCl4 = 4.0/1.0. (a) Survey, (b) Ti 2p, (c) O 1s and (d) F 1s. In (c), black trace shows the experimental data, blue and orange traces show the curves fitted to the experimental data, and the red traces show the envelope, i.e. the sum of the blue and orange traces. (e) F 1s spectrum of F-TiO2 NRs after removal of the F− ions absorbed on the surface by washing with 0.1 M NaOH solution. | ||
Light absorptions of commercial P25 nanoparticles, pure TiO2 nanoparticles and the TiO2 NRs prepared with different NaF/TiCl4 molar ratio were measured and the diffuse reflectance spectra are shown in Fig. 5a. The absorption spectrum of P25 only exhibits a stronger absorption in the UV range. However, pure TiO2 nanoparticles and all F-TiO2 NRs show UV and visible light absorption. To determine the value of energy gap of TiO2, the relation of (αhν)1/2 with the incident photon energy (hν) is shown in Fig. 5b. The band gap energies of these materials are about 2.94 (P25), 2.23 (pure TiO2 nanoparticles), 2.20 (NaF/TiCl4 = 2/1), 2.16 (NaF/TiCl4 = 4/1), 2.26 (NaF/TiCl4 = 6/1) and 2.30 eV (NaF/TiCl4 = 4/1, washing with 0.1 M NaOH solution). A smaller band gap of pure TiO2 is attributed to surface disorders, oxygen vacancies, etc.60 caused by the use of H2O2 during preparation process. It has been reported that the band gap TiO2/fly-ash cenospheres treated with H2O2 was reduced to 2.6 eV.62 In F-TiO2 NRs, the band gap energy of TiO2 NRs is further decreased, which is attributed to surface disorders, oxygen vacancies and Ti3+. It has been reported that the F doping can convert some Ti4+ to Ti3+. The Ti3+ surface states in TiO2 form a donor level between the bandgaps of TiO2, which would enhance the visible light absorption.60 In addition, the F doping can promote the creation of oxygen vacancies, which induces the enhancement of the visible light absorption.45 Such an enhancement in absorbance can increase the number of photo-generated electrons and holes to be involved in the photocatalytic reaction.63 If the F− ions absorbed on the surface were removed by washing with 0.1 M NaOH solution, F-TiO2 NRs also exhibited UV and visible light absorption, the band gap energy of the F-TiO2 NRs (NaF/TiCl4 = 4/1) was increased slightly. That is, and the F-doping in the lattice could improve the light absorption of TiO2 according to the UV-vis result (Fig. 5a).
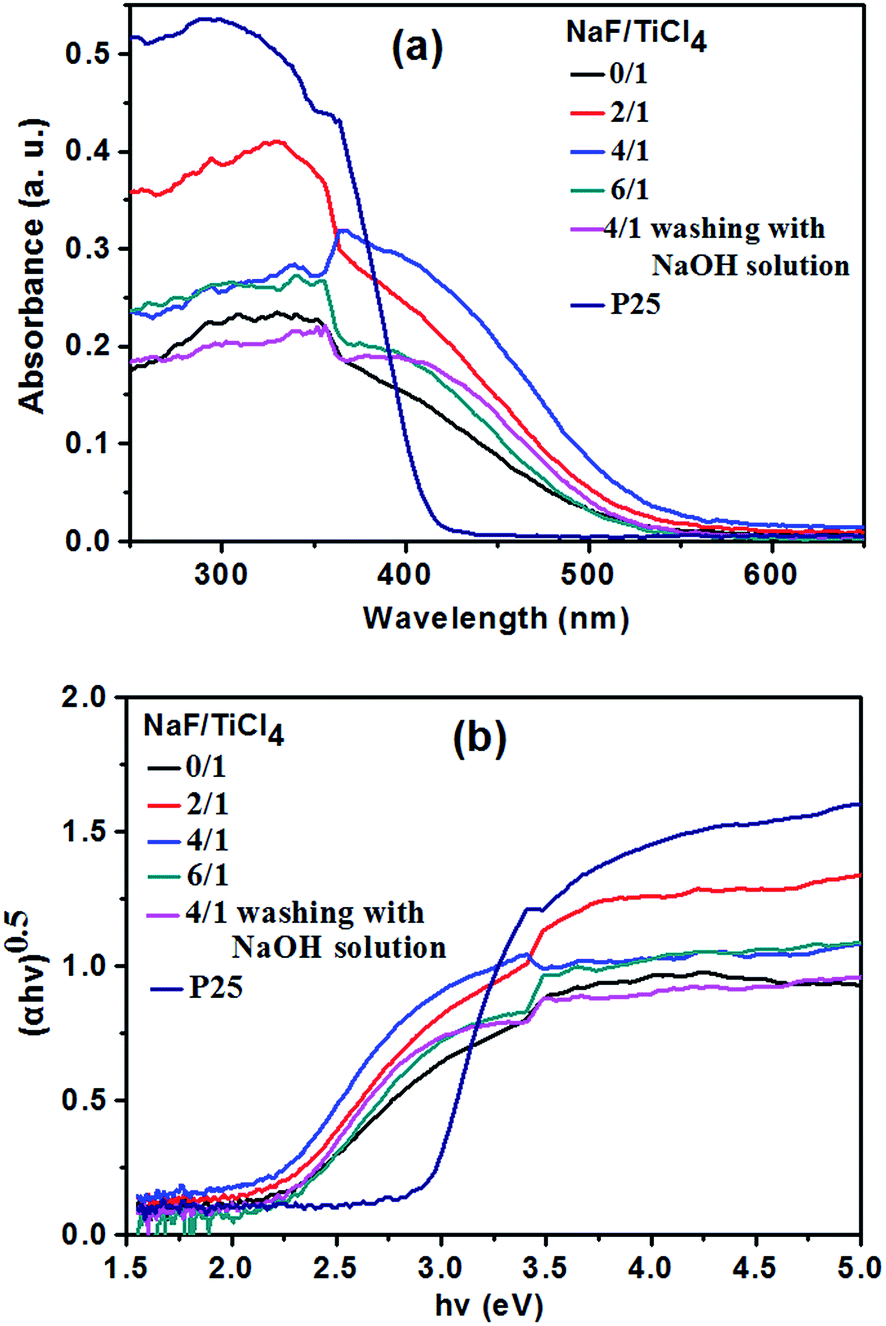 | ||
| Fig. 5 Diffuse reflectance UV-vis spectra of P25, pure TiO2 nanoparticles and F-TiO2 NRs prepared with different NaF/TiCl4 molar ratio. | ||
3.2 Formation mechanism of F-TiO2 NRs
To understand the formation process of the F-TiO2 NRs, time-dependent experiments were carried out. After the system was heated to 100 °C for 0 min, 20 min and 60 min, the morphologies of the products were observed with TEM. When the reaction time was 0 min, Fig. 6a and a′ indicate that the as-obtained product are a large quantity of aggregates of nanoparticles, and no lattice fringes were observed, which might be assigned as amorphous titania. When the reaction went on at 100 °C for 20 to 60 min, the very short rod-like structures with an average length of up to 2–3 nm formed (Fig. 6b and c′). Obvious lattice fringes were observed (Fig. 6b′ and c′), indicating the appearance of crystal structure of TiO2. When the reaction time was prolonged to 2 h, willow-leaf-like TiO2 NRs with length of ∼30 nm were observed (Fig. 6d). Further increasing the reaction time (4 h, 6 h and 24 h), there was no significant changes of the F-TiO2 nanorod morphology (Fig. 6e–g), indicating prolonged heating did not further influence the particle growth.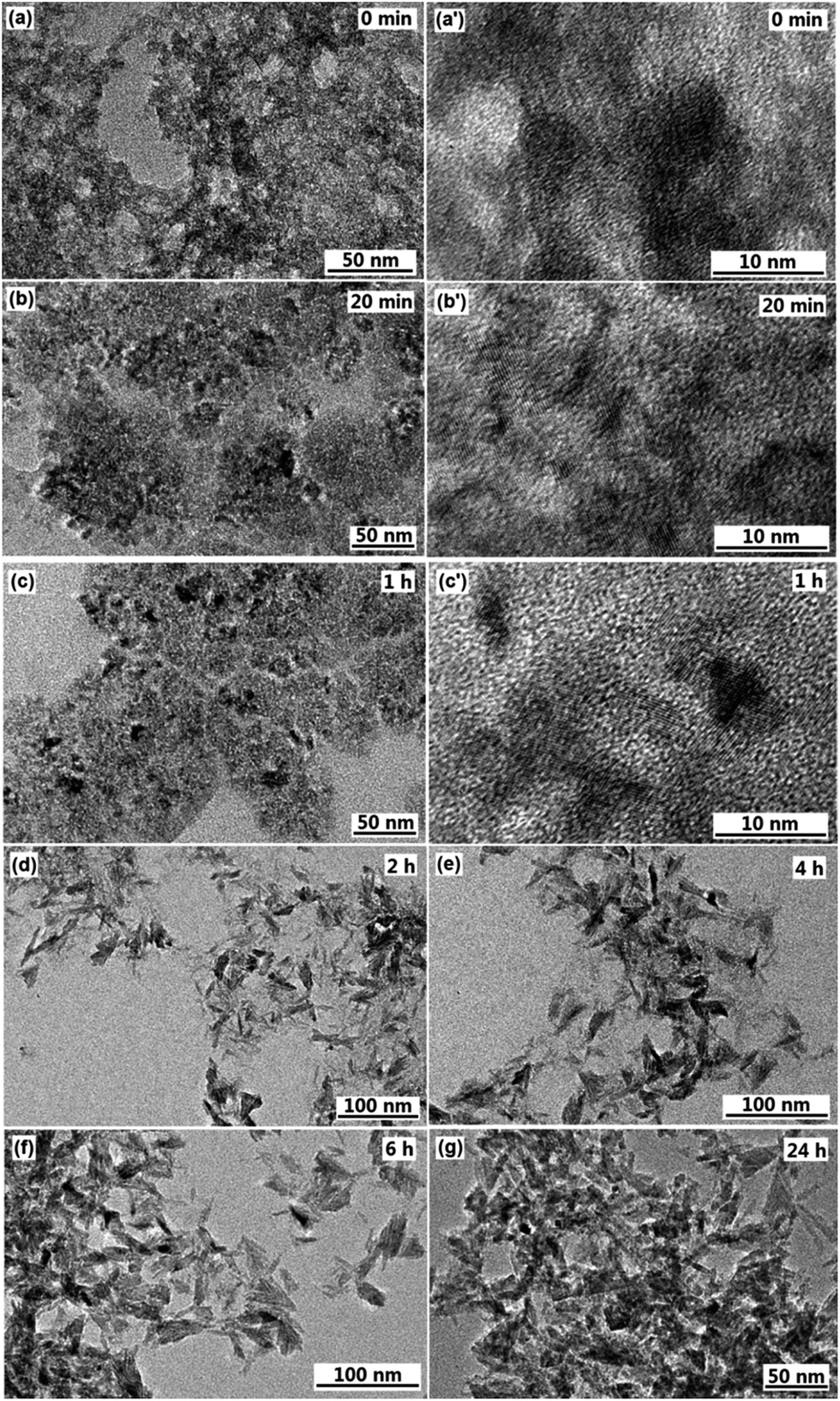 | ||
| Fig. 6 TEM images of F-TiO2 nanoparticles synthesized under a refluxing process at 100 °C for 0 min (a), 20 min (b), 1 h (c), 2 h (d), 4 h (e), 6 h (f) and 24 h (g), the NaF/TiCl4 molar ratio is 2.0/1.0. (a′, b′ and c′ are high magnification images). | ||
In terms of the above results, the nanorod formation may be understood as follows: after addition of H2O2 to the titanium hydrate dispersion, peroxotitanium acid is produced. Upon refluxing at 100 °C, dehydration reaction of the peroxotitanium acid to form TiO6 octahedra along with the condensation of TiO6 octahedra to TiO2 takes place. The nucleus of crystalline nanocrystals are formed first in the solution when the temperature is increased from 50 °C to 100 °C. With the increase of the reaction time, amorphous phase of TiO2 gradually transforms to crystalline phase, collision of nanoparticles along with the generation of one-dimensional metastable aggregates may occur. The adsorption of F− ions might lead to the growth of the metastable aggregates along a certain direction to create nanorod-like TiO2 crystals, which may be based on the Ostwald ripening mechanism. When the reaction time is longer, the anatase TiO2 NRs will not change due to the low supersaturation degree in this case (<9 mmol L−1). It has been reported that F− ions adsorbed on the surface of TiO6 octahedra can catalyze the bridging of adjacent TiO6 octahedra, facilitating the face-sharing rearrangement of the octahedra and leading to the spiral chain growth,64,65 as a result of condensation reaction anatase TiO2 is formed. In addition, it has been reported that the average crystallite size and degree of crystallinity of anatase nanoparticles increased with increasing concentration of fluoride.66 It is also proposed that the mediating role of F− ions in the dissolution and re-crystallization processes of metastable TiO2 intermediates results in the facilitation of the crystal growth and crystallization, accounting for the greater crystal size and higher degree of crystallinity.36
3.3 Nitrogen adsorption/desorption experiments
The specific surface areas and the pore volumes of pure TiO2 the F-TiO2 NRs with different NaF/TiCl4 molar ratio were determined by nitrogen gas adsorption–desorption isotherms measurement. Fig. 7 shows the typical isotherm and the pore size distribution. According to the IUPAC classification, the nitrogen adsorption–desorption isotherm is attributed to the Langmuir IV type. The formation of the hysteresis loops in the plot of Fig. 7a reveal the presence of mesopores, which may be ascribed to agglomerates of TiO2 nanoparticles. The BET specific surface area, the average pore size and pore volume of the samples are listed in Table 1. Compared to spherical nanoparticles, the larger BET specific surface area of F-TiO2 NRs may be due to the high surface-to-volume ratio and the reduced aggregation of the nanoparticles.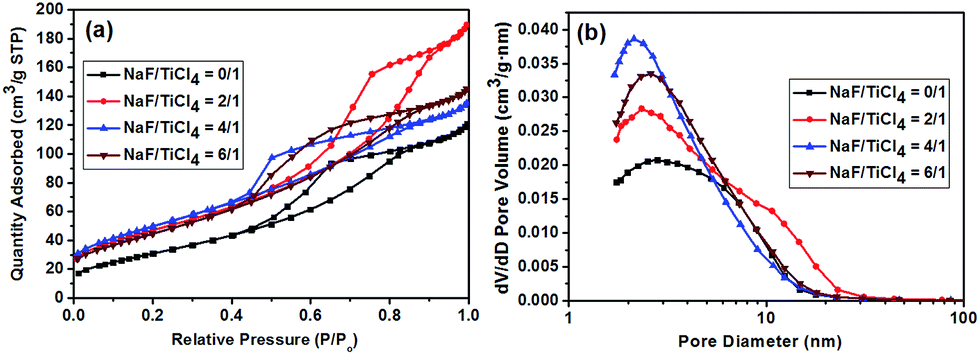 | ||
| Fig. 7 (a) Nitrogen adsorption/desorption isotherm. (b) Corresponding BJH pore size distribution curve of pure TiO2 nanoparticles and F-TiO2 NRs with different NaF/TiCl4 molar ratios. | ||
| NaF/TiCl4(molar ratio) | 0.0/1.0 | 2.0/1.0 | 4.0/1.0 | 6.0/1.0 |
|---|---|---|---|---|
| BET surface area (m2 g−1) | 117.4 | 172.0 | 181.2 | 165.9 |
| Pore volume (cm3 g−1) | 0.20 | 0.29 | 0.21 | 0.22 |
| Average pore size (nm) | 4.7 | 5.6 | 4.1 | 4.2 |
3.4 Photocatalytic activity of F-TiO2 NRs
In this experiment, the photocatalytic activities of F-TiO2 NRs were tested by the photocatalytic degradation of MB in aqueous solution under UV and visible light. Fig. 8a shows the evolution of MB absorption spectra in the presence of TiO2 NRs catalyst, from which it can be see that the absorbance peaks of MB decreased quickly with irradiation time. Under UV light, one can see from Fig. 8b that the photocatalytic activity is greatly improved by fluorine doping, which is in agreement with previous report.44 NaF/TiCl4 molar ratio has a great influence on the photocatalytic activity of the F-TiO2 NRs samples. Before and after removal of the F− ions absorbed on the surface, the optimal NaF/TiCl4 molar ratio always is 4.0/1.0. When the NaF/TiCl4 molar ratio is 4.0/1.0, 80.5% MB was eliminated within 100 min (Fig. 8b, curve 4/1). It should be noted that the photocatalytic activities of F-TiO2 NRs still lower than that of P25 under UV light though F-TiO2 NRs have higher adsorption capability. However, the photocatalytic activity of F-TiO2 NRs is improved when the F− ions absorbed on the surface are removed by washing with 0.1 M NaOH solution, in which 96.8% MB was eliminated within 100 min (Fig. 8b, curve 4/1 (r)). This can be attributed to the reduced recombination of electrons and holes at the surface of F-TiO2 NRs. Under visible light, similar results were obtained when NaF/TiCl4 molar ratio was varied (Fig. 8c). It was found that the degradation rate of MB for pure TiO2 and P25 was very low (Fig. 8c). However, 72% of MB was degraded in the presence of F-TiO2 NRs (NaF/TiCl4 molar ratio of 4.0/1.0) after 260 min (Fig. 8c, curve 4/1). After removal of the F− ions absorbed on the surface by washing with NaOH solution, 90.0% MB was eliminated within 130 min (Fig. 8c, curve 4/1 (r)). These results indicated that F-TiO2 NRs show much higher visible light catalytic activity than that of P25 or pure TiO2 nanoparticles.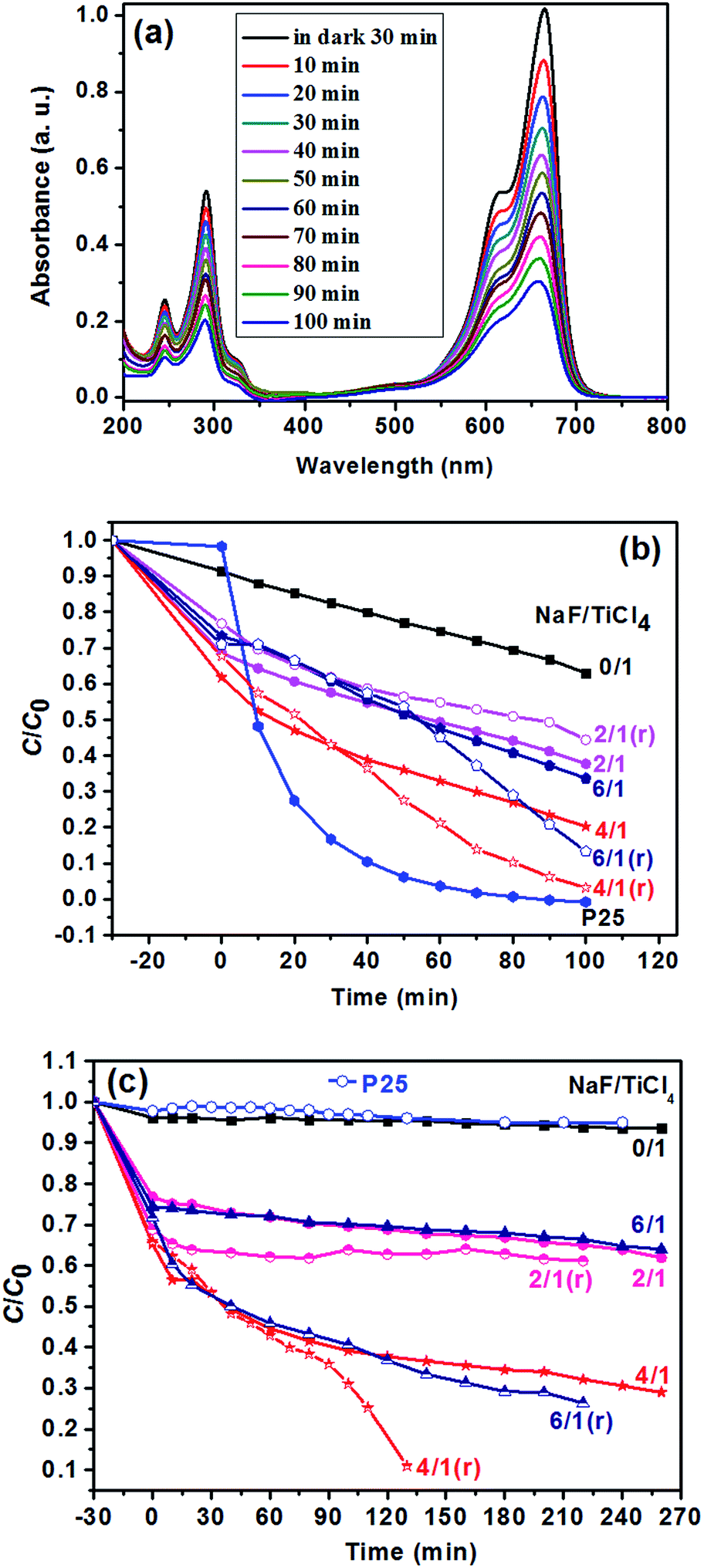 | ||
| Fig. 8 (a) UV-vis spectra of MB solution under UV irradiation for different times using F-TiO2 NRs as photocatalyst prepared with NaF/TiCl4 molar ratio of 4.0/1.0. (b) The change in the concentration of MB as a function of UV light irradiation time using P25 and the F-TiO2 photocatalysts prepared with different NaF/TiCl4 molar ratios. For the curves of 2/1 (r), 4/1 (r) and 6/1 (r), the F− ions absorbed on the surface were removed by washing with 0.1 M NaOH aqueous solution. (c) The change in the concentration of MB as a function of visible light irradiation time using P25 and the F-TiO2 photocatalysts prepared with different NaF/TiCl4 molar ratios. For the curve s of 2/1 (r), 4/1 (r) and 6/1 (r), the F− ions absorbed on the surface were removed by washing with 0.1 M NaOH aqueous solution. C0 = 10 mg L−1, mass of F-TiO2 NRs = 2.0 mg, solution volume = 40 mL. | ||
The effect of the content of catalysts on the photocatalytic activities of TiO2 NRs was also performed. As shown in Fig. 9, the photocatalytic degradation rate of MB increases with the amount of catalysts. The MB was removed almost completely (Fig. 9c) when the solution was irradiated under UV-light for 20 min. In addition, it is noted that prior to UV irradiation, obvious colour change was observed for the MB solution when the content of TiO2 NRs was increased to 10.0 mg in 40 mL MB solution, and the C/C0 value of MB solution after equilibrium in the dark was 0.06 (Fig. 9c), indicating that the adsorptive capacity of TiO2 NRs for MB is very large. The adsorption rate and the amount of adsorbed dye with contact time for the solution of MB (10 mg L−1) is illustrated in Fig. 10. As can be seen from Fig. 10 that the adsorption process is very rapid. When the amount of TiO2 NRs is 10.0 mg, the equilibrium was achieved and eliminated almost 95% of MB from the solution after 10 min. If the amount of TiO2 NRs is increased 20.0 mg, the equilibrium was achieved very quickly and eliminated almost 98% of MB from the solution after 5 min. We observed previously that N-doped TiO2 nanosheets with larger adsorption capability exhibited enhanced photocatalytic activity for the decomposition of MB.52
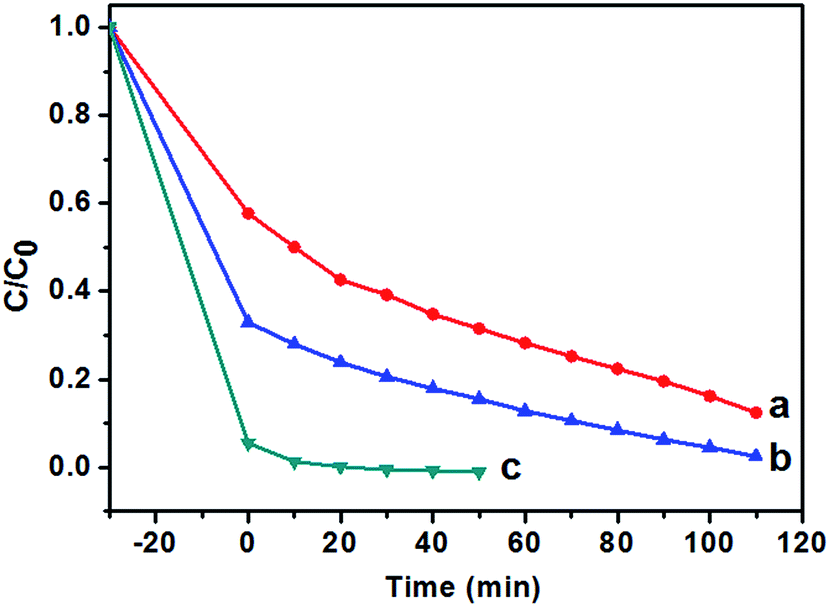 | ||
| Fig. 9 Photocatalytic degradation (UV light) of MB solution (40 mL, C0 = 10 mg L−1) with different amount of F-TiO2 NRs. (a) 2.0 mg, (b) 6.0 mg and (c) 10.0 mg. F-TiO2 NRs photocatalysts were prepared with NaF/TiCl4 molar ratio of 4.0/1.0. | ||
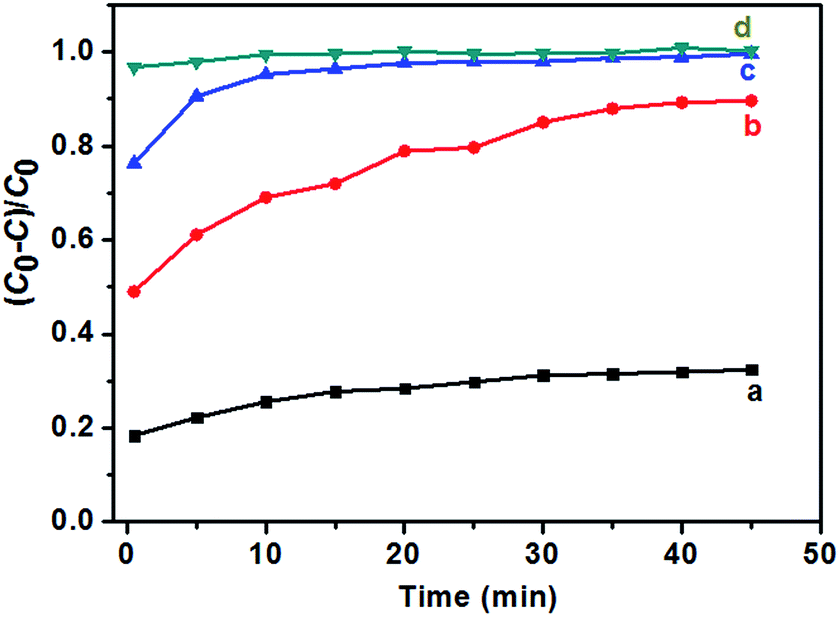 | ||
| Fig. 10 Adsorption ratio for the solutions of MB (10 mg L−1, 40 mL) in the presence of F-TiO2 NRs prepared with NaF/TiCl4 molar ratio of 4.0/1.0. (a) 2.0 mg, (b) 6.0 mg, (c) 10.0 mg and (d) 20.0 mg. | ||
It has been reported that fluorine doping can increase the specific surface area of TiO2 and enhance the adsorption of MB, increase the visible light absorption and decrease the recombination of the photoinduced electrons and holes, which is beneficial to the photocatalytic performance.66,67 The synergistic effect of fluorine doping and hierarchical nanostructure are responsible for the improved photocatalytic activity under visible light illumination.68 Therefore, the enhanced photocatalytic activity of F-doped TiO2 NRs in this case is mainly ascribed to the increase in the visible light absorption and the reduction in electron–hole pair recombination (Fig. 5). Furthermore, F-TiO2 NRs with the high adsorption capacity to MB (Fig. 10) and mesostructure (Fig. 7) can exhibit more effective photocatalytic performance.52,69,70 The photocatalytic activity of F-TiO2 NRs depends on the F doping concentration, the optimal NaF/TiCl4 molar ratio is 4.0/1.0, which is in agreement with previous report that, when the F doping concentration is more than a special value, the degradation rate decreases due to the promotion of the recombination of electron–hole pairs.53,57
4 Conclusions
Anatase F-TiO2 NRs were synthesized through a hydrolysis TiCl4 and a refluxing process at 100 °C and atmospheric pressure in the presence of NaF. During the synthesis procedure no surfactant or capping agents were added. F− ions are incorporated to the lattice of TiO2 and absorbed on the surface of TiO2 NRs. The NaF/TiCl4 molar ratio is found to plays an important role in the formation of the anatase F-TiO2 NRs. The prepared F-doped TiO2 NRs show the high photocatalytic activity for the degradation of MB under UV and visible light irradiation, showing that F-TiO2 NRs are efficient photocatalytic degradation catalysts of pollutant MB in wastewater treatment.Acknowledgements
This work was funded by the National Natural Science Foundation of China (51173069, 51473068).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- S. Sakthivel, M. Janczarek and H. Kisch, J. Phys. Chem. B, 2004, 108, 19384 CrossRef CAS.
- (a) B. Bayarri, J. Gimenez, D. Curco and S. Esplugas, Catal. Today, 2005, 101, 227 CrossRef CAS; (b) J. L. Vivero-Escoto, Y. D. Chiang, K. C. W. Wu and Y. Yamauchi, Sci. Technol. Adv. Mater., 2012, 13, 013003 CrossRef PubMed.
- M. Umadevi, R. Parimaladevi and M. Sangari, Spectrochim. Acta, Part A, 2014, 120, 365 CrossRef CAS PubMed.
- M.-H. Yang, M.-C. Tsai, Y.-W. Chang, Y.-C. Chang, H.-T. Chiu and C.-Y. Lee, ChemCatChem, 2013, 5, 1871 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231 CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- J. H. Pan, H. Dou, Z. Xiong, C. Xu, J. Ma and X. S. Zhao, J. Mater. Chem., 2010, 20, 4512 RSC.
- S. Sánchez-Muñoz, D. Pérez-Quintanilla and S. Gómez-Ruiz, Mater. Res. Bull., 2013, 48, 250 CrossRef.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- U. Vukičević, S. C. Ziemian, A. Bismarck and M. S. P. Shaffer, J. Mater. Chem., 2008, 18, 3448 RSC.
- Q. Zhang, S.-J. Liu and S.-H. Yu, J. Mater. Chem., 2009, 19, 191 RSC.
- R. Menzel, B. F. Cottam, S. Ziemian and M. S. P. Shaffer, J. Mater. Chem., 2012, 22, 12172 RSC.
- B. Liu, A. Khare and E. S. Aydil, Chem. Commun., 2012, 48, 8565 RSC.
- R. S. Chen, C. A. Chen, H. Y. Tsai, W. C. Wang and Y. S. Huang, Appl. Phys. Lett., 2012, 100, 123108 CrossRef.
- R. S. Chen, C. A. Chen, W. C. Wang, H. Y. Tsai and Y. S. Huang, Appl. Phys. Lett., 2011, 99, 222107 CrossRef.
- S. K. Pradhan, P. J. Reucroft, F. Yang and A. Dozier, J. Cryst. Growth, 2003, 256, 83 CrossRef CAS.
- S. Z. Chu, S. Inoue, K. Wada, S. Hishita and K. Kurashima, Adv. Funct. Mater., 2005, 15, 1343 CrossRef CAS.
- J.-M. Wu, T.-W. Zhang, Y.-W. Zeng, S. Hayakawa, K. Tsuru and A. Osaka, Langmuir, 2005, 21, 6995 CrossRef CAS PubMed.
- Q. Zhang and L. Gao, Langmuir, 2003, 19, 967 CrossRef CAS.
- R. Penn, Geochim. Cosmochim. Acta, 1999, 63, 1549 CrossRef CAS.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539 CrossRef CAS PubMed.
- A. Vioux, Chem. Mater., 1997, 9, 2292 CrossRef.
- T. J. Trentler, T. E. Denler, J. F. Bertone, A. Agrawal and V. L. Colvin, J. Am. Chem. Soc., 1999, 121, 1613 CrossRef CAS.
- Y.-W. Jun, M. F. Casula, J.-H. Sim, S. Y. Kim, J. Cheon and A. P. Alivisatos, J. Am. Chem. Soc., 2003, 125, 15981 CrossRef CAS PubMed.
- B. Liu and E. S. Aydil, J. Am. Chem. Soc., 2009, 131, 3985 CrossRef CAS PubMed.
- J. Jiu, S. Isoda, F. Wang and M. Adachi, J. Phys. Chem. B, 2006, 110, 2087 CrossRef CAS PubMed.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751 CrossRef CAS PubMed.
- (a) I. S. Grover, S. Singh and B. Pal, Appl. Surf. Sci., 2013, 280, 366 CrossRef CAS; (b) J. Joo, S. G. Kwon, T. Yu, M. Cho, J. Lee, J. Yoon and T. E. Coli, J. Phys. Chem. B, 2005, 109, 15297 CrossRef CAS PubMed; (c) W. Yawen, Z. Lizhi, D. Kejian, C. Xinyi and Z. Zhigang, J. Phys. Chem. C, 2007, 111, 2709 CrossRef.
- (a) R. K. Wahi, W. W. Yu, Y. Liu, M. L. Mejia, J. C. Falkner, W. Nolte and V. L. Colvin, J. Mol. Catal. A: Chem., 2005, 242, 48 CrossRef CAS; (b) D. P. Macwan, P. N. Dave and S. Chaturvedi, J. Mater. Sci., 2011, 46, 3669 CrossRef CAS; (c) M. Qamar, C. R. Yoon, H. J. Oh, N. H. Lee, K. Par, D. H. Kim, K. S. Lee, W. J. Lee and S. J. Kim, Catal. Today, 2008, 131, 3 CrossRef CAS; (d) J. Yu, H. Yu, B. Cheng and C. Trapalis, J. Mol. Catal. A: Chem., 2006, 249, 135 CrossRef CAS; (e) E. Jr-Morgado, M. A. S. De-Abreu, O. R. C. Pravia, B. A. Marinkovic, P. M. Jardim, F. C. Rizzo and A. S. Araujo, Solid State Sci., 2006, 8, 888 CrossRef.
- (a) G. D. Yang, T. C. Xiao, J. Sloan, G. Q. Li and Z. F. Yan, Chem.–Eur. J., 2011, 17, 1096–1100 CrossRef CAS PubMed; (b) J. K. Zhou, L. Lv, J. Yu, H. L. Li, P.-Z. Guo, H. Sun and X. S. Zhao, J. Phys. Chem. C, 2008, 112, 5316–5321 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed; J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Y. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124 CrossRef PubMed.
- S. Liu, J. Yu, B. Cheng and M. Jaroniec, Adv. Colloid Interface Sci., 2012, 173, 35 CrossRef CAS PubMed.
- J. Song, H. B. Yang, X. Wang, S. Y. Khoo, C. C. Wong, X.-W. Liu and C. M. Li, ACS Appl. Mater. Interfaces, 2012, 4, 3712 CAS.
- C. L. Yu, J. C. Yu and M. Chan, J. Solid State Chem., 2009, 182, 1061 CrossRef CAS.
- C. L. Yu, W. Q. Zhou, K. Yang and G. Rong, J. Mater. Sci., 2010, 45, 5756 CrossRef CAS.
- A. S. Ichimura, B. M. Mack, S. M. Usmani and D. G. Mars, Chem. Mater., 2012, 24, 2324 CrossRef CAS PubMed.
- G. Wu, J. Wang, D. F. Thomas and A. Chen, Langmuir, 2008, 24, 3503 CrossRef CAS PubMed.
- M. Liu, L. Y. Piao, W. M. Lu, S. T. Ju, L. Zhao, C. L. Zhou, H. L. Li and W. J. Wang, Nanoscale, 2010, 2, 1115 RSC.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946 RSC.
- Y. Dong, M. Kapilashrami, Y. Zhang and J. Guo, CrystEngComm, 2013, 15, 10657 RSC.
- W. Ho, J. C. Yu and S. Lee, Chem. Commun., 2006, 1115 RSC.
- W. Q. Fang, Z. Huo, P. Liu, X. L. Wang, M. Zhang, Y. Jia, H. Zhang, H. Zhao, H. G. Yang and X. Yao, Chemistry (Weinheim an der Bergstrasse, Germany), 2014, 20(36), 11439 CAS.
- H. Ichinose, M. Terasaki and A. H. Katsuki, J. Sol-Gel Sci. Technol., 2001, 22, 33 CrossRef CAS.
- J. Mühlebach, K. Müller and G. Schwarzenbach, Inorg. Chem., 1970, 9, 2381 CrossRef.
- P. D. Christy, N. Melikechi, N. S. N. Jothi, A. R. B. Suganthi and P. Sagayaraj, J. Nanopart. Res., 2010, 12, 2875 CrossRef CAS.
- J. N. Nian and H. Teng, J. Phys. Chem. B, 2006, 110, 4193 CrossRef CAS PubMed.
- L. Wu, B. X. Yang, X. H. Yang, Z. G. Chen, Z. Li, H. J. Zhao, X. Q. Gong and H. G. Yang, CrystEngComm, 2013, 15, 3252 RSC.
- W. Li, C. Shang and X. Li, Appl. Surf. Sci., 2015, 357, 2223 CrossRef CAS.
- J. C. Yu, W. Ho, J. G. Yu, S. K. Hark and K. Iu, Langmuir, 2003, 19, 3889 CrossRef CAS.
- Z. Lai, F. Peng, Y. Wang, H. Wang, H. Yu, P. Liu and H. Zhao, J. Mater. Chem., 2012, 22, 23906 RSC.
- G. Liu, C. H. Sun, H. G. Yang, S. C. Smith, L. Z. Wang, G. Q. Lu and H. M. Cheng, Chem. Commun., 2010, 755 RSC.
- J. Pan, G. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2011, 123, 2181 CrossRef.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078 CrossRef CAS PubMed.
- Q. Wang, C. C. Chen, D. Zhao, W. H. Ma and J. C. Zhao, Langmuir, 2008, 24, 7338 CrossRef CAS PubMed.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086 CrossRef CAS.
- J. C. Yu, J. G. Yu, W. K. Ho, Z. T. Jiang and L. Z. Zhang, Chem. Mater., 2002, 14, 3808 CrossRef CAS.
- K. L. Lv, Q. J. Xiang and J. G. Yu, Appl. Catal., B, 2011, 104, 275 CrossRef CAS.
- P. Huo, Y. Yan, S. Li, H. Li, W. Huang, S. Chen and X. Zhang, Desalination, 2010, 263, 258 CrossRef CAS.
- W. Yu, X. Liu, L. Pan, J. Li, J. Liu, J. Zhang, P. Li, C. Chen and Z. Sun, Appl. Surf. Sci., 2014, 319, 107 CrossRef CAS.
- (a) D. Li, H. Haneda, N. K. Labhsetwar, S. Hishita and N. Ohashi, Chem. Phys. Lett., 2005, 401, 579 CrossRef CAS; (b) F. Izumi, Bull. Chem. Soc. Jpn., 1978, 51, 1771 CrossRef CAS.
- J. Yu, G. Dai and B. Cheng, J. Phys. Chem. C, 2010, 114, 19378 CAS.
- J. G. Yu, W. G. Wang, B. Cheng and B.-L. Su, J. Phys. Chem. C, 2009, 113, 6743 CAS.
- Y. Cong, J. L. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976 CAS.
- L. G. Gai, X. Q. Duan, H. H. Jiang, Q. H. Mei, G. W. Zhou, Y. Tian and H. Liu, CrystEngComm, 2012, 14, 7662 RSC.
- S. Liu, J. Yu and W. Wang, Phys. Chem. Chem. Phys., 2010, 12, 12308 RSC.
- (a) S. W. Liu, J. G. Yu and S. Mann, J. Phys. Chem. C, 2009, 113, 10712 CrossRef CAS; (b) J. G. Yu, S. W. Liu and H. G. Yu, J. Catal., 2007, 249, 59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00850c |
| This journal is © The Royal Society of Chemistry 2017 |