
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent evaporation induced self-assembly of graphene foam for thermally conductive polymers†
Jingbo Maab,
Xufeng Zhou *a,
Shiyun Dinga and
Zhaoping Liu*a
*a,
Shiyun Dinga and
Zhaoping Liu*a
aKey Laboratory of Graphene Technologies and Applications of Zhejiang Province and Advanced Li-ion Battery Engineering Lab, Ningbo Institute of Materials Technology and Engineering (NIMTE), Chinese Academy of Sciences (CAS), Ningbo, 315201, China. E-mail: liuzp@nimte.ac.cn
bCollege of Material Science and Chemical Engineering, Ningbo University, Ningbo 315211, China
First published on 8th March 2017
Abstract
Graphene, a new carbon material with the highest thermal conductivity (TC) in known materials, is a good candidate for polymer-based thermally conductive material applications. However, the homogeneous dispersion of graphene and effective construction of graphene-based thermally conductive network in the polymer matrix still remains a big challenge. In this paper, we report an effective way to avoid aggregation of graphene in polymers through the fabrication of 3D porous graphene foam (GF) in advance by a simple solvent evaporation induced self-assembly method. The as-prepared GF is proved to be an effective thermally conductive network after the epoxy perfusion, giving rise to a high TC of 11.58 W (m−1 K−1) for the GF/epoxy composite. In addition, anisotropic TC in the GF/epoxy composites is observed because of the oriented arrangement of graphene sheets in the GF due to solvent evaporation. Besides, further improvement of TC to 16.69 W (m−1 K−1) can be achieved by addition of polyvinyl pyrrolidone (PVP) during the preparation of GF, which can be ascribed to the reduction of interfacial thermal resistance by amorphous carbon generated from pyrolysis of PVP.
Introduction
With the rapid development of modern electronic technology, the integration level and the assembly density of electronic components have been continuously improved, giving rise to constant upgrade of the device performance, however, it also leads to rising power consumption and sharp increase of heat output.1–3 Consequently, the heat removal by thermally conductive polymer which fills the voids and grooves created by imperfect surface finish of mating surfaces between heat sources and heat sink plays a vital role for high performance electronic devices.Thermally conductive filler dispersed in polymer matrix is the key factor to improve the thermal conductivity (TC) of polymer-based composites.4 By employing traditional thermally conductive fillers with relatively low TC (30–360 W (m−1 K−1)),5 such as aluminum oxide, aluminum nitride, magnesium oxide, etc., the improvement of the TC of composites is quite limited (less than 8 W (m−1 K−1)).6–9 The thermally conductive filler with higher TC thus is urgently required to further enhance TC of composites for applications in next-generation electronics. In recent years, the research of high thermally conductive composites utilizing graphene as the filler has been widely carried out.10–12 The TC of graphene has been reported to be 5300 W (m−1 K−1) at room temperature,13 which is the highest among the known materials. Besides, graphene possesses large specific surface area, light weight and other excellent characteristics,14 all of which make it one of the most ideal thermally conductive fillers.
Mechanical mixing which has been widely adopted to prepare traditional thermally conductive composites has also been employed in most researches to obtain graphene-based thermally conductive polymers. For instance, Stirling et al. fabricated graphene/silicone composites by conventional mechanical mixing with graphene nanoplatelets loading of 20 wt%, and the TC of 1.91 W (m−1 K−1) was obtained.15 Haddon et al. mixed graphene nanoplatelets and epoxy resin in acetone solution, and TC of 6.44 W (m−1 K−1) was obtained at the graphene loading of ∼25 vol%.16 Balandin et al. fabricated nanocomposite using both single-layer graphene and multi-layer graphene by high-sheer mixing for few hours to days, and TC of 5.5 W (m−1 K−1) was achieved with the graphene loading of 10 vol%.11 Examples above and many other reports reveal that a large amount of graphene is usually needed to obtain thermally conductive composites with high TC by mechanical mixing. This is because the high specific surface area and strong π–π interaction of graphene sheets make it difficult to achieve homogeneous dispersion of graphene in polymer matrices just by mechanical mixing. The frequently encountered aggregation or restacking of graphene sheets strongly affects the formation of effective thermally conductive network, resulting in insufficient enhancement of TC of polymers.
Considering the drawbacks of mechanical mixing, it is essential to develop novel methods to fully utilize the advantages of graphene as thermally conductive filler by construction of three dimensionally (3D) interconnected graphene network to avoid aggregation. Such foam-like 3D graphene monoliths with porous structure have drawn much attraction in recent years due to its interconnected structure and mechanical strength,17–20 and their application as pre-constructed thermally conductive network for further perfusion of polymers to obtain graphene-based thermally conductive composites has been recently spotlighted. For instance, Zhao et al. used template-directed chemical vapor deposition (CVD) method to produce macroscopic graphene foam (GF) which was then infiltrated by polydimethylsiloxane (PDMS). Due to the interconnected structure of GF, the TC of GF/PDMS composite was 20% higher than that of PDMS/graphene composite prepared by mechanical mixing of PDMS with graphene powder with the same graphene loading of 0.7 wt%, and 300% higher than that of pure PDMS.21 However, due to the ultralow density of GF prepared by CVD method, the low graphene loading of 0.7 wt% in GF/PDMS composite is ineffective to enhance the TC (0.56 W (m−1 K−1)). Wong et al. fabricated composites by perfusion of epoxy into vertically aligned and interconnected graphene networks prepared by thermal reduction of orientedly freeze-casted graphene oxide liquid crystals. TC of 2.13 W (m−1 K−1) has been obtained at an ultralow graphene loading of 0.92 vol%.22 Similarly, because of the low graphene loading, the improvement of TC is relatively limited.
In this study, we propose a novel solvent evaporation induced self-assembly method to construct GF with connected pores and certain trend of oriented arrangement of graphene sheets as an effective thermally conductive network. Moreover, the density of GF and the degree of oriented arrangement of graphene can be simply adjusted by regulating the composition of the solvents. Because of the relatively high density of GF, a high TC of 11.58 W (m−1 K−1) can be achieved for the GF/epoxy composite, and anisotropic TC of the composite is observed due to the orientation of graphene sheets in the foam. The TC of GF/epoxy composite can be further increased to 16.69 W (m−1 K−1) by reducing the interfacial thermal resistance between graphene sheets using amorphous carbon derived from pyrolysis of polyvinyl pyrrolidone (PVP).
Experimental
Preparation of GF and GF/epoxy composites
30 g graphene aqueous slurry (5 wt%, kindly provided by Ningbo Morsh Tech. Co., Ltd.) was dispersed in a 500 mL mixture of ethanol and water with different volume ratios (Vethanol/Vwater = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, 3:1, 5:1 and 9:1, respectively) by stirring and ultra-sonication to form uniform and stable graphene suspensions. Then the suspensions were dried by evaporating the solvents at 80 °C in an oven, and monolithic graphene foams (GF) whose volume and density depend on the composition of the solvents were obtained. The foams were named GF/1, GF/2, GF/3, GF/5 and GF/9, respectively, according to the volume ratio of ethanol to water in the solvents. In addition, graphene film and graphene foam obtained by evaporating graphene suspension in pure water and pure ethanol, respectively, were also prepared for structural comparison. Then, GF was immersed in the mixture of epoxy monomer (EPIKOT Resin RIMR135, Momentive) and curing agent (RIMH1366, Momentive) with a weight ratio of 100:30 ± 2 for 2 h under vacuum at room temperature. After curing at 80 °C for 7 h, the GF/epoxy composites were obtained, and named GF/E-2, GF/E-3, GF/E-5 and GF/E-9, respectively (due to the compact structure of GF/1, the subsequent experiment has not been implemented). In addition, the preparation of GF with polyvinyl pyrrolidone (PVP, with a weight ratio of PVP/graphene = 1/5 and named GF/PVP) was also conducted by adding PVP in the original mixed solution, and followed by the same evaporation process. Afterwards, the GF/PVP was annealed at 500 °C for an hour and 800 °C for two hours in argon to carbonize PVP, and then perfused with epoxy using the same procedure as that for the preparation of GF/epoxy composite, and the obtained sample is named GF/E/PVP. For comparison, with the same content of graphene in GF/E-2 sample, graphene/epoxy composite was prepared by mechanical mixing of graphene powder (obtained by freeze-drying of graphene slurry) and epoxy using a planetary vacuum mixer (ARV-310). All the composite samples were finally cut and polished to form a square piece with a size of 1 cm × 1 cm and a thickness of ∼1 mm for the measurement of TC.
:1, 2:1, 3:1, 5:1 and 9:1, respectively) by stirring and ultra-sonication to form uniform and stable graphene suspensions. Then the suspensions were dried by evaporating the solvents at 80 °C in an oven, and monolithic graphene foams (GF) whose volume and density depend on the composition of the solvents were obtained. The foams were named GF/1, GF/2, GF/3, GF/5 and GF/9, respectively, according to the volume ratio of ethanol to water in the solvents. In addition, graphene film and graphene foam obtained by evaporating graphene suspension in pure water and pure ethanol, respectively, were also prepared for structural comparison. Then, GF was immersed in the mixture of epoxy monomer (EPIKOT Resin RIMR135, Momentive) and curing agent (RIMH1366, Momentive) with a weight ratio of 100:30 ± 2 for 2 h under vacuum at room temperature. After curing at 80 °C for 7 h, the GF/epoxy composites were obtained, and named GF/E-2, GF/E-3, GF/E-5 and GF/E-9, respectively (due to the compact structure of GF/1, the subsequent experiment has not been implemented). In addition, the preparation of GF with polyvinyl pyrrolidone (PVP, with a weight ratio of PVP/graphene = 1/5 and named GF/PVP) was also conducted by adding PVP in the original mixed solution, and followed by the same evaporation process. Afterwards, the GF/PVP was annealed at 500 °C for an hour and 800 °C for two hours in argon to carbonize PVP, and then perfused with epoxy using the same procedure as that for the preparation of GF/epoxy composite, and the obtained sample is named GF/E/PVP. For comparison, with the same content of graphene in GF/E-2 sample, graphene/epoxy composite was prepared by mechanical mixing of graphene powder (obtained by freeze-drying of graphene slurry) and epoxy using a planetary vacuum mixer (ARV-310). All the composite samples were finally cut and polished to form a square piece with a size of 1 cm × 1 cm and a thickness of ∼1 mm for the measurement of TC.
Characterization
Scanning electron microscopy (SEM) images were acquired by a field emission SEM (S-4800, Hitachi). Transmission electron microscope (TEM, Tecnai F20) was used to determine the layer numbers of graphene raw materials. The content of graphene in the composites was determined by thermogravimetric analysis (TGA, Pyris Diamond) under nitrogen atmosphere with a heating rate of 10 °C min−1 up to 600 °C. The TC was measured by light flash method using Netzsch LFA447 NanoFlash instrument. The Raman spectroscopy (Renishaw inVia Reflex) was taken to determine the defects of graphene, as well as the X-ray photoelectron spectroscopy (AXIS ULTRA DLD).Results and discussion
TC of graphene depends on phonon transmission.23,24 The higher quality of graphene is, the less interference of the phonon in the transmission process will be.25–28 Therefore, it is necessary to use graphene with high crystallinity and few defects to achieve high TC. In our experiments, commercial graphene (Ningbo Morsh Technology Co., Ltd.) produced by intercalation and exfoliation of graphite without oxidation process was selected as the raw material due to its high quality. Typical SEM image of graphene is shown in Fig. 1a. With some wrinkles, the lateral dimensions of graphene are ranged from a few microns to ten microns. TEM image focusing on the edge of a graphene sheet (Fig. 1b) clearly shows that this individual sheet has a layer number of 5, and examination of dozens of graphene sheets reveals that the majority have layer numbers between 3–10. Both Raman and XPS analysis (Fig. S1a and b†) reveals that the graphene sample possesses few defects, which is suitable as thermally conductive filler. | ||
| Fig. 1 (a) SEM and (b) TEM micrographs of graphene. | ||
The preparation process of epoxy-based thermally conductive composite using GF as the filler is shown in Fig. 2. In the first step, graphene was dispersed in a mixed solution of ethanol and water with different volume ratios to form a uniform suspension with the graphene concentration of 3 mg mL−1. Then the mixed solvent was evaporated at 80 °C, during which the concentration of graphene gradually increased, and finally, monolithic GF was obtained after complete removal of the solvents. Afterwards, the GF was immersed in the mixture of epoxy resin and curing agent under vacuum. After final curing process, the GF/epoxy composite was successfully prepared. Raman and XPS analyses (Fig. S1c and d†) confirm that the high crystallinity of raw graphene material is well preserved in the GF sample, which ensures high thermal conductivity of GF.
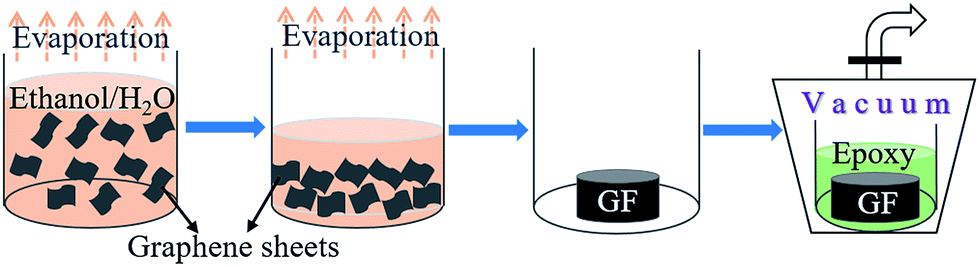 | ||
| Fig. 2 Schematic representation of the formation of GF by the evaporation of solvents, as well as the following process of the perfusion of epoxy resin in a vacuum suction device. | ||
The evaporation of the solvent results in the formation of GF, whose typical morphology is shown in Fig. 3a. Although the self-assembly is a spontaneous organization process of graphene sheets through non-covalent interactions, the GF shows good mechanical strength. As we can see in Fig. 3a, GF (with a diameter of 5 cm and a thickness of 2.5 cm) has no deformation under 500 g weight, showing good capability to withstand a certain pressure, which ensures that the microstructure of GF can be maintained during the filling process of epoxy resin. Furthermore, the morphology and the density of GF vary with the volume ratio of ethanol to water (Vethanol/Vwater). As is shown in Fig. S2a,† the volume of GF rises with the increase of Vethanol/Vwater. The trend of the density of GFs shown in Fig. S2b† is contrary to the trend of the volume, which declines with the increase of Vethanol/Vwater. A dramatic decrease of the density from 1.32 g cm−3 of graphene film from pure water to 0.24 g cm−3 of the sample from Vethanol/Vwater = 1 is observed. Afterwards, the density of GF decreases gradually by increasing the volume ratio of ethanol, and a density of 0.1 g cm−3 was obtained for the sample from Vethanol/Vwater = 9:1, which is reduced by 13 times compared with that of the graphene film.
 | ||
| Fig. 3 (a) Photograph of GF, showing no deformation under 500 g weight. (b and c) Cross-sectional SEM images of GF/2 under different magnifications. (d) SEM image of GF obtained by the evaporation of graphene suspension in pure ethanol. (e) Cross-sectional SEM image of graphene film obtained by evaporation of graphene suspension in pure water. | ||
The variation of the macroscopic morphology and density of the GFs along with the volume ratio of ethanol to water is ascribed to the change of their microstructure. Fig. 3b and c show the SEM images along the cross-sectional view of a typical sample with Vethanol/Vwater = 2:1 (GF/2). Relatively loose stacking of graphene sheets can be seen in the sample, and many voids whose sizes range from a few microns to ten microns exist among graphene sheets, which forms porous structure and is conducive to the perfusion of epoxy resin. Moreover, the arrangement of graphene in the sample exhibits a certain oriented trend, i.e., graphene sheets tend to align horizontally in the foam, yet the bending and curling of flexible graphene sheets, as well as partial vertically aligned graphene sheets finally result in a wave-like microstructure and constructs numerous macropores. The SEM image under higher magnification in Fig. 3c further shows uniform dispersion of graphene sheets without severe aggregation in the foam, which ensures homogeneous dispersion of graphene in the GF/epoxy composites. The SEM image (Fig. 3d) of graphene foam obtained by evaporation of graphene suspension in pure ethanol also shows porous structure but with no discernable oriented arrangement of graphene sheets as that in the GF/2 sample. Instead, only random distribution of graphene sheets is observed. In great contrast, non-porous and layered structure composed of densely stacked graphene sheets (Fig. 3e) is obtained in the sample from graphene suspension in pure water. Further examination of the GFs prepared from different Vethanol/Vwater ratios using SEM (Fig. S3a–d†) indicates an obvious evolution trend of their microstructures. By increasing the volume ratio of ethanol in the mixed solvent, the stack of graphene sheets becomes looser, and the size of pores among graphene sheets becomes larger. Besides, the oriented arrangement of graphene becomes less obvious.
The solvent dependent microstructure of GFs is mainly caused by capillary force,29 which is generated by the solvents and is proportional to the surface tension of solvents. During solvent evaporation, the shrinkage of the solvent volume continuously draws graphene sheets closer, and the capillary force between the solvent and graphene sheets begins to play an important role when the distance between graphene sheets is close enough. In case of pure water solvent, the large capillary force due to the high surface tension of water (62.75 N m−1 at 80 °C) generates driving force strong enough to conquer free movement of graphene in the solvent and causes close restacking of graphene sheets in a plane-to-plane way, resulting in dense layered structure and thin-film like morphology after water evaporation, which has been extensively reported.30–33 Comparing with water, the capillary force generated by ethanol is much weaker because of its lower surface tension (17.15 N m−1 at 80 °C). As a result, the weaker capillary force in ethanol is insufficient to overcome the free movement of graphene in the solvent, resulting in disordered stack of graphene sheets and loose porous structure. The surface tension of mixed solvent of ethanol and water lies between the ones in pure water and in pure ethanol, and it increases with the decrease of the volume ratio of ethanol, and so is the capillary force. Consequently, when the volume ratio of ethanol is low, the relatively large capillary force overwhelms the free movement of graphene sheets to a large extent, resulting in an obvious oriented arrangement, but the existence of ethanol still generates porous structure rather than dense layered structure in pure water. By increasing the volume ratio of ethanol in the mixed solvent, the gradually reduced capillary force is becoming more difficult to overcome the free movement of graphene, so the arrangement of graphene sheets becomes less dense and oriented.
Fig. 4a shows the photograph of a GF/epoxy composite disc with a diameter of 4 cm and a thickness of 1.5 cm, and small pieces with a size of 1 × 1 cm and a thickness of ∼1 mm after cut and polishing for the measurement of TC. Due to the good mechanical strength of GF, its macroscopic morphology and microstructure can be well maintained after perfusion of epoxy under vacuum. Fig. 4b is the SEM image showing the microstructure of fracture surface of the sample GF/E-2. A compact structure with no pores can be clearly seen, which indicates that all macropores in GF are accessible and can be completely filled with epoxy. Though it is not easy to differentiate two components in the composite under SEM, the oriented arrangement of graphene sheets along the horizontal direction can still be discerned, and typical thermally conductive paths composed of aligned graphene sheets are marked with red dashed lines. The compact filling of epoxy and the well maintenance of graphene network can also be observed from the fracture of GF/E-3, GF/E-5 and GF/E-9 samples as shown in Fig. S4a–c.† In contrast, severe restacking and aggregation of graphene sheets can be seen in the composite prepared by mechanical mixing of epoxy and graphene powder (Fig. S4d†), implying that mechanical blending is not able to achieve homogeneous distribution of graphene in epoxy matrix, which is harmful to enhancement effect of graphene as the thermally conductive filler.
 | ||
| Fig. 4 (a) Photograph of a GF/epoxy composite disc and square pieces. (b) SEM image of GF/E-2 from the fracture surfaces. | ||
TC of the composites after perfusion of epoxy resin along two directions (indicated in the inset in Fig. 5) has been explored respectively. As seen in Fig. 5, when Vethanol/Vwater is 2/1, the composite has the highest TC of 11.58 W (m−1 K−1) along X-axis and 7.55 W (m−1 K−1) along Y-axis. And as the increase of Vethanol/Vwater, the TC along both directions exhibits a declining trend, which is mainly due to the decreased graphene content in composites as the density of GF decreases with the increase of Vethanol/Vwater (Fig. S2†). The quantitative measurement of graphene content was conducted by thermo-gravimetric analysis and the results are shown in Fig. S5.† It is obvious that the weight content of graphene decreases with the increase of Vethanol/Vwater, which is in good accordance with the densities of the GFs obtained by evaporation with different volume ratios of ethanol to water. On the other hand, the composites exhibit a distinct anisotropic thermally conductive behavior, that is, at any proportion between ethanol and water, the TC along X-axis is higher than that along Y-axis. This is because the oriented arrangement of graphene sheets along X-axis in the composites constructs more effective thermally conductive pathways comparing with that along Y-axis. Besides, along with the increase of Vethanol/Vwater, the ratio of TC along X-axis to that along Y-axis reduces gradually from 1.53 (Vethanol/Vwater = 2:1) to 1.21 (Vethanol/Vwater = 9:1), which can be ascribed to the decline of oriented arrangement degree of graphene sheets in GFs with the increase of the volume ratio of ethanol. For comparison, the TC of graphene/epoxy composite prepared by mechanical mixing only reaches 1.46 W (m−1 K−1) which is much lower than the TC of GF/epoxy composites, mainly due to inhomogeneous dispersion and aggregation of graphene sheets in the composite. It evidently suggests the advantage of pre-prepared graphene network in enhancing the TC of polymer matrix.
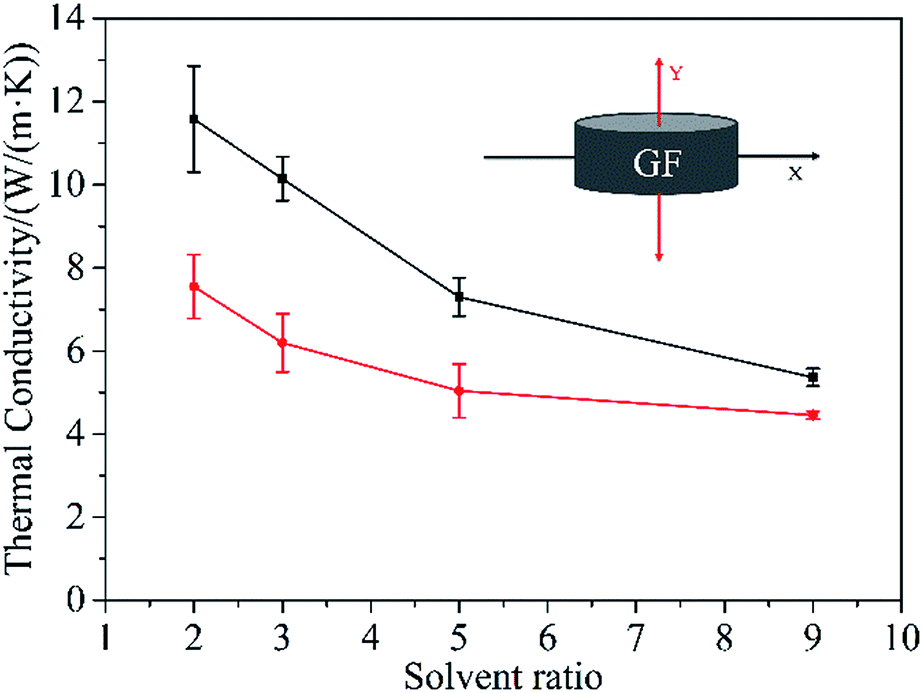 | ||
| Fig. 5 TC of GF/epoxy composites along X-axis (black) and Y-axis (red). The inset is the schematic diagram indicating two axial directions. | ||
In order to further improve the TC of composites, PVP was added into the original graphene suspension to help forming a more effective thermally conductive network. The morphology and micro-structure (Fig. S6†) of the as-prepared GF/E is almost identical to pure graphene foam without PVP. The orientation of graphene sheets along the X-axis can still be observed, and large amounts of cavities among graphene sheets still exist. The GF/PVP was then calcined in Ar to turn PVP into amorphous carbon. The calcined foam was afterwards applied as the substrate for epoxy infiltration using the same process as that in the case of pure graphene foam. Based on TG analysis (Fig. S7†), the content of carbon derived from PVP is only ∼0.4 wt% in the composite after calcination, however, it is surprising to find that the TC of the composite is remarkably improved after the addition of PVP as shown in Fig. 6. The GF/epoxy composite with the addition of PVP reaches the TC of 16.69 W (m−1 K−1), which is ∼40% higher than that of the GF/epoxy without PVP, 1140% higher than that of the epoxy/graphene composite prepared by simple mechanical mixing, and 8300% higher than that of pure epoxy.
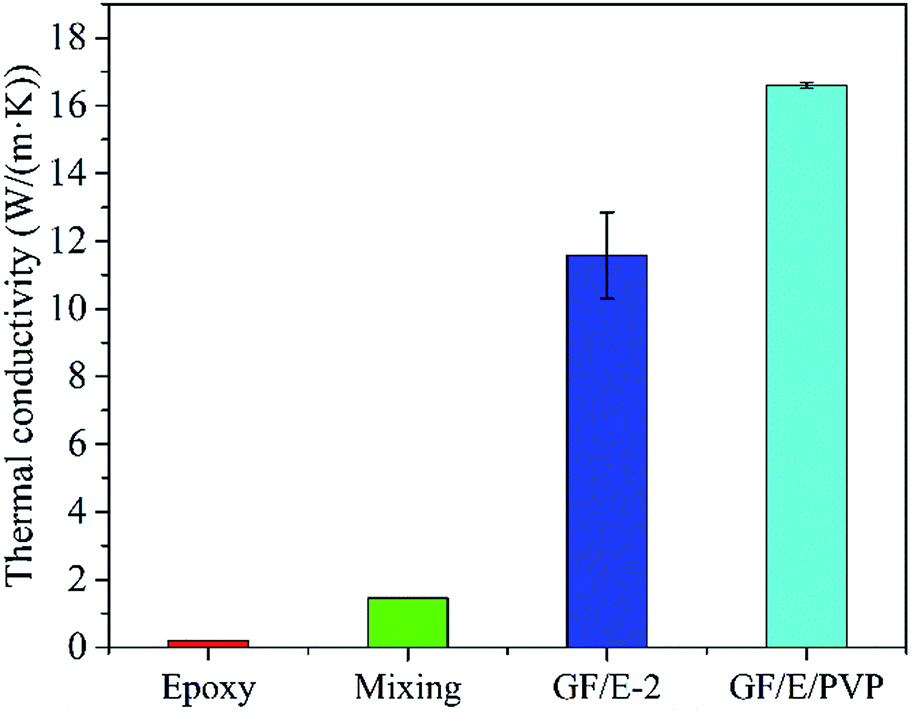 | ||
| Fig. 6 Histogram comparing the TC of pure epoxy, graphene/epoxy composite obtained by mechanical mixing, GF/E-2 and the composite after the addition of PVP (GF/E/PVP). | ||
We have also tried other polymers instead of PVP, such as polyvinyl alcohol (PVA) and starch, however, no enhancement of the TC of the composite is acquired. TC of 8.71 W (m−1 K−1) and 9.61 W (m−1 K−1) are obtained by the addition of PVA and starch, respectively. The advantage of PVP over other water soluble polymers can be ascribed to strong π–π interaction between PVA and graphene, which does not exist in the case of PVA and starch.34,35 Consequently, PVP molecules can disperse uniformly on graphene sheets after solvent evaporation, which then turn to be amorphous carbon locally by calcination. Comparing with pure graphene foam in which graphene sheets are only connected by weak physical interaction, the PVP derived carbon can bridge graphene sheets more tightly, which can further reduce the interfacial thermal resistance. Consequently, the TC can be effectively improved by the addition of PVP.
Conclusions
An effective way was utilized to construct a favorable graphene-based thermally conductive network for thermally conductive polymer applications. Due to the interconnection of graphene sheets and numerous macropores in this network, a relatively high TC of 11.58 W (m−1 K−1) can be achieved for the graphene/epoxy composite, which is much higher than the TC of the sample prepared by conventional mechanical mixing. In addition, the oriented arrangement of graphene sheets in the network generated by solvent evaporation results in anisotropic TC of the composite. The TC can be further enhanced to 16.69 W (m−1 K−1) by introducing amorphous carbon derived from PVP in the graphene network, which is ascribed to the reduced interfacial thermal resistance between graphene sheets by additional carbon. The use of commercially available graphene and the high performance of such graphene network make it potentially suitable for thermal management applications.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21371176) and Ningbo Key Science and Technology Projects (Grant No. 2016S1002).Notes and references
- M. J. Yim and K. W. Paik, Int. J. Adhes. Adhes., 2006, 26, 304–313 CrossRef CAS.
- D. G. Cahill, W. K. Ford, K. E. Goodson, G. D. Mahan, A. Majumdar, H. J. Maris, R. Merlin and P. Sr, J. Appl. Phys., 2003, 93, 793–818 CrossRef CAS.
- W. L. Song, L. M. Veca, A. Anderson, M. S. Cao, L. Cao and Y. P. Sun, Nanotechnol. Rev., 2012, 1, 363–376 CAS.
- M. Shtein, R. Nadiv, M. Buzaglo, K. Kahil and O. Regev, Chem. Mater., 2015, 27, 2100–2106 CrossRef CAS.
- I. Jo, M. T. Pettes, J. Kim, K. Watanabe, T. Taniguchi, Z. Yao and L. Shi, Nano Lett., 2013, 13, 550–554 CrossRef CAS PubMed.
- P. Bujard, G. Kuhnlein, S. Ino and T. Shiobara, IEEE Trans. Compon., Packag., Manuf. Technol., Part A, 1994, 17, 527–532 CrossRef CAS.
- G. W. Lee, M. Park, J. Kim, J. I. Lee and H. G. Yoon, Composites, Part A, 2006, 37, 727–734 CrossRef.
- Z. Q. Shi, M. Radwan, S. Kirihara, Y. Miyamoto and Z. H. Jin, Appl. Phys. Lett., 2009, 95, 224104 CrossRef.
- J. R. Wang, J. L. Qiao, J. F. Wang, Y. Zhu and L. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 9281–9286 CAS.
- Q. Z. Liang, X. X. Yao, W. Wang, Y. Liu and C. P. Wong, ACS Nano, 2011, 5, 2392–2401 CrossRef CAS PubMed.
- K. M. Shahil and A. A. Balandin, Nano Lett., 2012, 12, 861–867 CrossRef CAS PubMed.
- S. H. Song, K. H. Park, B. H. Kim, Y. W. Choi, G. H. Jun, D. J. Lee, B. S. Kong, K. W. Paik and S. Jeon, Adv. Mater., 2013, 25, 732–737 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- M. A. Raza, A. Westwood, A. Brown, N. Hondow and C. Stirling, Carbon, 2011, 49, 4269–4279 CrossRef CAS.
- A. P. Yu, P. Ramesh, M. E. Itkis, E. Bekyarova and R. C. Haddon, J. Phys. Chem. C, 2007, 111, 7565–7569 CAS.
- J. Tang and Y. Yamauchi, Nat. Chem., 2016, 8, 638–639 CrossRef CAS PubMed.
- R. R. Salunkhe, J. Tang, N. Kobayashi, J. Kim, Y. Ide, S. Tominaka, J. H. Kim and Y. Yamauchi, Chem. Sci., 2016, 7, 5704–5713 RSC.
- R. R. Salunkhe, C. Young, J. Tang, T. Takei, Y. Ide, N. Kobayashi and Y. Yamauchi, Chem. Commun., 2016, 52, 4764–4767 RSC.
- J. Tang, J. Liu, R. R. Salunkhe, T. Wang and Y. Yamauchi, Chem. Commun., 2016, 52, 505–508 RSC.
- Y.-H. Zhao, Z.-K. Wu and S.-L. Bai, Composites, Part A, 2015, 72, 200–206 CrossRef CAS.
- G. Lian, C. C. Tuan, L. Y. Li, S. L. Jiao, Q. L. Wang, K. S. Moon, D. L. Cui and C. P. Wong, Chem. Mater., 2016, 28, 6096–6104 CrossRef CAS.
- L. A. Falkovsky, Phys. Lett. A, 2008, 372, 5189–5192 CrossRef CAS.
- L. Lindsay, D. A. Broido and N. Mingo, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 115427 CrossRef.
- J. N. Hu, X. L. Ruan and Y. P. Chen, Nano Lett., 2009, 9, 2730–2735 CrossRef CAS PubMed.
- D. L. Nika, E. P. Pokatilov, A. S. Askerov and A. A. Balandin, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 155413 CrossRef.
- A. V. Savin, Y. S. Kivshar and B. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195422 CrossRef.
- E. Pop, V. Varshney and A. K. Roy, MRS Bull., 2012, 37, 1273–1281 CrossRef CAS.
- L. A. Richards, J. Appl. Phys., 1931, 1, 318–333 Search PubMed.
- C. M. Chen, Q. H. Yang, Y. G. Yang, W. Lv, Y. F. Wen, P. X. Hou, M. Z. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3007–3011 CrossRef CAS.
- B. Shen, W. T. Zhai and W. G. Zheng, Adv. Funct. Mater., 2014, 24, 4542–4548 CrossRef CAS.
- M. Zhang, Y. L. Wang, L. Huang, Z. P. Xu, C. Li and G. Q. Shi, Adv. Mater., 2015, 27, 6708–6713 CrossRef CAS PubMed.
- L. Y. Dong, C. G. Hu, L. Song, X. K. Huang, N. Chen and L. T. Qu, Adv. Funct. Mater., 2016, 26, 1470–1476 CrossRef CAS.
- S. J. Guo, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- S. J. Guo, S. J. Dong and E. Wang, J. Phys. Chem. C, 2008, 112, 2389–2393 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01670k |
| This journal is © The Royal Society of Chemistry 2017 |