
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the one-electron reduction behavior of tetrachloro-o-benzoquinone: a DFT and molecular dynamics study
Chao Guo,
Weihua Wang*,
Wenling Feng and
Ping Li *
*
Key Laboratory of Life-Organic Analysis, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, P. R. China. E-mail: wwh78@163.com; lignip@163.com
First published on 23rd February 2017
Abstract
In this study, the one-electron reduction behavior of tetrachloro-o-benzoquinone (o-TCBQ) has been systematically investigated at the B3LYP/6-311++G** level of theory in combination with the ab initio molecular dynamics. It was found that the explicit water molecules have slight effects on the geometry of o-TCBQ. On the contrary, the introduction of an electron can make the geometry change significantly. Moreover, the C2v symmetry of neutral and anionic o-TCBQ in the gas phase has been changed to be C2 symmetry in solution. All the electron affinity and vertical detachment energies are positive in the gas phase and in solution, increasing with the increasing of the dielectric constant of the bulk solvent. Therefore, explicit water molecules and bulk solvents can efficiently enhance the electron-accepting ability of the o-TCBQ, reflecting the intrinsic nature of o-TCBQ as a good electron acceptor in different media.
1. Introduction
As a class of toxic intermediates, halogenated quinones can initiate many harmful effects in vivo, such as acute hepatoxicity, nephrotoxicity, and carcinogenesis.1,2 Some of them, such as tetrachloro-p-benzoquinone (p-TCBQ), tetrachloro-o-benzoquinone (o-TCBQ), and 2,5-dichloro-1,4-benzoquinone (DCBQ), are the major genotoxic and carcinogenic quinoid metabolites of the widely used pesticides chlorinated phenols like pentachlorophenol (PCP), where PCP has been classified as a group 2B environmental carcinogen by the International Agency for Research on Cancer (IARC). Meanwhile, they can also be produced during the oxidation or destruction of halophenols and other polyhalogenated persistent organic pollutants (POPs).3–5 Moreover, some chloro- and bromo-benzoquinones have been identified as new disinfection byproducts in drinking water.6,7Recently, more and more studies have shown that chlorinated benzoquinones, such as p-TCBQ and DCBQ, can react with the organic hydroperoxides or benzohydroxamic acids to produce the hydroxyl radical, organic alkoxyl radicals, and quinone ketoxy radicals experimentally,8–17 which can be used to partially elucidate the potential carcinogenicity of the polyhalogenated aromatic environmental pollutants since these active radicals can cause oxidative damage to the DNA, protein, and lipids.13
Despite the extensive studies on the structure and chemical reactivity of p-TCBQ experimentally and theoretically,8–26 however, its isomer, namely o-TCBQ, has been paid less attentions although o-TCBQ has been involved in the toxicity of the bleached kraft chlorination effluent many years ago.27–32 Moreover, considering the fact that p-TCBQ behaves as a good electron acceptor generally and its chemical reactivity is governed by its intrinsic property, so some questions remain unclear for o-TCBQ. For example, is it a good electron acceptor like p-TCBQ? If so, how about the molecular dynamics for its electron-accepting process? How about the influences of the explicit water and bulk solvent on its electron-accepting ability?
To address these questions, in this study, the electron affinity (EA) associated with the one-electron reduction behavior of o-TCBQ as well as its F- and Br-substitution species has been systematically investigated employing the density functional theory (DFT) and ab initio molecular dynamics. To our best knowledge, no relevant theoretical studies have been reported on the one-electron reduction behavior of o-TCBQ in different media. Expectedly, the present results not only can provide new insights into the electron-accepting processes of o-TCBQ and its derivatives in the redox reactions, but also can pave the way for the future study of the chemical reactivity of o-TCBQ and its derivatives with other species.
2. Computational methods
All the geometries have been fully optimized at the B3LYP/6-311++G** level of theory, where the reliability and efficiency of the method has been verified by a number of investigations on the various systems.20,33–41 Subsequently, vibrational frequency analysis has also been performed at the same level of theory to identify the nature of the optimized structures.To better clarify the nature of the intermolecular H-bonds between o-TCBQ and water molecules, the atoms in molecules (AIM) theory has been employed on the basis of the optimized structures.42 In terms of the AIM theory, the interatomic interaction is indicated by the location of a bond critical point (BCP). Once the BCP has been localized, various properties can be evaluated at this position in space. For example, the interatomic interaction strength can be estimated from the magnitude of the electron density (ρbcp) at the BCP. Similarly, the ring structures are characterized by the location of a ring critical point (RCP). Furthermore, the nature of the interatomic interaction can be predicted from the topological parameters at the BCP, such as the Laplacian of electron density (∇2ρbcp) and energy density (Hbcp), where the Hbcp is composed of the potential energy density (Vbcp) and kinetic energy density (Gbcp). Generally, the sign of ∇2ρbcp reveals whether charge is concentrated (∇2ρbcp < 0) as in covalent bonds (shared interaction) or depleted (∇2ρbcp > 0) as in ionic bonds, H-bonds, and van der Waals interactions (closed-shell interaction).
To evaluate the electron redistribution behavior for o-TCBQ upon capturing an electron, natural bond orbital (NBO) analyses have also been carried out on the basis of the optimized geometries at the B3LYP/6-311++G** level of theory.43
To gain the microscopic details of the one-electron reduction process for o-TCBQ, electron capture dynamics of o-TCBQ has been investigated by means of Atom Centered Density Matrix Propagation (ADMP) molecular dynamics44–46 at the B3LYP/6-311G* level of theory on the basis of the optimized neutral state. As for the ADMP approach, it belongs to the extended Lagrangian approach to molecular dynamics using Gaussian basis functions and propagating the density matrix. Unlike plane wave Car–Parrinello (CP) molecular dynamics, it is not necessary to use pseudopotentials on hydrogen or to use deuterium rather than hydrogen in the dynamics. Fictitious masses for the electronic degrees of freedom are set automatically and can be small enough that thermostats are not required for good energy conservation. Here, the dynamics calculations were performed under condition of constant total energy. The time stepsize was 0.1 fs and a total of 5000 steps were calculated. The fictitious electronic mass is 0.1 amu and the initial nuclear and electron kinetic energy are zero. No thermostats are chosen to control the nuclear temperature.
To characterize the one-electron reduction behavior of o-TCBQ, the main terms discussed below have been defined as follows:
(a) Adiabatic electron affinity (AEA):
| AEA = H(neutral) − H(anion) |
(b) Vertical electron affinity (VEA):
| VEA = E(neutral) − E(anion/neutral) |
(c) Vertical detachment energy (VDE):
| VDE = E(neutral/anion) − E(anion) |
To qualitatively assess the degree of the structural changes for neutral state upon electron attachment and solvation, deformation energy has been calculated as the energy difference between the neutral state at the geometry of its optimized states in different media and the optimized neutral state.
To investigate the solvent effects on the relevant energy quantities mentioned above, the polarizable continuum model (PCM)47,48 has been employed within the framework of the self-consistent reaction filed (SCRF) theory. All the geometries have been fully optimized in a series of bulk solvents characterized by different dielectric constants from 1.43 to 78.36. Additionally, to investigate the effects of the adjacent explicit water molecules, the mono- and dihydrated o-TCBQ complexes have been constructed.
All the calculations have been carried out using Gaussian 09 program.49
3. Results and discussion
3.1 Structural features
Unlike p-TCBQ possessing D2h symmetry, as displayed in Fig. 1, the optimized o-TCBQ has C2v symmetry in the gas phase, which is consistent with the experimental result.50 Subsequently, on the basis of its optimized structure, ADMP molecular dynamics calculations have been carried out to provide more details of the electron-accepting process on the assumption of vertical electron attachment. | ||
| Fig. 1 The optimized isolated and hydrated o-TCBQ before and after capturing an electron, where the selected intermolecular distances are given in Å. | ||
As displayed in Fig. 2, the potential energy decreases suddenly from point a to b, corresponding to the electron-accepting process. Obviously, structural changes occur for the selected bonds although the C2v symmetry is still kept upon capturing an electron. As a result, all the C–X (X = C, O, Cl) bonds have been weakened except for the C–C single bond within the six-membered ring. In other words, the single- and double-bond difference in the six-membered ring of o-TCBQ decreases upon capturing an electron. Moreover, those structural changes can be further confirmed by the optimized anionic o-TCBQ. For example, the C1–O7, C3–C4, and C3–Cl9 bonds have been increased by about 0.034, 0.025, and 0.030 Å after introduction of an electron, respectively. On the other hand, the opposite is true for the C1–C2, C2–C3, and C4–C5 single bonds, which has been decreased by about 0.045, 0.028, and 0.056 Å, respectively. Especially, these structural changes can be further confirmed by the corresponding electron densities at the BCPs of the C–X bonds. As shown in Table 1, the electron densities at the BCPs for the C–X and C–C single bonds have been decreased and increased upon electron capture, respectively.
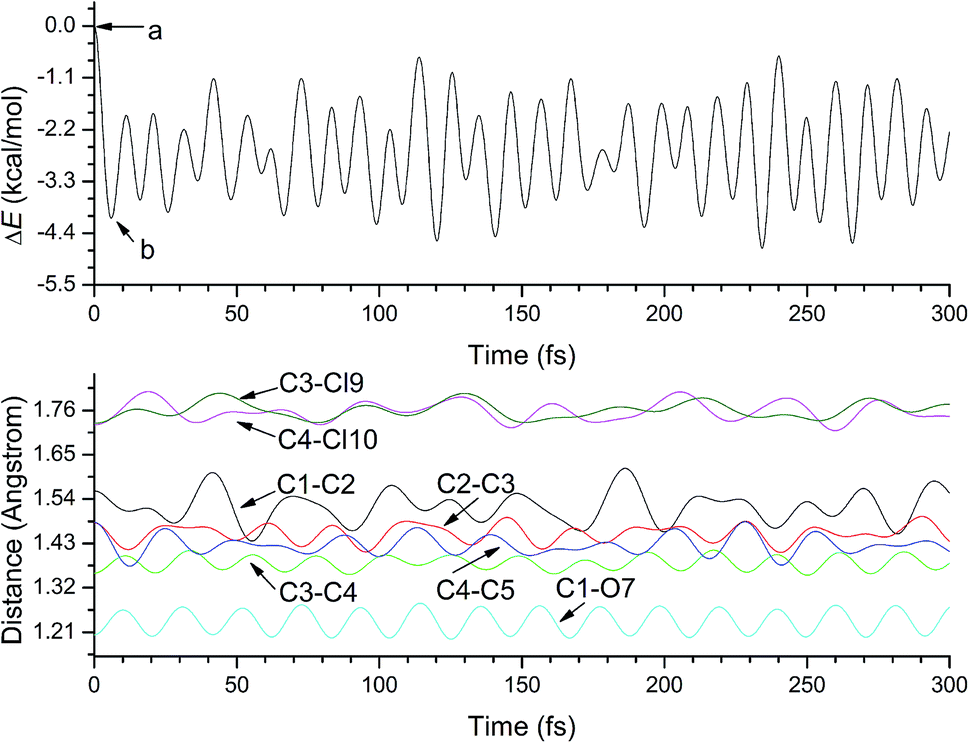 | ||
| Fig. 2 The evolution of the potential energy relative to that of o-TCBQ at 0 fs (top) and the selected bond lengths of o-TCBQ (bottom) upon capturing an electron. | ||
| Species | BCP | ρbcp | ∇2ρbcp | Vbcp | Gbcp | Hbcp |
|---|---|---|---|---|---|---|
| a The data in italics refer to the results of the corresponding anionic state. | ||||||
| o-TCBQ | C1–C2 | 0.2380 | −0.5270 | −0.2357 | 0.0520 | −0.1837 |
| 0.2549 | −0.5953 | −0.2114 | 0.0626 | −0.1488 | ||
| C2–C3 | 0.2687 | −0.6628 | −0.3047 | 0.0695 | −0.2352 | |
| 0.2786 | −0.6975 | −0.2568 | 0.0824 | −0.1744 | ||
| C3–C4 | 0.3292 | −0.9306 | −0.4846 | 0.1260 | −0.3586 | |
| 0.3123 | −0.8434 | −0.3228 | 0.1119 | −0.2109 | ||
| C4–C5 | 0.2629 | −0.6335 | −0.2922 | 0.0669 | −0.2253 | |
| 0.2878 | −0.7342 | −0.2725 | 0.0890 | −0.1836 | ||
| C1–O7 | 0.4153 | 0.0152 | −1.4081 | 0.7060 | −0.7022 | |
| 0.3860 | −0.2339 | −0.6326 | 0.5741 | −0.0585 | ||
| C3–Cl9 | 0.2062 | −0.2996 | −0.2189 | 0.0720 | −0.1469 | |
| 0.1914 | −0.2582 | −0.1305 | 0.0659 | −0.0646 | ||
| C4–Cl10 | 0.2059 | −0.3001 | −0.2174 | 0.0712 | −0.1462 | |
| 0.1907 | −0.2577 | −0.1292 | 0.0648 | −0.0644 | ||
| o-TCBQ1w1 | C1–C2 | 0.2386 | −0.5302 | −0.2363 | 0.0519 | −0.1844 |
| 0.2585 | −0.6127 | −0.2811 | 0.0640 | −0.2172 | ||
| C2–C3 | 0.2713 | −0.6764 | −0.3099 | 0.0704 | −0.2395 | |
| 0.2809 | −0.7101 | −0.3426 | 0.0826 | −0.2601 | ||
| C3–C4 | 0.3287 | −0.9284 | −0.4824 | 0.1252 | −0.3573 | |
| 0.3134 | −0.8501 | −0.4373 | 0.1124 | −0.3249 | ||
| C4–C5 | 0.2628 | −0.6329 | −0.2917 | 0.0667 | −0.2250 | |
| 0.2874 | −0.7335 | −0.3598 | 0.0882 | −0.2716 | ||
| C1–O7 | 0.4128 | 0.0116 | −1.3944 | 0.6987 | −0.6958 | |
| 0.3804 | −0.2408 | −1.1769 | 0.5583 | −0.6186 | ||
| C2–O8 | 0.4119 | 0.0096 | −1.3890 | 0.6957 | −0.6933 | |
| 0.3804 | −0.2409 | −1.1768 | 0.5583 | −0.6185 | ||
| C3–Cl9 | 0.2069 | −0.3010 | −0.2198 | 0.0723 | −0.1475 | |
| 0.1933 | −0.2645 | −0.1991 | 0.0665 | −0.1326 | ||
| C4–Cl10 | 0.2072 | −0.3042 | −0.2197 | 0.0718 | −0.1479 | |
| 0.1926 | −0.2635 | −0.1967 | 0.0654 | −0.1313 | ||
| C5–Cl11 | 0.2071 | −0.3037 | −0.2194 | 0.0718 | −0.1477 | |
| 0.1926 | −0.2635 | −0.1967 | 0.0654 | −0.1313 | ||
| O8–H14 | 0.0131 | 0.0476 | −0.0084 | 0.0101 | 0.0018 | |
| 0.0201 | 0.0726 | −0.0143 | 0.0162 | 0.0019 | ||
| O7–H15 | 0.0089 | 0.0310 | −0.0058 | 0.0068 | 0.0010 | |
| 0.0201 | 0.0725 | −0.0143 | 0.0162 | 0.0019 | ||
| o-TCBQ1w2 | C1–C2 | 0.2385 | −0.5295 | −0.2360 | 0.0518 | −0.1842 |
| 0.2577 | −0.6090 | −0.2794 | 0.0636 | −0.2158 | ||
| C2–C3 | 0.2693 | −0.6663 | −0.3057 | 0.0696 | −0.2361 | |
| 0.2784 | −0.6978 | −0.3376 | 0.0816 | −0.2560 | ||
| C3–C4 | 0.3293 | −0.9309 | −0.4844 | 0.1258 | −0.3586 | |
| 0.3137 | −0.8509 | −0.4384 | 0.1129 | −0.3256 | ||
| C4–C5 | 0.2629 | −0.6335 | −0.2920 | 0.0668 | −0.2252 | |
| 0.2876 | −0.7339 | −0.3604 | 0.0885 | −0.2720 | ||
| C1–C6 | 0.2717 | −0.6777 | −0.3108 | 0.0707 | −0.2401 | |
| 0.2826 | −0.7171 | −0.3471 | 0.0839 | −0.2632 | ||
| C5–C6 | 0.3283 | −0.9267 | −0.4814 | 0.1249 | −0.3565 | |
| 0.3123 | −0.8444 | −0.4341 | 0.1115 | −0.3226 | ||
| C1–O7 | 0.4103 | 0.0089 | −1.3803 | 0.6913 | −0.6891 | |
| 0.3781 | −0.2286 | −1.1681 | 0.5554 | −0.6126 | ||
| C2–O8 | 0.4155 | 0.0166 | −1.4097 | 0.7069 | −0.7028 | |
| 0.3863 | −0.2315 | −1.2087 | 0.5754 | −0.6333 | ||
| C3–Cl9 | 0.2067 | −0.3012 | −0.2198 | 0.0722 | −0.1475 | |
| 0.1930 | −0.2635 | −0.1987 | 0.0664 | −0.1323 | ||
| C4–Cl10 | 0.2065 | −0.3017 | −0.2183 | 0.0715 | −0.1469 | |
| 0.1919 | −0.2612 | −0.1957 | 0.0652 | −0.1305 | ||
| C5–Cl11 | 0.2073 | −0.3047 | −0.2199 | 0.0718 | −0.1480 | |
| 0.1925 | −0.2632 | −0.1967 | 0.0654 | −0.1312 | ||
| C6–Cl12 | 0.2073 | −0.3013 | −0.2205 | 0.0726 | −0.1479 | |
| 0.1929 | −0.2628 | −0.1985 | 0.0664 | −0.1321 | ||
| O7–H14 | 0.0165 | 0.0662 | −0.0109 | 0.0137 | 0.0028 | |
| 0.0264 | 0.1045 | −0.0213 | 0.0237 | 0.0024 | ||
| o-TCBQ2w1 | C1–C2 | 0.2384 | −0.5295 | −0.2357 | 0.0517 | −0.1840 |
| 0.2586 | −0.6137 | −0.2811 | 0.0638 | −0.2173 | ||
| C2–C3 | 0.2699 | −0.6692 | −0.3070 | 0.0699 | −0.2372 | |
| 0.2781 | −0.6969 | −0.3363 | 0.0811 | −0.2553 | ||
| C3–Cl9 | 0.2063 | −0.2999 | −0.2190 | 0.0720 | −0.1470 | |
| 0.1939 | −0.2666 | −0.2001 | 0.0667 | −0.1334 | ||
| C3–C4 | 0.3292 | −0.9305 | −0.4841 | 0.1258 | −0.3584 | |
| 0.3143 | −0.8543 | −0.4400 | 0.1132 | −0.3268 | ||
| C4–C5 | 0.2631 | −0.6343 | −0.2925 | 0.0669 | −0.2255 | |
| 0.2876 | −0.7344 | −0.3602 | 0.0883 | −0.2719 | ||
| C1–C6 | 0.2724 | −0.6809 | −0.3119 | 0.0709 | −0.2411 | |
| 0.2842 | −0.7250 | −0.3510 | 0.0849 | −0.2661 | ||
| C5–C6 | 0.3278 | −0.9248 | −0.4801 | 0.1245 | −0.3557 | |
| 0.3119 | −0.8433 | −0.4328 | 0.1110 | −0.3218 | ||
| C1–O7 | 0.4078 | 0.0051 | −1.3670 | 0.6842 | −0.6829 | |
| 0.3749 | −0.2362 | −1.1508 | 0.5459 | −0.6049 | ||
| C2–O8 | 0.4151 | 0.0141 | −1.4070 | 0.7053 | −0.7017 | |
| 0.3869 | −0.2277 | −1.2124 | 0.5777 | −0.6347 | ||
| C4–Cl10 | 0.2065 | −0.3019 | −0.2184 | 0.0715 | −0.1469 | |
| 0.1925 | −0.2628 | −0.1964 | 0.0654 | −0.1311 | ||
| C5–Cl11 | 0.2072 | −0.3044 | −0.2198 | 0.0719 | −0.1480 | |
| 0.1936 | −0.2668 | −0.1984 | 0.0659 | −0.1326 | ||
| C6–Cl12 | 0.2093 | −0.3025 | −0.2230 | 0.0737 | −0.1493 | |
| 0.1921 | −0.2603 | −0.1972 | 0.0661 | −0.1311 | ||
| O7–H14 | 0.0190 | 0.0770 | −0.0132 | 0.0162 | 0.0030 | |
| 0.0356 | 0.1281 | −0.0317 | 0.0318 | 0.0002 | ||
| O13–H18 | 0.0275 | 0.1023 | −0.0215 | 0.0236 | 0.0020 | |
| 0.0303 | 0.1048 | −0.0241 | 0.0251 | 0.0011 | ||
To elucidate these phenomena, the lowest unoccupied molecular orbital (LUMO) of o-TCBQ has been constructed considering the fact that the additional electron may enter its LUMO. As displayed in Fig. 3, all the LUMOs are characterized by π antibonding orbital except for the π bonding characters between the C–C single bonds (e.g., C1–C2 bond). Therefore, these bonds mentioned above should be weakened and strengthened since the additional electron enters the antibonding and bonding orbitals, respectively. As a result, as shown in Table 2, the corresponding deformation energy for o-TCBQ upon capturing an electron is 5.46 kcal mol−1.
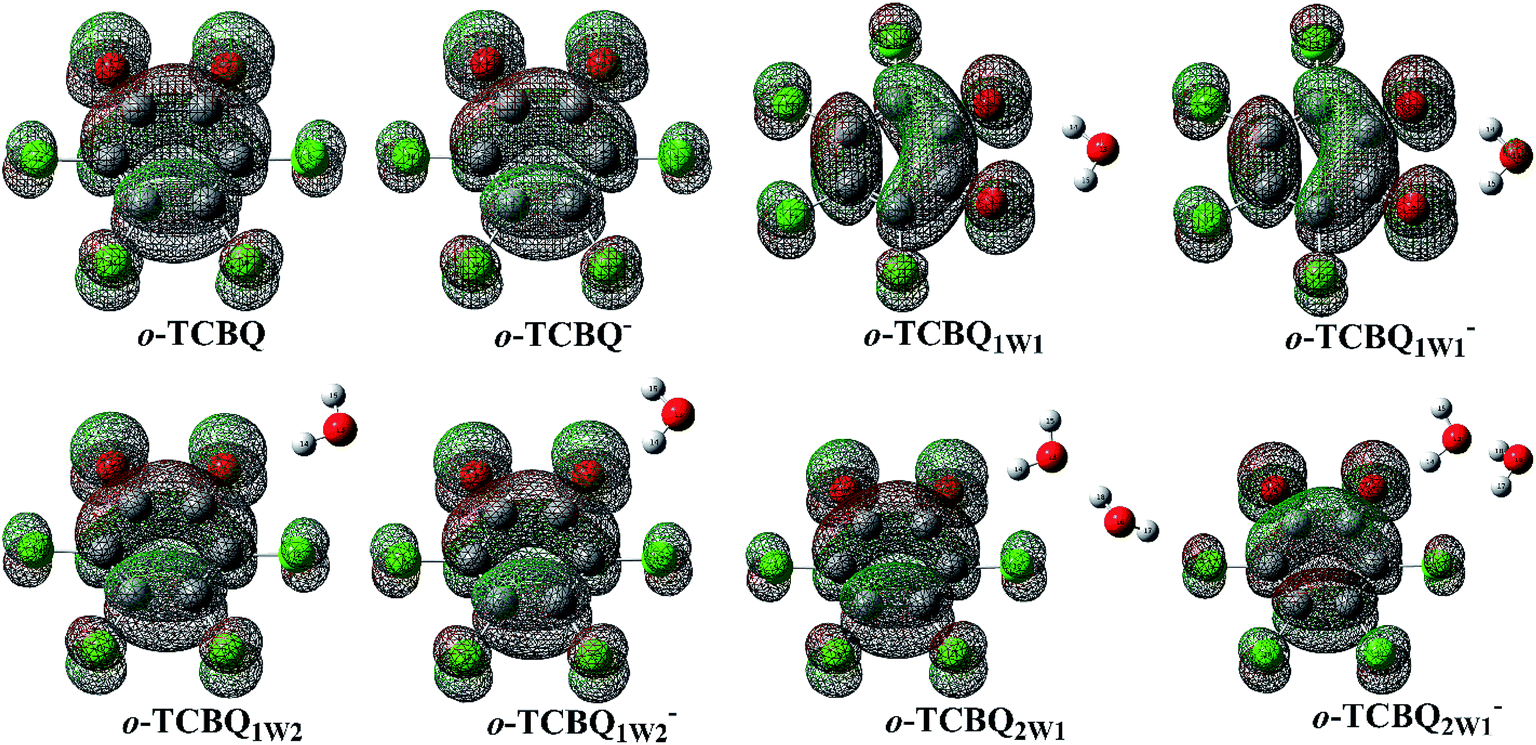 | ||
| Fig. 3 The LUMOs/HOMOs for the neutral/anionic o-TCBQ and its hydrated forms. The isodensity contours are 0.02 electron per bohr3. | ||
| ε | o-TCBQ | o-TCBQ1w1 | o-TCBQ1w2 | o-TCBQ2w1 |
|---|---|---|---|---|
| a All the units are in kcal mol−1. The data before and after the slash refer to the results of the neutral and anionic states, respectively. The data for ε = 1.00 refer to the results in the gas phase. | ||||
| 1.00 | 0.00/5.46 | 0.09/5.84 | 0.10/5.63 | 0.15/5.81 |
| 78.36 | 0.34/7.29 | 0.34/7.50 | 0.39/7.57 | 0.35/7.56 |
In bulk solvents, the original C2v symmetry of o-TCBQ has been changed to C2 symmetry before and after capturing an electron, exhibiting the solvent effects on the geometry. However, as for the p-TCBQ, its D2h symmetry is still kept in solvents before and after capturing an electron at the same level of theory.20 Moreover, as shown in Fig. 4, the structural changes increase with the increasing of the dielectric constant. Moreover, the geometry of o-TCBQ is no longer changed at ε = 10.
 | ||
| Fig. 4 The dependence of the deformation energy of o-TCBQ before and after capturing an electron on the ε of the solvents. | ||
To investigate the influences of the adjacent water molecules on the EA of o-TCBQ, microhydrated complexes containing one and two water molecules have been constructed. As shown in Fig. 1, o-TCBQ interacts with the water molecules via intermolecular H-bonds, which can be reflected from the presence of the bond critical points for those H-bonds as shown in Fig. 5. For the monohydrated case, two complexes named o-TCBQ1W1 and o-TCBQ1W2 have been located. In o-TCBQ1W1 complex, two intermolecular H-bonds have been observed, where water molecule acts as proton donor. Similarly, the same is also true for the other complex o-TCBQ1W2. As can be seen from the large H-bond distances ranging from 2.075 to 2.447 Å, these H-bonds should be weak. Actually, these H-bonds are mainly predominated by the electrostatic interactions in nature since the corresponding Laplacians of the electron density and energy densities at the BCPs of these H-bonds are positive within the framework of the AIM theory. As for the dihydrated case, the second water molecule mainly interacts with the first water molecule via intermolecular H-bond. Moreover, the H-bond distance between two water molecules is shorter than that of the H-bond between o-TCBQ and water molecule. Therefore, the H-bond interaction between water molecules should be stronger than the interaction of o-TCBQ with water molecule, which can be further confirmed by the larger electron density at the BCP of the former H-bond than the latter. As expected, the influences of water molecules on the geometry of o-TCBQ are small due to the weak interaction between them. This point can be further confirmed by the calculated small deformation energy of o-TCBQ upon microhydration, where the deformation energies are 0.09, 0.10, and 0.15 kcal mol−1 in o-TCBQ1W1, o-TCBQ1W2, and o-TCBQ1W2, respectively. However, all the intermolecular H-bonds have been strengthened more or less upon capturing an electron although the interaction modes are still kept, which can be reflected from the shorter H-bond distances relative to those of the neutral cases. This point can be understood since the negative charges on the proton acceptor increase upon capturing an electron as mentioned below.
 | ||
| Fig. 5 The molecular graphs of the neutral and hydrated o-TCBQ before and after capturing an electron, where the BCP and RCP are denoted as small red and yellow dots, respectively. | ||
3.2 Electron redistribution and spin density analyses
To further confirm the residence of the additional electron, the highest occupied molecular orbitals (HOMOs) of the anionic o-TCBQ and its hydrated complexes have been constructed to compare with the LUMOs of the neutral o-TCBQ. As displayed in Fig. 3, both the HOMOs and LUMOs are similar to each other. Therefore, similar to p-TCBQ,20 the additional electron should enter the HOMO of o-TCBQ, resulting in the corresponding structural changes mentioned above.To gain the information for the electron density redistribution upon capturing an electron, electron density difference maps have been constructed on the basis of the geometry of o-TCBQ before and after capturing an electron vertically. As displayed in Fig. 6, the loss of the electron density is mainly observed in the regions along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (e.g., C3C4 and C5C6 bonds) as well as the peripheral regions of the C and O atoms. This point is consistent with the elongation of the double bonds upon capturing an electron. However, the gained electron density is mainly distributed on the regions along the C–C single bond and the carbonyl O atom and four Cl atoms. As a result, on the basis of the NBO analyses, it was found that all the atoms have gained several electrons ranging from 0.10 to 0.15 except for those C atoms associated with the CC double bonds.
C double bonds (e.g., C3C4 and C5C6 bonds) as well as the peripheral regions of the C and O atoms. This point is consistent with the elongation of the double bonds upon capturing an electron. However, the gained electron density is mainly distributed on the regions along the C–C single bond and the carbonyl O atom and four Cl atoms. As a result, on the basis of the NBO analyses, it was found that all the atoms have gained several electrons ranging from 0.10 to 0.15 except for those C atoms associated with the CC double bonds.
 | ||
| Fig. 6 Electron density difference maps for o-TCBQ and its monohydrated form upon capturing an electron (left and right) and the neutral o-TCBQ upon monohydration (middle). The blue and violet regions represent the depleted and increased electron density, respectively. The isodensity contours are 0.004 electron per bohr3. | ||
As for the hydrated o-TCBQ, as displayed in Fig. 6, the electron density distribution is similar to that of the isolated o-TCBQ, suggesting slight effects of adjacent water molecules on the electron density distribution upon accepting an electron. On the other hand, the effect of the water molecule can be also reflected from the electron density distribution of o-TCBQ upon monohydration. As shown in Fig. 6, the loss and gain of the electron density are concentrated on the positions of the H atom of the H-bond donors and the O atom of the H-bond acceptors, respectively. This point is consistent with the formed intermolecular H-bonds between water molecules and o-TCBQ mentioned above.
To confirm the distribution of the additional electron, we have performed the spin density analyses for the anionic o-TCBQ and its hydrated complexes on their optimized structures. As displayed in Fig. 7, nearly half of the introduced electrons are localized on the six-membered ring except for the regions containing C3–C4 and C5–C6 bonds. For the other half of the unpaired electrons, they have been observed on the two carbonyl O atoms. Similarly, the same is also true for the hydrated o-TCBQ, implying that the adjacent water molecules have no effects on the distributions of the unpaired electron.
 | ||
| Fig. 7 The spin density maps for o-TCBQ and its hydrated complexes after accepting an electron. The isodensity contours are 0.004 electron per bohr3. | ||
3.3 Electron affinity
As mentioned above, from the molecular dynamics results in Fig. 2, the time scale is about 6 fs in the whole process of the electron capturing, accompanying with the corresponding structural relaxation. The energy decreases from point a to b suggest that the electron capturing process is favorable thermodynamically. Actually, the corresponding Gibbs free energy change is −72.2 kcal mol−1 in the electron-accepting process.Table 3 gives the calculated vertical and adiabatic EAs of o-TCBQ in the gas phase and in different solvents. In the gas phase, the calculated VEA and AEA are 2.85 and 3.12 eV. Here, the large difference between VEA and AEA (about 0.27 eV) is consistent with the structural relaxation mentioned above. Obviously, the positive value of EA suggests that anionic o-TCBQ is more stable than the neutral state. In bulk solution, as displayed in Fig. 8, all the EAs have been increased by about 0.50–1.65 eV with the increasing of dielectric constant, suggesting that the EA of o-TCBQ is solvent-sensitive. Similarly, the same phenomenon has also observed for p-TCBQ.20 Thus, the bulk solvents can efficiently enhance the electron-accepting ability of o-TCBQ significantly.
| ε | AEA | VEA | VDE |
|---|---|---|---|
| a All the units are in eV. The data in parentheses refer to the results of the p-TCBQ.20 | |||
| 1.00 | 3.12(3.07) | 2.85(2.75) | 3.32(3.31) |
| 1.43 | 3.63 | 3.35 | 3.82 |
| 1.52 | 3.70 | 3.42 | 3.89 |
| 1.91 | 3.92 | 3.65 | 4.12 |
| 4.71 | 4.45 | 4.17 | 4.65 |
| 10.13 | 4.63 | 4.35 | 4.85 |
| 24.85 | 4.73 | 4.44 | 4.95 |
| 36.56 | 4.75 | 4.46 | 4.97 |
| 78.36 | 4.77 | 4.48 | 4.99 |
 | ||
| Fig. 8 The calculated AEA, VEA, and VDE as a function of the ε of the solvents. | ||
As for the hydrated o-TCBQ, as shown in Table 4, the calculated VEA and AEA are all positive and larger than those of the results in the gas phase. Therefore, the adjacent water molecules around o-TCBQ are favorable to the capture of an electron although the presence of water molecules has a slight effect on the geometry of o-TCBQ as mentioned above. Moreover, as shown in Table 4, all the EAs are further significantly increased with the increasing of the ε in bulk solvents. For example, the calculated VEA (3.22 eV) and AEA (3.53 eV) for o-TCBQ1w1 have been increased to 4.54 and 4.84 eV in aqueous solution, respectively. Additionally, note that the interaction modes of water molecules in the hydrated o-TCBQ have a large effect on the EA in the gas phase. However, this phenomenon disappears in aqueous solution. For example, the EA of TCBQ1w1 is larger by about 0.12 eV than that of TCBQ1w2 in the gas phase. In aqueous solution, the EAs of them are equal to each other.
| Complexes | AEA | VEA | VDE |
|---|---|---|---|
| a All the units are in eV. The data in parentheses refer to the results in aqueous solution employing PCM model. | |||
| TCBQ1w1 | 3.53(4.84) | 3.22(4.54) | 3.86(5.07) |
| TCBQ1w2 | 3.41(4.84) | 3.09(4.53) | 3.73(5.07) |
| TCBQ2w1 | 3.53(4.84) | 3.04(4.54) | 3.91(5.08) |
| F-TCBQ | 3.05 | 2.67 | 3.32 |
| Cl-TCBQ | 3.12 | 2.85 | 3.32 |
| Br-TCBQ | 3.15 | 2.90 | 3.32 |
Compared with the p-TCBQ, o-TCBQ has a larger EA value, implying its strong electron-accepting ability. Moreover, the halogen-substitution effects on the EA have been investigated. As a general rule, as shown in Table 4, the EA increases in the order of F-, Cl-, and Br-substituted case.
As also shown in Tables 3 and 4, on the basis of the anionic o-TCBQ, all the calculated VDEs are positive in the gas phase and in solution. Moreover, as displayed in Fig. 8, all of them increase with the increasing of the ε of bulk solvents, which is also true for the p-TCBQ.20 Similarly, the explicit water molecules also make the VDE significantly increase. Therefore, the anionic state of o-TCBQ should be stable with respect to the detachment of an electron.
In summary, similar to p-TCBQ, o-TCBQ should behave as a good electron acceptor in the charge-transfer reactions in different environmental media.
4. Conclusions
In this study, the one-electron reduction behavior of o-TCBQ has been systematically investigated theoretically. The main conclusions are summarized as follows.(1) Large geometrical changes occur for the o-TCBQ if an additional electron has been introduced. Meanwhile, the C2v symmetry of neutral and anionic o-TCBQ in the gas phase has been changed to be C2 symmetry in solution. Moreover, the intermolecular H-bonds between o-TCBQ and water molecules have been strengthened upon accepting an electron.
(2) The electron-accepting ability of o-TCBQ can be enhanced by the adjacent explicit water molecules although they have slight effects on the geometry of o-TCBQ.
(3) Similar to p-TCBQ, the EA and VDE of o-TCBQ are positive in the gas phase and in bulk solution, which increase with the increasing of the dielectric constant of the bulk solvent. Therefore, o-TCBQ is a good electron acceptor in different media.
Acknowledgements
This work is supported by NSFC (21577076, 21303093, and 21003082) and the NSF of Shandong Province (ZR2014BM020). The State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences (KF2013-05) is also acknowledged.References
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Chem. Res. Toxicol., 2000, 13, 135–160 CrossRef CAS PubMed.
- Y. Song, B. A. Wagner, J. R. Witmer, H. J. Lehmler and G. R. Buettner, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9725–9730 CrossRef CAS PubMed.
- B. Meunier, Science, 2002, 296, 270–271 CrossRef CAS PubMed.
- S. S. Gupta, M. Stadler, C. A. Noser, A. Ghosh, B. Steinhoff, D. Lenoir, C. P. Horwitz, K. Schramm and T. J. Collins, Science, 2002, 296, 326–328 CrossRef PubMed.
- A. Sorokin, J. L. Seris and B. Meunier, Science, 1995, 268, 1163–1166 CAS.
- Y. L. Zhao, F. Qin, J. M. Boyd, J. Anichina and X. F. Li, Anal. Chem., 2010, 82, 4599–4605 CrossRef CAS PubMed.
- F. Qin, Y. Y. Zhao, Y. L. Zhao, J. M. Boyd, W. J. Zhou and X. F. Li, Angew. Chem., Int. Ed., 2010, 49, 790–792 CrossRef CAS PubMed.
- D. H. Sarr, C. Kazunga, M. J. Charles, J. G. Pavlovich and M. D. Aitken, Environ. Sci. Technol., 1995, 29, 2735–2740 CrossRef CAS PubMed.
- B. Z. Zhu, N. Kitrossky and M. Chevion, Biochem. Biophys. Res. Commun., 2000, 270, 942–946 CrossRef CAS PubMed.
- B. Z. Zhu, H. Zhao, B. Kalyanaraman and B. Frei, Free Radical Biol. Med., 2002, 32, 465–473 CrossRef CAS PubMed.
- B. Z. Zhu, B. Kalyanaraman and G. B. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17575–17578 CrossRef CAS PubMed.
- B. Z. Zhu and G. Q. Shan, Chem. Res. Toxicol., 2009, 22, 969–977 CrossRef CAS PubMed.
- R. Yin, D. Zhang, Y. Song, B. Z. Zhu and H. Wang, Sci. Rep., 2013, 3, 1269 Search PubMed.
- B. Z. Zhu, H. T. Zhao, B. Kalyanaraman, J. Liu, G. Q. Shan, Y. G. Du and B. Frei, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3698–3702 CrossRef CAS PubMed.
- B. Z. Zhu, G. Q. Shan, C. H. Huang, B. Kalyanaraman, L. Mao and Y. G. Du, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11466–11471 CrossRef CAS PubMed.
- G. Q. Shan, A. Yu, C. F. Zhao, C. H. Huang, L. Y. Zhu and B. Z. Zhu, J. Org. Chem., 2014, 80, 180–189 CrossRef PubMed.
- B. Z. Zhu, J. G. Zhu, L. Mao, B. Kalyanaraman and G. Q. Shan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20686–20690 CrossRef CAS PubMed.
- C. H. Huang, F. R. Ren, G. Q. Shan, H. Qin, L. Mao and B. Z. Zhu, Chem. Res. Toxicol., 2015, 28, 831–837 CrossRef CAS PubMed.
- P. Li, W. H. Wang, Q. Sun, Z. Li, A. J. Du, S. W. Bi and Y. Zhao, ChemPhysChem, 2013, 14, 2737–2743 CrossRef CAS PubMed.
- P. Li, W. H. Wang, H. T. Sun and S. W. Bi, Comput. Theor. Chem., 2013, 1006, 127–132 CrossRef CAS.
- P. Ranzieri, M. Masino and A. Girlando, J. Phys. Chem. B, 2007, 111, 12844–12848 CrossRef CAS PubMed.
- C. Katan, P. E. Blöchl, P. Margl and C. Koenig, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 12112–12120 CrossRef CAS.
- S. E. Boesch and R. A. Wheeler, J. Phys. Chem. A, 1997, 101, 8351–8359 CrossRef CAS.
- S. E. Boesch and R. A. Wheeler, J. Phys. Chem., 1995, 99, 8125–8134 CrossRef CAS.
- A. Girlando, I. Zanon, R. Bozio and C. Pecile, J. Chem. Phys., 1978, 68, 22–31 CrossRef CAS.
- D. A. Horke and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2011, 13, 19546–19552 RSC.
- B. S. Das, S. G. Reid, J. L. Betts and K. Patrick, J. Fish. Res. Board Can., 1969, 26, 3055–3067 CrossRef CAS.
- X. W. Guo and H. Mayr, J. Am. Chem. Soc., 2014, 136, 11499–11512 CrossRef CAS PubMed.
- S. Maddila, V. D. B. C. Dasireddy and S. B. Jonnalagadda, Appl. Catal., B, 2013, 138–139, 149–160 CrossRef CAS.
- M. Freytag, P. G. Jones, R. Schmutzler and M. Yoshifuji, Heteroat. Chem., 2001, 12, 300–308 CrossRef CAS.
- M. Kot and W. Zaborska, J. Enzyme Inhib. Med. Chem., 2006, 21, 537–542 CrossRef CAS PubMed.
- H. R. Zare, M. Eslami, M. Namazian and M. L. Coote, J. Phys. Chem. B, 2009, 113, 8080–8085 CrossRef CAS PubMed.
- M. T. Huynh, C. W. Anson, A. C. Cavell, S. S. Stahl and S. Hammes-Schiffer, J. Am. Chem. Soc., 2016, 138, 15903–15910 CrossRef CAS PubMed.
- S. Arulmozhiraja and M. Morita, J. Phys. Chem. A, 2004, 108, 3499–3508 CrossRef CAS.
- Y. Y. Zhao, F. M. Tao and E. Y. Zeng, J. Phys. Chem. A, 2007, 111, 11638–11644 CrossRef CAS PubMed.
- M. Melicherčík, L. F. Pašteka, P. Neogrády and M. Urban, J. Phys. Chem. A, 2012, 116, 2343–2351 CrossRef PubMed.
- A. Gupta, H. M. Jaeger, K. R. Compaan and H. F. Schaefer III, J. Phys. Chem. B, 2012, 116, 5579–5587 CrossRef CAS PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer III, S. Nandi and G. B. Ellison, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed.
- W. L. Feng, C. Ren, W. H. Wang, C. Guo, Q. Sun and P. Li, Theor. Chem. Acc., 2016, 135, 190 CrossRef.
- W. H. Wang, X. X. Zhang, P. Li, Q. Sun, Z. Li, C. Ren and C. Guo, J. Phys. Chem. A, 2015, 119, 796–805 CrossRef CAS PubMed.
- W. L. Feng, C. Ren, W. H. Wang, C. Guo, Q. Sun and P. Li, RSC Adv., 2016, 6, 48099–48108 RSC.
- R. F. W. Bader, Atoms in molecules: A quantum theory. Oxford University Press, Oxford, UK, 1990 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 114, 9758–9763 CrossRef CAS.
- S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 115, 10291–10302 CrossRef CAS.
- H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2002, 117, 8694–8704 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
- K. Hagen and K. Hedberg, J. Mol. Struct., 1978, 49, 351–360 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |