
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA poly(amidoamine) dendrimer-based drug carrier for delivering DOX to gliomas cells†
Yan Liab,
Hai Heb,
Wanliang Lu*c and
Xinru Jia *b
*b
aCollege of Materials Science and Engineering, Hebei University of Engineering, Handan 056038, China
bBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of the Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: xrjia@pku.edu.cn
cState Key Laboratory of Natural and Biomimetic Drugs and School of Pharmaceutical Science, Peking University, Beijing, China. E-mail: luwl@bjmu.edu.cn
First published on 8th March 2017
Abstract
A drug delivery carrier G4–FA–PEG with PEG and FA modified on the periphery of G4.0 poly(amidoamine) (PAMAM) dendrimer was synthesized, and doxorubicin (DOX) was encapsulated in the interior. The in vitro cytotoxicity and cellular uptake in gliomas cells were both enhanced via a FR-mediated endocytosis pathway.
Introduction
Gliomas, as the most common primary brain tumors, often result in the death of patients within several years.1,2 In spite of the extensive application of surgery, radiotherapy and chemotherapy in the treatment of malignant gliomas, the therapeutic effect is always poor.3 Therefore, it is crucial to develop safe and effective drug/gene delivery systems. Over the past decades, the development of nonviral drug delivery carriers, such as liposomes,4,5 quantum dots,6 polymeric nanoparticles,7 gold nanoparticles8 and micelles9 has been the focus of contemporary drug delivery research.10Dendrimers are a novel class of macromolecules with controllable nano-size, easily modifiable periphery groups and interior cavities. Particularly, poly(amidoamine) (PAMAM) dendrimers closely match in sizes and contours with some important proteins and bioassemblies.11 Such macromolecules are recognized as the unique nanoscale carriers and currently being widely studied for their applications in diagnostics, imaging and treatment of cancers.12 To meet the pharmaceutical and biomedical requirements, various strategies have been employed to endow the dendrimers with multi-functions.13 Commonly, the peripheral modification with functional moieties, including targeting ligands, biocompatible linkers, drug molecules and image contrast agents is considered as an effective way to tune and manipulate the properties of dendrimers.
As reported, covalent attachment of poly(ethylene glycol) (PEG), a nontoxic and nonimmunogenic hydrophilic polymer, to the exterior of dendrimers could reduce the toxicity, improve biocompatibility and prolong circulation time of the dendrimer-based drug delivery systems due to the decrease of surface positive charges and increase of the size and molecular weight.14,15 In addition, these PEGylated carriers were expected to have better permeability and retention (EPR) effect so as to afford the enhanced ability to enter tumor tissues.16 A. D'Emanuele et al. studied the cytotoxicity of PAMAM peripherally modified with lauroyl or PEG chains. They reported that such modification showed particularly effective for decreasing cytotoxicity due to the reduction/shielding of the positive charges on the exterior of PAMAM dendrimers.17 Kono et al. designed and synthesized a PAMAM-based nanocapsule with an amino acid-based shell that was further modified by linking PEG chains. They found that such dendrimers displayed high ability for encapsulating guest molecules.18,19 To provide guidelines in designing appropriate dendritic scaffolds for drug delivery, Kim et al. synthesized a series of PEGylated G3 PAMAM dendrimers. They suggested that a lower degree of substitution with shorter PEG chains was sufficient for them as efficient universal scaffolds for drug delivery.20
It is well known that the site-specific delivery of anticancer drugs remains a great challenge because it is difficult to control the drugs mainly accumulated in the tumor site rather than permeating in the normal tissues in vivo, which leads to the harmful side-effects and even death of the patients. Many targeting agents have been tried to conjugate on the surface of the dendrimers to explore the targeting effects.21,22 Folic acid (FA), a necessary vitamin for the proliferation of cells, has been wildly studied for drug targeting because the receptors (FRs) of FA are over expressed on cancer cells and activated macrophages, while it shows a limited distribution in normal tissues.23,24 The advantages of using FA as a ligand are due to (1) the high affinity of FA to FRs (Kd = 10−10 M) was maintained even after FA was conjugated via the γ-carboxyl group to other molecules;25 (2) the deeper penetration effect than antibodies because of its small size and low molecular weight.26,27
Herein, we report a PAMAM dendrimer-based drug delivery carrier (G4–FA–PEG/DOX) with PEG2000 partly linked to the exterior, doxorubicin (DOX) encapsulated in the interior and FA directly conjugated on the periphery of the carrier for taking the advantage of “cluster” effect.28 Our aim is to endow the carrier with good biocompatibility and enhanced endocytosis through both the targeting of FA and electrostatic interaction between the residual positive charges of PAMAM and negative cell membrane. DOX was an effective and widely used anti-cancer drug, which was also reported to treat gliomas.1 For comparison, temozolomide (TMZ), an alkylating agent which was able to cross the blood–brain barrier (BBB) and commonly used for the treatment of aggressive brain tumor, was also encapsulated in the carrier of G4–FA–PEG. Various measurements, including 1H NMR, FTIR, UV-visible spectra and DLS were performed to characterize the properties of resulting carrier. The murine C6 glioma cells were particularly chosen to evaluate the cytotoxicity, the targeting effect and the intracellular localization of the drugs delivered by the carriers. To the best of our knowledge, the report referred to the carriers with FA targeting to glioma cells is still rare.
Materials and methods
Materials
Folic acid from Sigma-Aldrich and doxorubicin from Nanjing Tianzunzezhong Co., Ltd. were both used as received. O-(2-Carboxyethyl) polyethylene glycol (HO–PEG–COOH, Mw: 2000) was purchased from Biomatrik Inc. Hydrochloride salt of 1-(3-dimethylaminopropyl)-3-ethylcarbo-diimide (EDC) was from GL Biochem (Shanghai) Ltd. Dimethylsulfoxide (DMSO), dimethylformamide (DMF), tetrahydrofuran (THF), methanol (CH3OH), acetone and anhydrous diethyl ether (all from Beijing Chemical Reagents) were purified following the standard procedures. G4 PAMAM dendrimers were prepared and purified according to the literature procedures.29For cell culture experiments, murine C6 glioma cells were from Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing). Folate-free RPMI 1640 medium was purchased from BioInd. Dulbecco's Modified Eagle's Medium (DMEM) was from Macgene technology Co., Ltd.
Synthesis of G4–FA
FA was conjugated to the periphery of G4.0 PAMAM dendrimers via EDC coupling reaction as described in the literature.30,31 Briefly, FA (0.335 g, 760 μmol) was dissolved in the mixed solvent of 18 mL DMF and 6 mL DMSO. To the above solution, EDC (2.06 g, 10.6 mmol) was added and stirred at room temperature for 1 h. The activated FA was added dropwise to 90 mL aqueous solution of G4.0 PAMAM (0.337 g, 23.7 μmol) and the solution was stirred in dark under nitrogen atmosphere at room temperature for 3 days. The solution was concentrated by rotary evaporation, and then precipitated in acetone. The crude product was dissolved in water and dialyzed against deionized water for 2 days to remove the unreacted FA. After lyophilization, the product was stored in dark. The number of FA linked to the dendrimer surface was estimated by 1H NMR (400 MHz). 1H NMR (D2O, 400 MHz, ppm): δ 8.66 (br, FA, protons at 7 position of pterin ring), 7.61 (br, FA, protons of phenyl next to amide group), 6.77 (br, FA, protons of phenyl next to amino group), 3.50–3.05 (m, PAMAM, –CONHCH2CH2–), 2.98–2.76 (m, PAMAM, –CH2CH2NH– and –NCH2CH2CO–), 2.75–2.60 (m, PAMAM, –CH2CH2N![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ), 2.57–2.34 (m, PAMAM, 137-CH2CH2CONH–).
), 2.57–2.34 (m, PAMAM, 137-CH2CH2CONH–).
Synthesis of G4–FA–PEG
The attachment of PEG to the dendrimer surface was similar with that of FA. Briefly, HO–PEG–COOH (0.6 g, 300 μmol) was dissolved in 7.5 mL DMF. EDC (0.8 g, 4.17 mmol) in 2.5 mL DMSO was added to the above solution. After stirring for 2.5 h, the yielding solution was transferred to G4–FA (279 mg, 16.6 μmol) in deionized water drop by drop. The reaction was stirred in dark for 3 days and concentrated. After that, repeated precipitation in THF was carried out to remove unreacted PEG which could be proved by thin layer chromatography (TLC). The number of PEG conjugated was calculated by 1H NMR (400 MHz) as well. 1H NMR (d6-DMSO, 400 MHz, ppm): δ 8.66 (br, FA, protons at 7 position of pterin ring), 7.72 (br, FA, protons of phenyl next to amide group), 6.63 (br, FA, protons of phenyl next to amino group), 7.13, 6.17 (m, –CONH–), 3.67–2.88 (m, PAMAM, –CONHCH2CH2–, –CH2CH2NH– and –NCH2CH2CO–; PEG, –CH2CH2O–), 2.87–2.60 (m, PAMAM, –CH2CH2N), 2.59–2.39 (m, PAMAM, 137-CH2CH2CONH–).
Drug loading
G4–FA–PEG (153 mg, 5 μmol) was dissolved in 7 mL deionized water. DOX (58.1 mg, 100 μmol) in 7 mL deionized water was added dropwise to the dendrimer solution and stirred for 24 h in dark. The removal of free DOX was conducted by ultrafiltration and dialyzed twice against deionized water for 40 min. The absorbance at 482 nm from UV-vis spectra was used to calculate the percentage of DOX encapsulated.The encapsulation of TMZ was following the similar procedure except DMSO as the solvent.
Particle size measurement
The particle size was measured with a zeta PALS analyzer (Brookhaven Instruments Corporation, BIC) equipped with a 35 mW solid state laser (660 nm), using BIC particle sizing software (9kpsdw32, ver.2.3). All the samples including G4.0 PAMAM, G4–FA, G4–FA–PEG and G4–FA–PEG/DOX were dissolved in 10 mM PBS (pH 7.4, 1 mg mL−1) and filtered through a Minisart High-Flow, cellulose acetate 0.2 μm syringe filter (Sartorius Stedim Biotech GmbH, Germany) into the scattering cell. The measurement was carried out at 37 °C.In vitro DOX release
The DOX release was carried on in PBS (pH 7.4) at 37 °C. 10.2 mg G4–DOX–PEG/DOX in 5 mL buffer solution was sealed in a dialysis bag (MWCO 8000) and immersed in 35 mL of the same buffer solution. A portion of 3 mL dialyzate was taken out at various time and replaced by 3 mL fresh buffer solution. The release of free DOX was performed in the same condition as a control. The released DOXs from the carrier were calculated with a standard curve draw by the ultraviolet spectrometry with the absorption wavelength of DOX at 482 nm.Cell cultures
Murine C6 glioma cells were routinely grown in DMEM supplemented by 10% heated-inactivated fetal bovine serum (FBS), antibiotics (penicillin 100 U mL−1, and streptomycin 100 μg mL−1) (all from GIBCO-BRL Life Technologies, Beijing local agent, China). The cells were maintained at 37 °C with 5% CO2 and harvested after 80% confluence.Cytotoxicity assays
The cytotoxicity of free DOX and G4–FA–PEG/DOX was determined by sulforhodamine B (SRB) assay. C6 cells were seeded into 96-well plates at a density of 5 × 103 cells per well and cultured for 24 h before use. Then free DOX and G4–FA–PEG/DOX were added to the plates at a series of DOX concentrations from 0 to 10 μM. Blank G4–FA–PEG without DOX was used to test the cytotoxicity of carrier. After 48 h incubation, the cells were washed three times with PBS and treated by SRB staining assay. The absorbance at 540 nm was detected with a microplate reader and the cell viability was calculated using the following formula: survival% = (A540 nm for the treated cells/A540 nm for the control cells) × 100%, where the A540 nm was the absorbance value. Each assay was repeated for at least three times. IC50 values were calculated from the dose–effect curve and expressed as concentration (μM) of DOX-equiv.32The measurement of cytotoxicity of free TMZ and G4–FA–PEG/TMZ was following the same procedure as above.
Cellular uptake and competition assay of FA
Before the experiment, the cells were cultured in FA-free RPMI 1640 medium for some days. Then the cells were harvested and seeded into 6-well culture plates at a density of 5 × 105 cells per well and incubated for 24 h. Free DOX and G4–FA–PEG/DOX were added at a DOX concentration of 10 μM and the cells were incubated for 2 h. Cells without drugs were used as a blank control. Thereafter, the cells were washed with cold PBS for three times and harvested. Since DOX has a quite strong fluorescence, it can be easily detected by FAScan flow cytometer (Becton Dickinson FACSCalibur, Mountain View, CA, USA).33 FL2-H filter (585 ± 21 nm) was used for the collection of fluorescence intensity and the events collected were ten thousands.For the competition assay, FA in RPMI solution (1 mM) was added to the cells and incubated for 30 min in advance. Afterwards, free DOX and G4–FA–PEG/DOX were added to the cells for making the final DOX concentration of 10 μM and maintained for 2 h at 37 °C. The cells only with FA solution were utilized as a blank control.
The intracellular localization of DOX in C6 cells was detected with laser scanning confocal microscope (Leica SP2, Heidelberg, Germany). Cells were cultured in chambered coverslips for 24 h and treated with free DOX or G4–FA–PEG/DOX at a DOX concentration of 10 μM. After 2 h incubation, the cells were washed with cold PBS for three times, fixed with 4% (v/v) paraformaldehyde and finally stained with Hoechst 33258.
The competition assay was similar as the above.
Statistics analysis
Data were presented as mean ± standard deviation. One-way analysis of variance (ANOVA) was used to determine significance among groups following the Bonferroni's post-test.Results and discussion
Synthesis and characterization of G4–FA–PEG
To reduce the side effect of therapeutic agents, their targeting efficiency should be improved. One effective strategy is the modification of ligands onto the surface of the carrier. Here, we select FA as the functional ligand because of its comprehensive advantages of low immunogenicity, functional stability, over expression of receptors on cancer cells, and the nondestructive cellular internalization pathway.2 Furthermore, while multiple ligands are simultaneously bound on one carrier, the multivalent interactions with the receptors are much stronger than the corresponding monovalent interaction, which is called “cluster” effect.28 For example, Prof. S. K. Choi and coworkers detected higher binding avidity of FA to the receptors with a multivalent enhancement factor of 167 when six FA moieties were conjugated on G5 PAMAM dendrimers compared to free FA.34In present study, FA was conjugated on the periphery of the fourth generation PAMAM through a carbodiimide mediated amide linkage by the reaction of exterior primary amino groups of PAMAM with the γ-carboxyl group of FA (Fig. 1). 1H NMR measurement was used to analyze and calculate the number of FA conjugated on the periphery of PAMAM (Fig. 2a). From the integration ratio of the proton signals at 7.67 ppm (phenyl protons of FA) to those at 2.49 ppm corresponding to the protons of methylenes next to the carbonyl groups in PAMAM branches, the average number of FA attached on PAMAM exterior was 5.9 per PAMAM molecule. Similarly, the number of PEG conjugated on the periphery of PAMAM was evaluated to be 6.9 per PAMAM (Fig. 2b).
 | ||
| Fig. 1 Schematic illustration of preparation of G4–FA–PEG dendrimers and the DOX encapsulation within the dendrimers. | ||
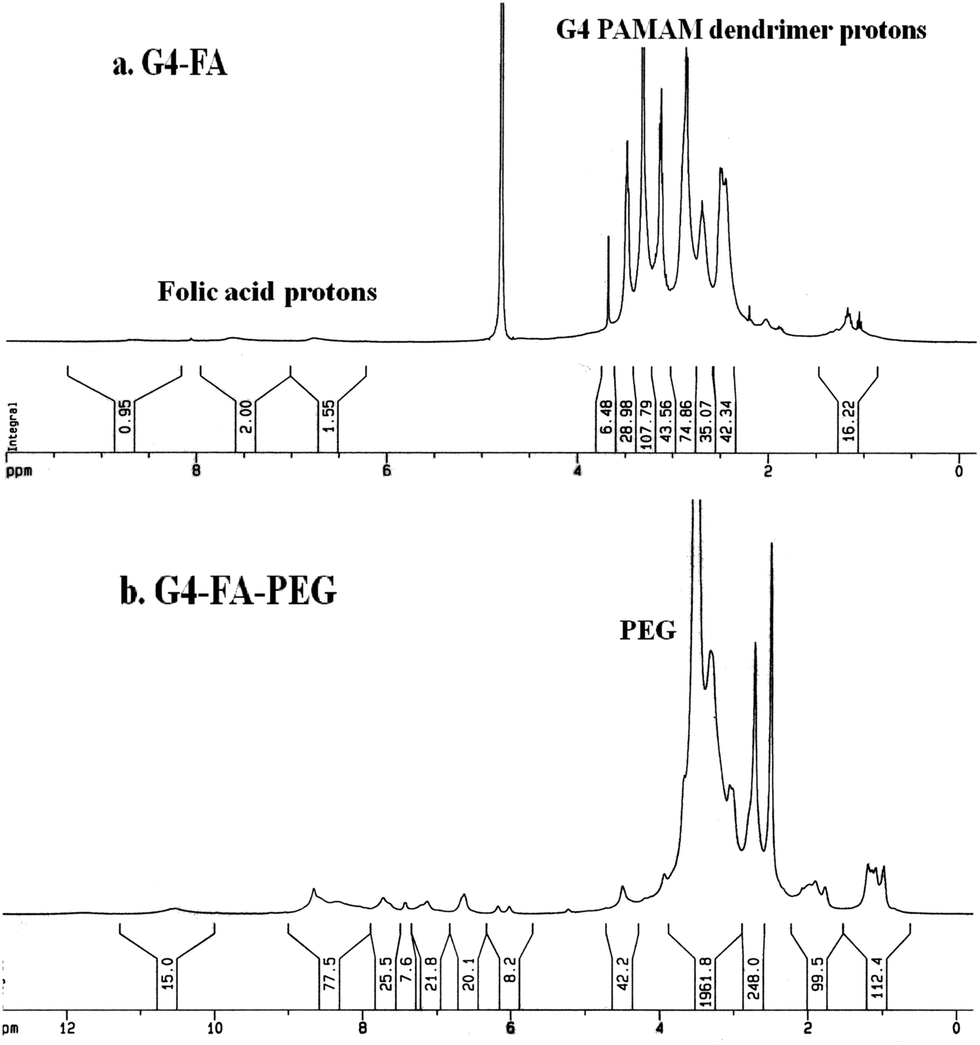 | ||
| Fig. 2 1H NMR spectra of (a) G4–FA in D2O; (b) G4–FA–PEG in d6-DMSO. | ||
Fourier transform infrared (FT-IR) measurement further proved the successful conjugation (Fig. 3). Firstly, the peaks at 3286 and 3345 cm−1 (primary amino groups of PAMAM) turned into a broad absorption band and shifted to 3346 cm−1, and the stretching vibration of –OH group (FA, 2925 cm−1) shifted to 2936 cm−1. Moreover, the characteristic absorption bands of amide I and II at 1639 and 1554 cm−1 shifted to 1645 and 1536 cm−1, respectively, further confirming the involvement of –COOH of FA and –NH2 of G4 PAMAM in the formation of the conjugates.30
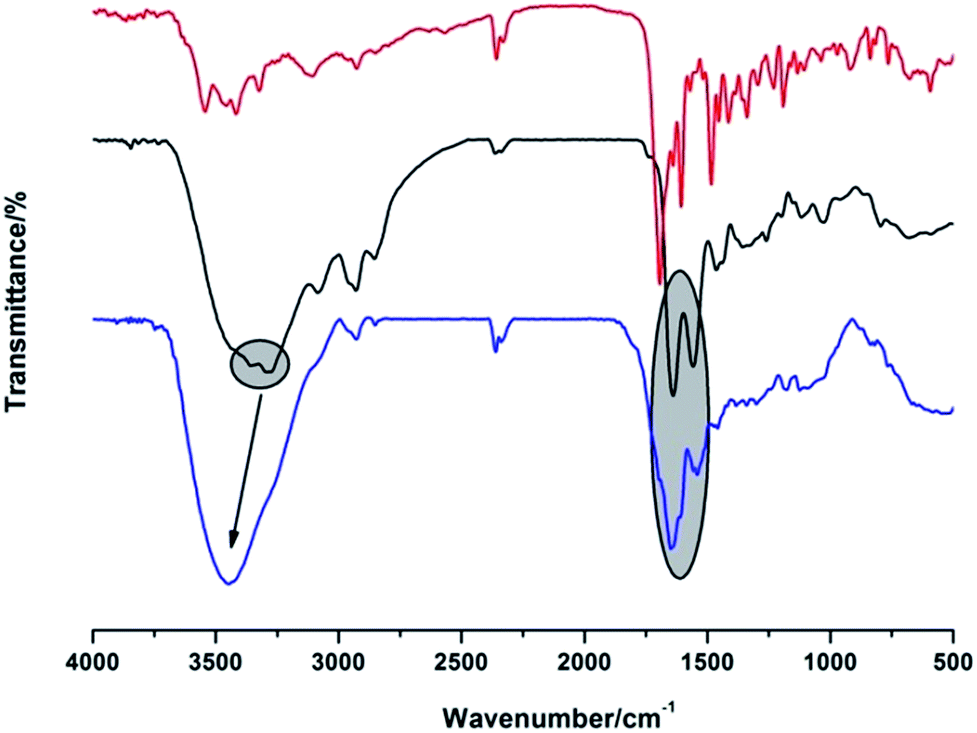 | ||
| Fig. 3 FT-IR spectra of FA, G4 PAMAM and G4–FA (from top to bottom). | ||
DLS measurement was used to detect the sizes of G4 PAMAM, G4–FA and G4–FA–PEG. It showed that the size appeared decrease in the order of G4–FA–PEG (12.1 nm) > G4–FA (5.4 nm) > G4 PAMAM (4.7 nm). The large size of G4–FA–PEG was due to the grafting of PEG chains. This result was similar to that reported by Jiang et al., who also found the size enlarged from 5.00 nm (G4 PAMAM) to 11.74 nm (PEG–PAMAM 16/1).35
Drug loading
The loading of DOX was carried out by adding the aqueous solution of DOX to G4–FA–PEG in deionized water. Excess DOX was removed by ultrafiltration and then followed by dialysis. Based on the UV-Vis spectra analysis (Fig. 4), the loading capacity of G4–FA–PEG was calculated to be 16.9% (w/w), which was much higher than the mPEG2000–G4 PAMAM reported by Kojima et al.19 It may be due to the conjugation of FA on the surface of PAMAM. The improvement of loading capacity by introduction of FA was also observed by Chandrasekar, whose work showed increased indomethacin encapsulation with more FA molecules conjugated on the exterior of G4 PAMAM dendrimers.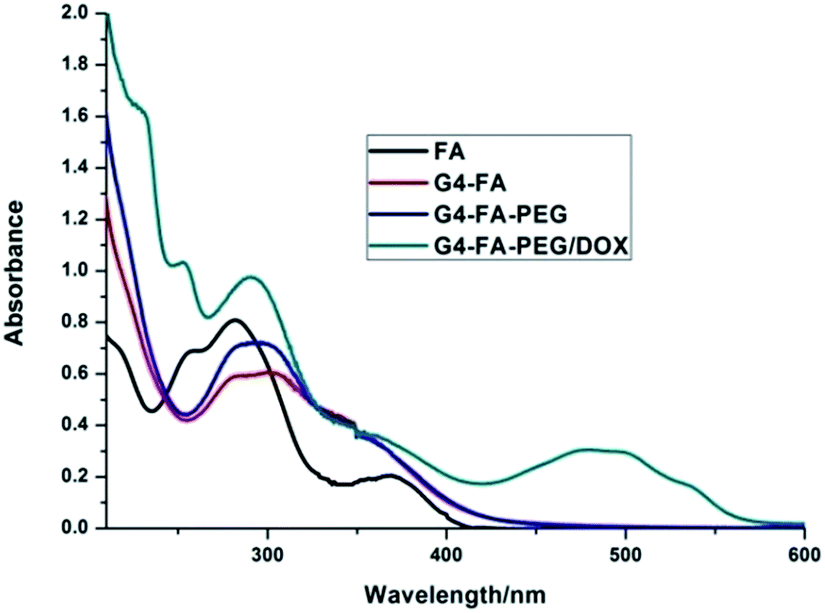 | ||
| Fig. 4 UV-vis spectra of FA, G4–FA, G4–FA–PEG and G4–FA–PEG/DOX. | ||
The encapsulated numbers of TMZ was calculated from 1H NMR (Fig. S1†). From the integration ratio of the proton signals at 8.76 ppm (TMZ) to those at 2.72 ppm corresponding to G4 dendrimer, the numbers of TMZ encapsulated were about 71.
In vitro DOX release
The encapsulated drugs should be able to release from the carriers to ensure the therapeutic effect. Therefore, we examined the in vitro release of DOX in PBS buffer (Fig. S2†). Free DOX was rapidly released within 2 hours. In contrast, the release of DOX from G4–FA–PEG/DOX followed a biphasic pattern, that was, an initial fast release followed by a sustained release. The release amount was about 50%.Cytotoxicity test
The cytotoxicity of free DOX and G4–FA–PEG/DOX against murine C6 cells was tested and compared with G4–FA–PEG as a blank. As a result, G4–FA–PEG was non-toxic at all tested concentrations even through 51 primary amino residues still remained on the periphery of PAMAM dendrimers. It indicated that the cytotoxicity of amine-terminated PAMAM was remarkably reduced by linking FA and PEG although the average attaching numbers were only 6 for FA and 7 for PEG per dendrimer. The similar phenomenon was also observed and reported by A. D'Emanuele et al. In their study, only six lauroyl or four PEG chains were conjugated on the exterior of cationic PAMAM dendrimers. G4–FA–PEG/DOX was basically nontoxic at low concentration (<0.01 μM), with the C6 cell viability above 90% (Fig. 5A). However, when the DOX concentration was increased to 0.1 μM, G4–FA–PEG/DOX exhibited obvious inhibitory effect to the proliferation of C6 cells and the IC50 value in DMEM was 0.528 μM (0.734 μM for free DOX). We deduced that the higher cytotoxicity of G4–FA–PEG/DOX was mainly caused by the enhanced cellular uptake which would be explained below.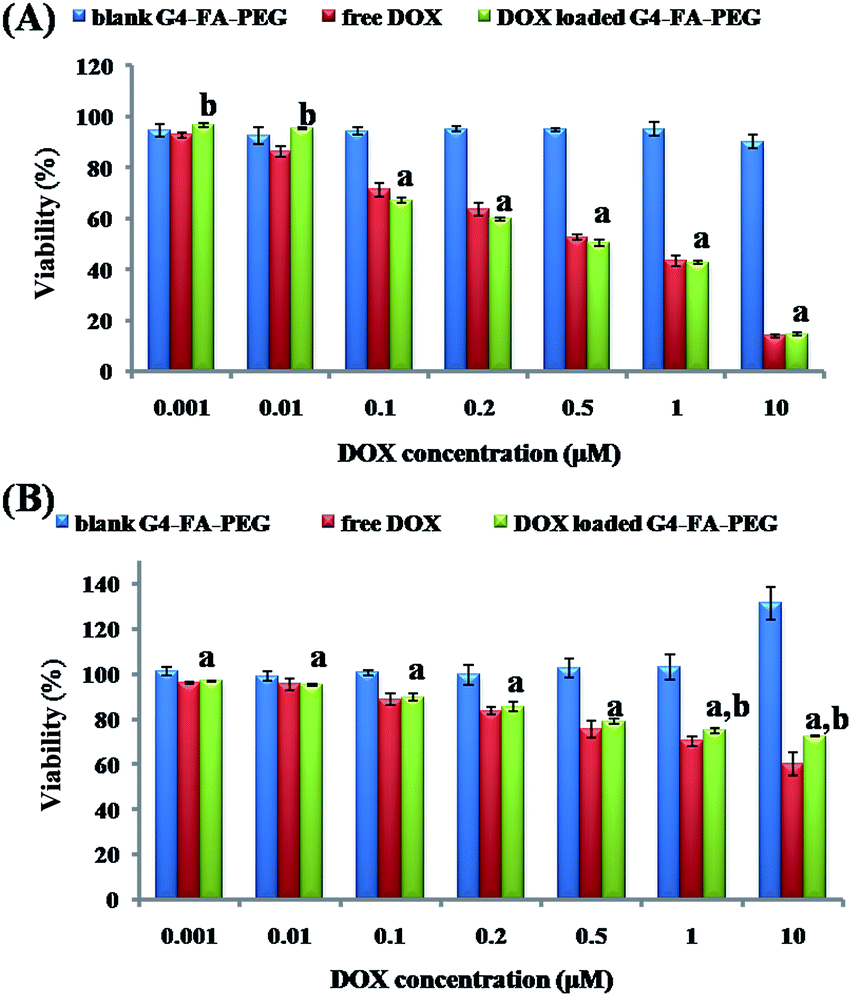 | ||
| Fig. 5 Cytotoxicity of free DOX and G4–FA–PEG/DOX to C6 cells in (A) DMEM medium or (B) FA-free RPMI 1640 medium. Each value is the mean ± SD of three determinations. (a) P < 0.05, versus G4–FA–PEG; (b) P < 0.05, versus free DOX. | ||
To explore the targeting effect of FA to the C6 cells, DMEM medium was replaced by FA-free RPMI medium to desaturate the FA receptors (FRs) on C6 cells.36 However, both free DOX and G4–FA–PEG/DOX showed lower cytotoxicity to C6 cells in RPMI medium as compared to that in DMEM medium (Fig. 5B). The cytotoxicity assay was repeated for many times, but the same result was obtained. The increased cell viability might be due to the growth of C6 cells stimulated by G4–FA–PEG in the FA-free RPMI medium because the higher cell viability was observed when increasing the concentration of G4–FA–PEG.
Although TMZ combined with adjuvant radiotherapy demonstrated survival benefit for the patients with high-grade gliomas,37 in our case, however, G4–FA–PEG/TMZ showed no obvious cytotoxicity until achieving extremely high concentration after incubation with C6 cells for 2 days (Fig. S2†). These results conformed with the previous work declaring that C6 cells were partially resistant to TMZ.38 Furthermore, a recent study revealed that TMZ with the concentration of 25 μM slightly reduced cell viability (by 9.5%) compared with 0.1% DMSO after 5 days.39 Thus we speculate that TMZ is not suitable for the treatment of C6 cells in our case.
Cellular uptake of DOX
DOXs show strong fluorescence which makes them useful markers. Fig. 6 shows the histogram profiles of the C6 cells treated by free DOX and G4–FA–PEG/DOX with the DOX concentration of 10 μM and the incubation time of 2 h. Cells without drugs exhibited only the auto-fluorescence and were set as the control. The amount of DOX internalized into cells was measured by using the fluorescence intensity as a ruler.40 The cells treated with G4–FA–PEG/DOX (curve A2) showed a slightly stronger fluorescence intensity than that with free DOX (curve A1), with the geometric mean fluorescence intensity of 82 for curve A2 and 76 for curve A1, indicating that the cellular uptake of DOX was increased by the conjugation of FA. This result was consistent with the report of Wang et al. They suggested that FA moieties promoted the cellular uptake of hybrid polymeric nanoparticles through FR-mediated endocytosis.41 However, PAMAM and FA–PAMAM showed similar efficiency in antisense oligonucleotides (ASODN) transfection in vitro according to another report. That might be owing to the limited FRs expression on C6 cells.4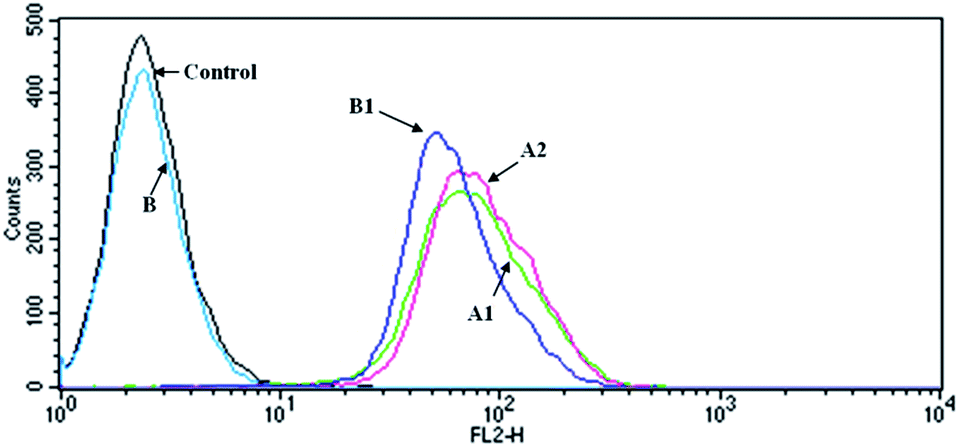 | ||
| Fig. 6 Flow cytometry histogram profiles of the C6 cells that were incubated with (A1) free DOX; (A2) G4–FA–PEG/DOX; (B) 1 mM FA; (B1) G4–FA–PEG/DOX + 1 mM FA (DOX concentration of 10 μM). | ||
In the competition assay, the fluorescence intensity of cells incubated with G4–FA–PEG/DOX was reduced after presaturation with free FA (62.44 for B2), illustrating that the decreased cellular uptake of G4–FA–PEG/DOX was due to the competitively binding of free FA with FRs on the C6 cell,42–44 which further confirmed the targeting effect of FA to C6 cells through FR-mediated endocytosis.
As a typical anticancer drug, DOXs bind and disrupt DNA in the cell nucleus, leading to the death of cells.45,46 In order to visualize the intracellular localization of DOX in C6 cells, laser scanning confocal microscopy was utilized to detect the fluorescence of DOX in the cells. Fig. 7 shows the confocal images of C6 cells treated with free DOX and G4–FA–PEG/DOX. After 2 h incubation, free DOX was mainly accumulated in the nuclei (Fig. 7a) while G4–FA–PEG/DOX exhibited retarded internalization with part of DOX transferred into nucleus and others distributed in the cytoplasm (Fig. 7b). This is reasonable because free DOX was rapidly transported into cells via passive diffusion.47,48 Differently, the internalization of G4–FA–PEG/DOX into C6 cells was via a FR-mediated endocytosis.49,50 In such a way, DOX was released from the carriers and then transported into the nuclei.
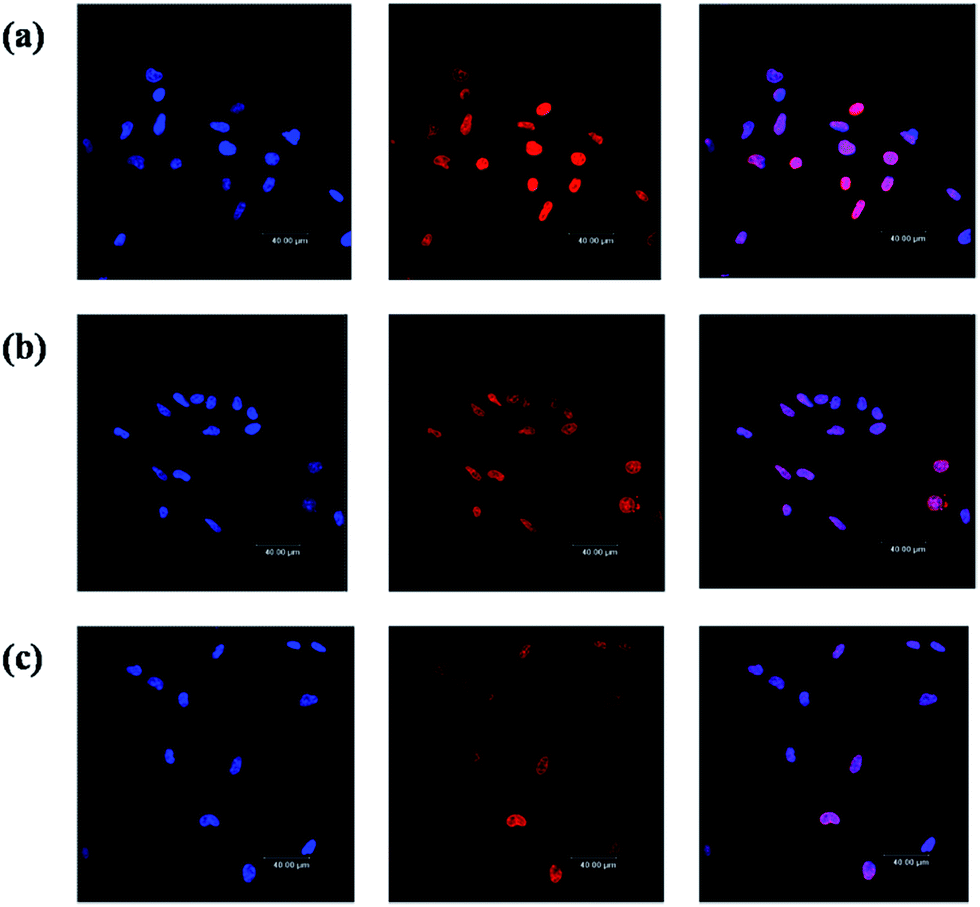 | ||
| Fig. 7 Confocal images of the C6 cells incubated 2 h with (a) free DOX; (b) G4–FA–PEG/DOX; and (c) G4–FA–PEG/DOX + 1 mM FA, respectively. For each panel, images from left to right showed the cells with nuclear staining by Hoechst 33258, with DOX fluorescence, and overlays of both images. | ||
Conclusions
Dendrimers have been proved to be potential drug carriers for their monodisperse hydrodynamic nanodiameter that facilitates the diffusion into tumors.51 However, the toxicity induced by surface charges hinders their biomedicine applications. In this regard, we prepared a G4.0 PAMAM dendrimer-based drug carrier by peripheral PEGylation to neutralize the positive charges of PAMAM dendrimers and FA modification to improve the site-targeting to tumors. The synthesized carrier showed increased size after conjugation of FA and PEG chains. In the in vitro test, G4–FA–PEG was nontoxic at measured concentrations as we expected, but G4–FA–PEG/DOX exhibited stronger cytotoxicity to C6 cells when compared to free DOX for the higher cellular uptake than free DOX. G4–FA–PEG/DOX showed the enhanced ability to enter the C6 cells by a FR-mediated endocytosis pathway. In addition, the cellular uptake could be inhibited by the addition of free FA, indicating the targeting effect of FA in the glioma tumor-specific delivery of drugs.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21274004) to X.-R. Jia and Natural Science Foundation of Hebei Province (B2016402030) to Tao Chang.Notes and references
- X. Xu, J. Li, S. Han, C. Tao, L. Fang, Y. Sun, J. Zhu, Z. Liang and F. Li, Eur. J. Pharm. Sci., 2016, 88, 178–190 CrossRef CAS PubMed
.
- C. Kang, X. Yuan, F. Li, P. Pu, S. Yu, C. Shen, Z. Zhang and Y. Zhang, J. Biomed. Mater. Res., Part A, 2010, 93, 585–594 Search PubMed
- J. Guo, X. Gao, L. Su, H. Xia, G. Gu, Z. Pang, X. Jiang, L. Yao, J. Chen and H. Chen, Biomaterials, 2011, 32, 8010–8020 CrossRef CAS PubMed
- J. M. Saul, A. Annapragada, J. V. Natarajan and R. V. Bellamkonda, J. Controlled Release, 2003, 92, 49–67 CrossRef CAS PubMed
- S. A. Shein, II. Kuznetsov, T. O. Abakumova, P. S. Chelushkin, P. A. Melnikov, A. A. Korchagina, D. A. Bychkov, I. F. Seregina, M. A. Bolshov, A. V. Kabanov, V. P. Chekhonin and N. V. Nukolova, Mol. Pharm., 2016, 13, 3712–3723 CrossRef CAS PubMed
- K. Manzoor, S. Johny, D. Thomas, S. Setua, D. Menon and S. Nair, Nanotechnology, 2009, 20, 065102 CrossRef PubMed
- Y. W. Yi, J. H. Kim, H. W. Kang, H. S. Oh, S. W. Kim and M. H. Seo, Pharm. Res., 2005, 22, 200–208 CrossRef CAS
- X. Y. Shi, S. H. Wang, I. Lee, M. W. Shen and J. R. Baker, Biopolymers, 2009, 91, 936–942 CrossRef CAS PubMed
- N. V. Cuong, M. F. Hsieh, Y. T. Chen and I. Liau, J. Appl. Polym. Sci., 2010, 117, 3694–3703 CAS
- J. A. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40 CrossRef CAS PubMed
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS
- M. A. Mintzer and M. W. Grinstaff, Chem. Soc. Rev., 2011, 40, 173–190 RSC
- A. Quintana, E. Raczka, L. Piehler, I. Lee, A. Myc, I. Majoros, A. K. Patri, T. Thomas, J. Mule and J. R. Baker, Pharm. Res., 2002, 19, 1310–1316 CrossRef CAS
- K. R. Vega-Villa, J. K. Takemoto, J. A. Yanez, C. M. Remsberg, M. L. Forrest and N. M. Davies, Adv. Drug Delivery Rev., 2008, 60, 929–938 CrossRef CAS PubMed
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS PubMed
- H. L. Crampton and E. E. Simanek, Polym. Int., 2007, 56, 489–496 CrossRef CAS PubMed
- R. Jevprasesphant, J. Penny, R. Jalal, D. Attwood, N. B. McKeown and A. D'Emanuele, Int. J. Pharm., 2003, 252, 263–266 CrossRef CAS PubMed
- K. Kono, T. Fukui, T. Takagishi, S. Sakurai and C. Kojima, Polymer, 2008, 49, 2832–2838 CrossRef CAS
- C. Kojima, K. Kono, K. Maruyama and T. Takagishi, Bioconjugate Chem., 2000, 11, 910–917 CrossRef CAS PubMed
- Y. Kim, A. M. Klutz and K. A. Jacobson, Bioconjugate Chem., 2008, 19, 1660–1672 CrossRef CAS PubMed
- A. Agarwal, S. Saraf, A. Asthana, U. Gupta, V. Gajbhiye and N. K. Jain, Int. J. Pharm., 2008, 350, 3–13 CrossRef CAS PubMed
- A. Beduneau, P. Saulnier and J. P. Benoit, Biomaterials, 2007, 28, 4947–4967 CrossRef CAS PubMed
- M. D. Salazar and M. Ratnam, Cancer Metastasis Rev., 2007, 26, 141–152 CrossRef CAS PubMed
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120–129 CrossRef CAS PubMed
- S. Hong, P. R. Leroueil, I. J. Majoros, B. G. Orr, J. R. Baker and M. M. B. Holl, Chem. Biol., 2007, 14, 107–115 CrossRef CAS PubMed
- C. P. Leamon, S. R. Cooper and G. E. Hardee, Bioconjugate Chem., 2003, 14, 738–747 CrossRef CAS PubMed
- C. P. Leamon and J. A. Reddy, Adv. Drug Delivery Rev., 2004, 56, 1127–1141 CrossRef CAS PubMed
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS
- D. Chandrasekar, R. Sistla, F. J. Ahmad, R. K. Khar and P. V. Diwan, Biomaterials, 2007, 28, 504–512 CrossRef CAS PubMed
- E. C. Wiener, S. Konda, A. Shadron, M. Brechbiel and O. Gansow, Invest. Radiol., 1997, 32, 748–754 CrossRef CAS PubMed
- Y. Yang, J. C. Wang, X. Zhang, W. L. Lu and Q. Zhang, J. Controlled Release, 2009, 135, 175–182 CrossRef CAS PubMed
- A. Beduneau, F. Hindre, A. Clavreul, J. C. Leroux, P. Saulnier and J. P. Benoit, J. Controlled Release, 2008, 126, 44–49 CrossRef CAS PubMed
- P. T. Wong, D. Chen, S. Tang, S. Yanik, M. Payne, J. Mukherjee, A. Coulter, K. Tang, K. Tao, K. Sun, J. R. Baker Jr and S. K. Choi, Small, 2015, 11, 6078–6090 CrossRef CAS PubMed
- S. J. Zhu, M. H. Hong, G. T. Tang, L. L. Qian, J. Y. Lin, Y. Y. Jiang and Y. Y. Pei, Biomaterials, 2010, 31, 1360–1371 CrossRef CAS PubMed
- X. Y. Shi, S. H. Wang, M. W. Shen, M. E. Antwerp, X. S. Chen, C. Li, E. J. Petersen, Q. G. Huang, W. J. Weber and J. R. Baker, Biomacromolecules, 2009, 10, 1744–1750 CrossRef CAS PubMed
- X. Deng, Z. Zheng, B. Lin, H. Su, H. Chen, S. Fei, Z. Fei, L. Zhao, X. Jin and C. Y. Xie, BMC Cancer, 2017, 17, 42 CrossRef PubMed
- X. Huang, C. Li, W. Zhang, Y. Lu, S. Fang and E. Wei, Pharmacology, 2008, 82, 1–9 CrossRef CAS PubMed
- R. S. Ignarro, G. Facchini, D. R. de Melo, K. J. Pelizzaro-Rocha, C. V. Ferreira, R. F. Castilho and F. Rogerio, Neurosci. Lett., 2017, 638, 189–195 CrossRef CAS PubMed
- Y. Choi, T. Thomas, A. Kotlyar, M. T. Islam and J. R. Baker, Chem. Biol., 2005, 12, 35–43 CrossRef CAS PubMed
- J. F. Wang, W. M. Liu, Q. Tu, J. C. Wang, N. Song, Y. R. Zhang, N. Nie and J. Y. Wang, Biomacromolecules, 2011, 12, 228–234 CrossRef CAS PubMed
- Y. J. Lu and P. S. Low, Adv. Drug Delivery Rev., 2002, 54, 675–693 CrossRef CAS PubMed
- H. J. Wang, P. Q. Zhao, X. F. Liang, X. Q. Gong, T. Song, R. F. Niu and J. Chang, Biomaterials, 2010, 31, 4129–4138 CrossRef CAS PubMed
- S. O. Loureiro, L. Heimfarth, B. A. Lacerda, L. F. Vidal, A. Soska, N. G. dos Santos, A. T. D. Wyse and R. Pessoa-Pureur, Cell. Mol. Neurobiol., 2010, 30, 557–568 CrossRef CAS PubMed
- M. Fritzer, T. Szekeres, V. Szuts, H. N. Jarayam and H. Goldenberg, Biochem. Pharmacol., 1996, 51, 489–493 CrossRef CAS PubMed
- B. Schott and J. Robert, Biochem. Pharmacol., 1989, 38, 167–172 CrossRef CAS PubMed
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Biomaterials, 2009, 30, 6065–6075 CrossRef CAS PubMed
- H. S. Yoo and T. G. Park, J. Controlled Release, 2004, 100, 247–256 CrossRef CAS PubMed
- H. Z. Zhao and L. Y. L. Yung, J. Biomed. Mater. Res., Part A, 2009, 91, 505–518 CrossRef PubMed
- L. Fan, F. Li, H. T. Zhang, Y. K. Wang, C. Cheng, X. Y. Li, C. H. Gu, Q. A. Yang, H. Wu and S. Y. Zhang, Biomaterials, 2010, 31, 5634–5642 CrossRef CAS PubMed
- J. Sinek, H. Frieboes, X. Zheng and V. Cristini, Biomed. Microdevices, 2004, 6, 297–309 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum of G4–FA–PEG/TMZ, release profiles of DOX and cytotoxicity of TMZ. See DOI: 10.1039/c7ra00713b |
| This journal is © The Royal Society of Chemistry 2017 |