
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface manipulation of atom substitution between cobalt phthalocyanine and the Cu(111) substrate†
Kongchao Shen‡
ab,
Bai Narsu‡c,
Gengwu Jib,
Haoliang Sunb,
Jinbang Hub,
Zhaofeng Liangb,
Xingyu Gaob,
Haiyang Lia,
Zheshen Li d,
Bo Song*e,
Zheng Jiang*b,
Han Huangf,
Justin W. Wellsg and
Fei Song*b
d,
Bo Song*e,
Zheng Jiang*b,
Han Huangf,
Justin W. Wellsg and
Fei Song*b
aDepartment of Physics, Zhejiang University, Hangzhou, 310027, China
bShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201204, China. E-mail: jiangzheng@sinap.ac.cn; songfei@sinap.ac.cn
cInner Mongolia Key Lab for Physics and Chemistry of Functional Materials, Department of Physics, Inner Mongolia Normal University, Hohhot, 010022, China
dISA, Department of Physics and Astronomy, University of Aarhus, 8000 Aarhus C, Denmark
eUniversity of Shanghai for Science and Technology, Shanghai, 200093, China. E-mail: bsong@usst.edu.cn
fSchool of Physics and Electronics, Central South University, Changsha, 410083, China
gDepartment of Physics, Norwegian University of Science and Technology, Trondheim N7491, Norway
First published on 2nd March 2017
Abstract
On-surface fabrication of controllable nanostructures is an appealing topic in the field of molecular electronics. Herein, the adsorption of cobalt phthalocyanine (CoPc) on a Cu(111) surface is investigated utilizing a combination of photoelectron spectroscopy (PES) and density functional theory (DFT). Interestingly, the scenario of atom exchange is discovered at the interface at room temperature (RT), namely the substitution of the cobalt atom in CoPc by a surface Cu adatom. Moreover, thermal annealing enhances the substitution process considerably which is demonstrated to be complete at about 573 K. As revealed by DFT calculations, the driving force for the observed interface transmetalation is most probably provided by the initial strong molecular-substrate interaction between Co atoms and the Cu(111) surface, the external thermodynamic energy gained from thermal sublimation and thermal annealing, and the tendency to form Co–Cu alloy at the interface. While CoPc has been successfully utilized in electrocatalysts for fuel cell applications and CuPc is commonly used as a leading material in organic solar cells, this report of interface transmetalation from CoPc to CuPc in a solid state environment may offer an encouraging approach towards the artificial engineering of organometallic nanostructures and related properties for surface catalysts, molecular electronics and so on.
Introduction
Controllable manipulation of organic nanostructures has been widely utilized as a promising approach for tuning the electronic, optical, chemical or physical properties at the surface/interface, with appealing application manifested in molecular electronics and biosensors.1 Fabrication of artificial on-surface structures is therefore a key procedure, including both the bottom-up method (for example, molecular self-assembly) and the top-down technique. The family of transition metal phthalocyanines (TMPcs, a π electron conjugated ring system complex) has been for a long period the subject of intense interest, due to their unique electronic and optical properties in biological processes,2,3 catalysis,4–8 field-effect transistors,9 solar cells10 and the dye industry.11 As realized, the performance of TMPc molecules in devices can be easily affected by the interface interactions.12–15 For example, the contact to specific substrate can alter or suppress the electronic properties of the TMPcs. Consequently, detailed investigations at molecular level of the interface between TMPcs and metallic substrates is necessary and will continually shed valuable insight into the understanding of various processes taking place at device interfaces.Due to the presence of partially empty d orbitals of the central metal atom in TMPcs, molecular substrate interaction can be systematically tuned depending on its contribution to the formation of interface structures. Therefore, well control on the transitional metal center incorporated into Pc macrocycles is of utmost importance to achieve tailored properties. For instance, the interface electronic structure has been studied for FePc, ZnPc, CuPc et al. on metallic substrates, and it was found that depending on the transition metal, the d-levels of metal atoms can significantly contribute to the highest occupied molecular orbital (HOMO) or lowest unoccupied molecular orbital (LUMO) of the TMPc.16–18 In particular, CoPc is a paramagnetic molecule with the cobalt ion interacts with its surrounding giving rise to different electronic and magnetic configurations,19 and is therefore a prototype candidate in view of a possible implementation of molecular magnetic junctions and electrocatalysts.20 Moreover, it has been repeatedly demonstrated in the literature that the cobalt atom in CoPc interacts strongly with metal substrates (Au, Cu or Ag),15,20–23 which might lead to the structural modification of the CoPc adsorbate, for example, the incorporation of surface adatom into the Pc macrocycle from the substrate and finally the substitution of central Co atom. This atom exchange process is the extension of on-surface metalation scenario, and usually called transmetalation in the form of M1Pc + M2 → M2Pc + M1 which has been demonstrated in solution before.24,25 As reported by Doyle et al. for nickel porphyrin compounds (NiDBrDPP and NiTBrPP) adsorbed on a Cu(111) surface, transmetalation at interface was discovered upon the contact with metal support and was enhanced by thermal annealing where nickel was completely removed from the porphyrin core and replaced by a substrate copper atom.26 However, on-surface transmetalation of transitional phthalocyanine macrocycle has not been observed before in solid-state conditions.
Herein we investigate the growth of CoPc on a Cu(111) surface with particular attention to explore the variation of interface structures and the related electronic properties. Surprisingly, it is shown in our study that CoPc interacts strongly with the Cu(111) substrate leading to the substitution of the Co ion by a Cu adatom from the substrate with the assistance of external thermodynamic energy, which subsequently results in the formation of Cu-metallated phthalocyanine and the releasing of metal cobalt atoms at interface.
Results and discussion
At the organic film/metal interfaces, various scenarios can be found: formation of the dipole layer due to charge transfer, redistribution of electron cloud, and interfacial chemical reaction,27 where the chemical interactions may result in new features/states at interface. As well known, XPS is a very powerful tool at detecting the changes in chemical state and environment of an atom, and will therefore provide fingerprint information regarding on the process occurring at CoPc/Cu(111) interface. First, 1 ML CoPc was prepared on the Cu(111) surface under UHV at RT followed by thermal annealing to 373 K, 473 K, 573 K, 673 K, 723 K for 30 minutes, respectively, while the corresponding core levels of Cu 2p3/2, Co 2p3/2, N 1s, and C 1s were recorded after each annealing treatment. Similarly, thick film of CoPc (around 3 ML) was also evaporated onto the Cu substrate afterwards and treated with the above-mentioned step-by-step annealing for direct comparison.To explore possible interactions at the interface involving the central metal atom of CoPc, Co 2p core level photoemission spectra was investigated in detail. In Fig. 1, the evolution of the Co 2p3/2 core level at the CoPc/Cu(111) interface are depicted as a function of annealing temperature for the thickness of 1 ML and 3 ML CoPc, respectively. At the first glance, a significant change of peak shape is observed by increasing annealing temperature. As seen, the second bottom spectrum in Fig. 1(a) for the as-deposited CoPc layer at RT is dominated by two components: a broad feature at high binding energy at 780.6 eV assigned to the Co(II) oxidation state, and an intense peak at lower binding energy of 778.5 eV, which can be assigned to the cobalt metal state (Co(0)).28 For convincing arguing, the spectrum of Co 2p3/2 from a Co metal film (around 4 Å) adsorbed on Cu(111) was also present at the bottom of Fig. 1(a) (red curve) as a reference, which matches well with the claimed cobalt(0) state from the CoPc/Cu(111) interface. Upon increasing annealing temperature, the Co(II) component attenuates in intensity and gets more and more faint, with a corresponding increase in the intensity of the Co(0) component from RT until to 573 K where the Co(II) component almost vanishes. Furthermore, no obvious change was observed afterwards when further annealing from 573 K to 723 K, except that the peak originated from Co(0) state turns broader which might be related to the desorption upon high temperature annealing, and shifts down to lower binding energy by about 0.3 eV. Similarly, in the case of 3 ML CoPc adsorbed on the Cu(111) substrate, Co(0) component was initially discovered at RT even though the signal of Co(II) state from multilayer is mostly dominating. Moreover, an additional component at 782.6 eV was resolved in Fig. 1(b), which can be assigned to the shake-up peak associated with Co(II) oxidation state at thick coverage,23 while in the case of monolayer thickness such satellite peak was not visible mostly due to the molecular-substrate interaction. Further annealing to 373 K and 473 K did not give any difference to the peak shape or relative intensity of Co(II) and Co(0) state, however, considerable enhancement of the peak intensity of Co(0) component was recorded when the annealing temperature went up to 573 K for multilayer of CoPc on Cu(111). Surprisingly, the peak contributed from the Co(II) oxidation state got much weaker and became equally intense to the peak related to the Co(0) state at 573 K, and shrunk further to about one third of the Co(0) component after annealed to 673 K, while the Co(0) peak was kept almost constant from 573 K to 673 K. Meanwhile, the satellite peak associated with the Co(II) component was also reduced in a similar manner.
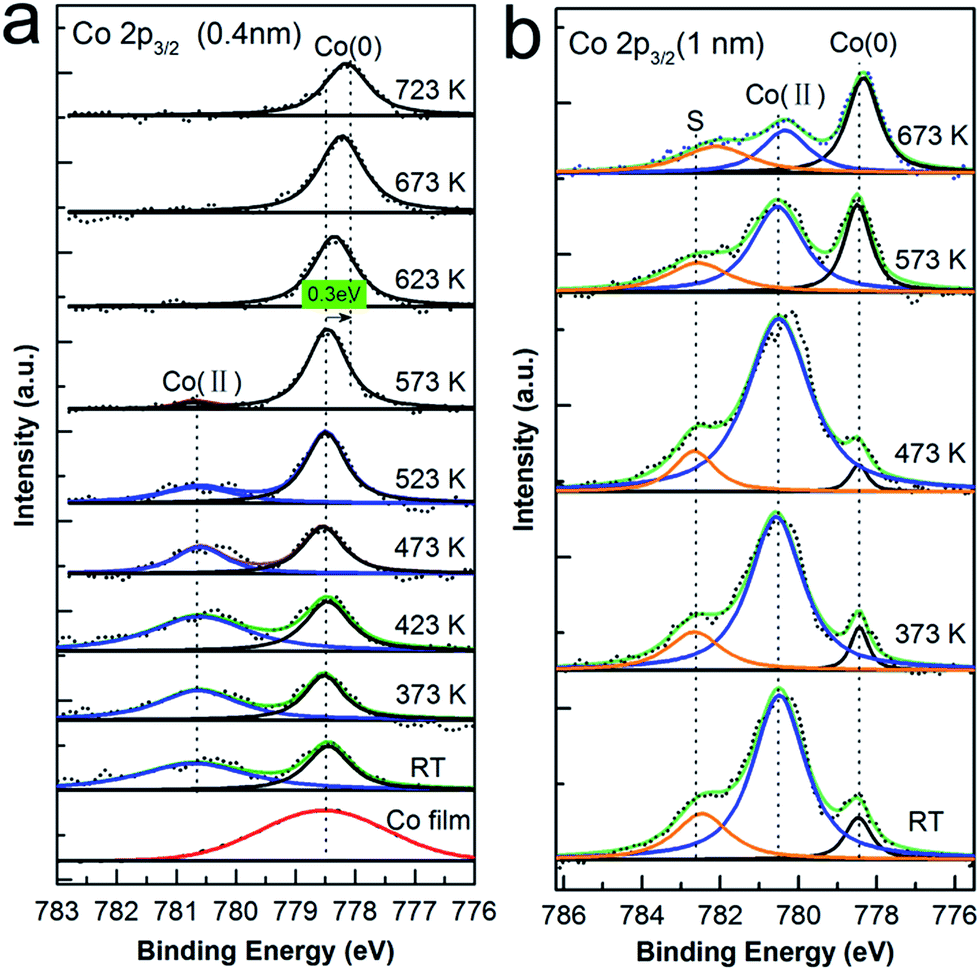 | ||
| Fig. 1 Co 2p3/2 XP spectra for the deposition of CoPc on Cu(111) kept at RT followed by subsequent annealing. (a) Co 2p3/2 core level of 1 ML and (b) 3 ML CoPc deposited on the Cu surface. Black dots: raw data, blue line: fitted component from Co(II) oxidation state, black line: fitted Co(0) component, orange line: the shake-up component related with the Co(II) state. As a reference, thin Co metal film (around 4 Å) was also prepared on the Cu(111) surface as shown in the bottom of (a) with red curve. | ||
For convenient comparison, spectrum of monolayer and multilayer CoPc adsorbed on the Cu(111) surface at RT is also stacked together as shown in Fig. S1 in ESI.† In the case of multilayer, relatively intense feature of Co(II) state was discovered as compared to the Co(0) state upon deposition at RT, since additional layers after the first layer are less affected by the Cu substrate. From the above analysis of Co 2p3/2 spectra between the monolayer and multilayer coverage, it is fairly visible that a Co(0) state is present upon adsorption regardless of the film thickness, which is located at 2.1 eV lower binding energy relative to the pristine Co(II) state from CoPc, and is almost identical to that of the metallic Co film on Cu(111). At this point, the presence of a Co(0) component in Fig. 1 implicating the appearance of metallic Co atom, might point out that the Co ion was displaced from the phthalocyanine macrocycle by Cu adatoms from substrate and turned to be metallic atoms at the interface. In addition, such replacement process on the Cu(111) surface begins at RT, enhanced by thermal annealing which probably provides adequate thermal dynamic energy for the surface Cu adatom, and finishes at about 573 K when all the Co ions have already been replaced by Cu atoms. Moreover, the vanishing of Co(II) peak at 573 K at the coverage of 1 ML and the remain of such peak after annealed to 673 K in the case of multilayer together suggest that the ion exchange process only happens for the first layer contacted with the copper substrate. Notably, the shift of Co(0) peak to lower binding energy by about 0.3 eV at the monolayer coverage from 573 K to 623 K is most probably induced by the polarization screening effect.29,30 As known, for atoms in a similar chemical state but adsorbed in the submonolayer, first and second layer, respectively, on a metal surface such shifts between the respective signals are always explained with polarization screening by metallic substrate.31
As reported by Chassé et al., charge transfer process involving mainly the Co atom was claimed from the significant changes of Co X-ray absorption spectra at the interface.23 Moreover, charge transfer induced strong chemical interaction between CoPc and various metals including silver, gold, and graphene/Ni(111) have also been demonstrated from previous reports,14,21,32–39 where it is claimed that such interactions directly affect the initial state of the metal ion and changes the effective oxidation state of the ion itself, resulting in a free Co metallic component shifted by about 2 eV to lower binding energy in the Co 2p2/3 spectrum.23,39,40 Since the interaction between CoPc and copper is expected to be much stronger compared to silver or gold, and the presence of cobalt metallic state (Co(0)) is undoubtedly evident, it can be concluded that the central atom Co in CoPc macrocycle is displaced by copper adatom from the Cu substrate, and therefore, atom exchange process-transmetalation is believed to occur at the CoPc/Cu(111) interface which forms the new CuPc organic and releasing metallic Co atoms. This procedure happens partially at RT, and gets greatly enhanced by thermal annealing which delivers free Cu adatoms to the interface from substrate and provides enough dynamic energy for barrier overcoming. However, due to the present of copper bulk substrate, no further information could be drawn from the Cu 2p core level since the spectra are kept unchanged during the whole annealing procedure except the intensity attenuation (as shown in Fig. S2†). Similarly, on-surface Ni–Cu ion exchange process has also been reported for Ni–porphyrinato on Cu(111),26 where an exchange of the Ni ion in the macrocycle with a substrate Cu atom results in Cu metallated porphyrins and metallic nickel atoms on the surface.
Since the desorption of CoPc molecule was recognized in Fig. 1, one may doubt about the intactness of the macrocycle after high temperature annealing and accordingly the conclusion of Co–Cu atom exchange at interface. To avoid such ambiguity, the N 1s and C 1s spectra were also recorded during annealing procedure, as shown in Fig. 2 and 3, respectively, meanwhile aiming to exploit the relative role of ligand atoms (C, N) in the interaction with the Cu substrate and the ion exchange process at the interface. The CoPc molecule has two different sites of nitrogen: the iminic nitrogen (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) and the Co bonded nitrogen (Co–N) in a chemically identical state.40 Previous studies show that the metallic nitrogen species is typically located at between 398.3 and 400.6 eV binding energy40–44 and the iminic N in MPc macrocycle is approximately in the binding energy range from 398.4 to 398.9 eV.40,41,45,46 In addition, there are 4 nitrogen atoms at iminic site and 4 metal-bonded nitrogen atoms in CoPc, which should give a 1
) and the Co bonded nitrogen (Co–N) in a chemically identical state.40 Previous studies show that the metallic nitrogen species is typically located at between 398.3 and 400.6 eV binding energy40–44 and the iminic N in MPc macrocycle is approximately in the binding energy range from 398.4 to 398.9 eV.40,41,45,46 In addition, there are 4 nitrogen atoms at iminic site and 4 metal-bonded nitrogen atoms in CoPc, which should give a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the corresponding peak intensity. As expected, the N 1s spectra of CoPc monolayer (Fig. 2(a)) and multilayer (Fig. 2(b)) on Cu(111) both show two well-resolved main peaks (blue and black curve) with approximately equal amplitude, the peak at 398.5 eV can be directly assigned to the iminic nitrogen while the peak at 400.1 eV is related to the Co-bonded nitrogen with the energy difference well consistent with the literature.40 Moreover, the evolution of N 1s peak with increasing annealing temperature is also presented. As seen, the peak shape was kept constant as well as the relative intensity ratio and the binding energy difference of the two components, except that both peak decrease and shift to lower binding energy by about 0.5 eV annealed from 523 K until to 723 K in the case of one monolayer CoPc deposition while the shift is almost negligible (less than 0.1 eV) at multilayer coverage. The attenuation of peak intensity is caused by desorption of CoPc molecules during annealing, which agrees well with the discussion in the Co 2p core level section. The peak shift can be explained again with the polarization screening effect induced by the metal substrate, which is distinct when the coverage varies from monolayer to submonolayer while ambiguous in the case of multilayer.31,47–52 However, there is still one point needed to be considered: the effect of the substitution of Co ion in CoPc by copper atom on the N 1s core level spectrum. As we pointed out before, the displacement of Co ion by Cu adatom results in the formation of CuPc, however, due to relative adjacency in binding energy of Co bonded and Cu bonded nitrogen41–45 and the chemical similarity of metal coordinated nitrogen atoms,31,52 it is fairly difficult to discriminate the difference between each other in the N 1s spectrum. For easy comparison, the N 1s of pristine CuPc prepared on Cu(111) with different thickness were also displayed in Fig. S3 in the ESI,† where it is quite visible that the binding energy of N 1s in CuPc is nearly same to that in CoPc on Cu(111). Although no further support could be drawn from N 1s spectra for the Co substitution process, it is still clear seen that typical N 1s peaks for the organic macrocycle in Fig. 2 were preserved well during thermal annealing. At this point, it can be concluded that either the original CoPc or the new formed CuPc is kept intact during the whole procedure.
:1 ratio of the corresponding peak intensity. As expected, the N 1s spectra of CoPc monolayer (Fig. 2(a)) and multilayer (Fig. 2(b)) on Cu(111) both show two well-resolved main peaks (blue and black curve) with approximately equal amplitude, the peak at 398.5 eV can be directly assigned to the iminic nitrogen while the peak at 400.1 eV is related to the Co-bonded nitrogen with the energy difference well consistent with the literature.40 Moreover, the evolution of N 1s peak with increasing annealing temperature is also presented. As seen, the peak shape was kept constant as well as the relative intensity ratio and the binding energy difference of the two components, except that both peak decrease and shift to lower binding energy by about 0.5 eV annealed from 523 K until to 723 K in the case of one monolayer CoPc deposition while the shift is almost negligible (less than 0.1 eV) at multilayer coverage. The attenuation of peak intensity is caused by desorption of CoPc molecules during annealing, which agrees well with the discussion in the Co 2p core level section. The peak shift can be explained again with the polarization screening effect induced by the metal substrate, which is distinct when the coverage varies from monolayer to submonolayer while ambiguous in the case of multilayer.31,47–52 However, there is still one point needed to be considered: the effect of the substitution of Co ion in CoPc by copper atom on the N 1s core level spectrum. As we pointed out before, the displacement of Co ion by Cu adatom results in the formation of CuPc, however, due to relative adjacency in binding energy of Co bonded and Cu bonded nitrogen41–45 and the chemical similarity of metal coordinated nitrogen atoms,31,52 it is fairly difficult to discriminate the difference between each other in the N 1s spectrum. For easy comparison, the N 1s of pristine CuPc prepared on Cu(111) with different thickness were also displayed in Fig. S3 in the ESI,† where it is quite visible that the binding energy of N 1s in CuPc is nearly same to that in CoPc on Cu(111). Although no further support could be drawn from N 1s spectra for the Co substitution process, it is still clear seen that typical N 1s peaks for the organic macrocycle in Fig. 2 were preserved well during thermal annealing. At this point, it can be concluded that either the original CoPc or the new formed CuPc is kept intact during the whole procedure.
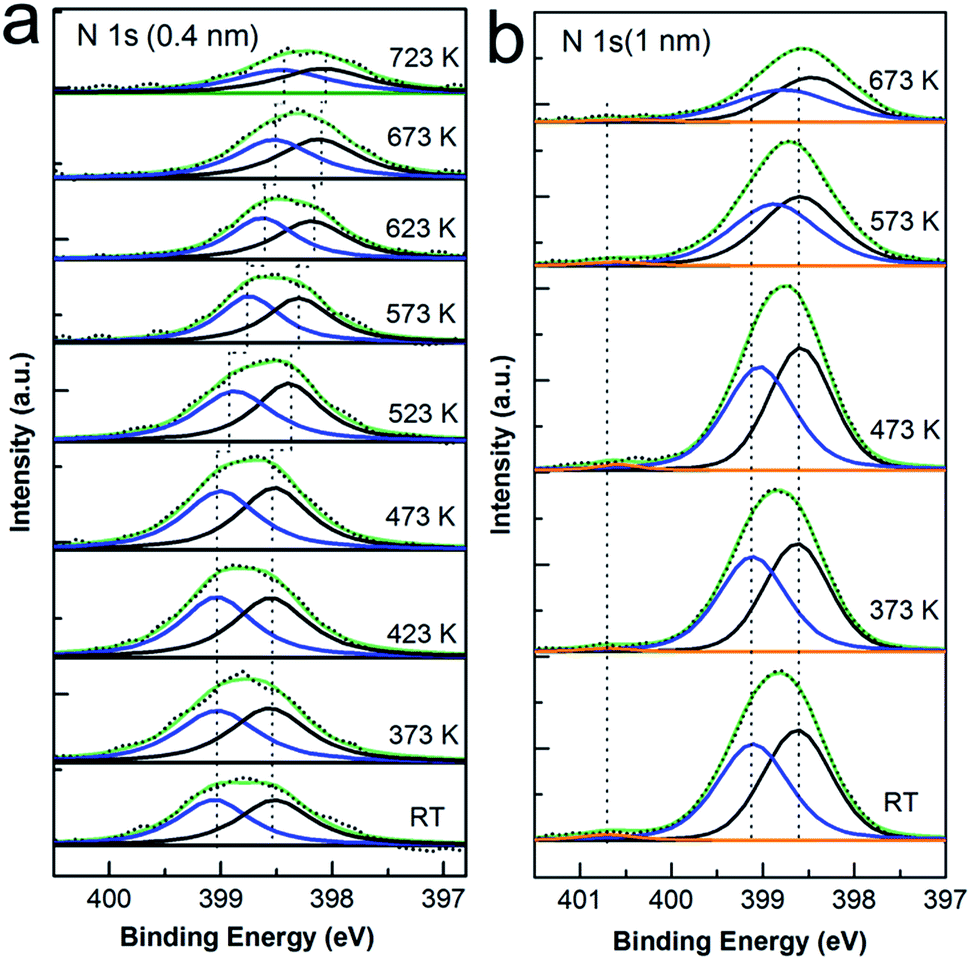 | ||
| Fig. 2 N 1s XP spectra of CoPc deposited onto Cu(111) with different thickness, followed by annealing to the temperatures as indicated. (a) N 1s core level spectra for 1 ML CoPc and (b) 3 ML CoPc as a function of annealing temperature. Black dots: raw data, black curve: the fitted iminic N (–N) component, blue curve: the fitted metal coordinated N component, orange curve: the shake-up feature. | ||
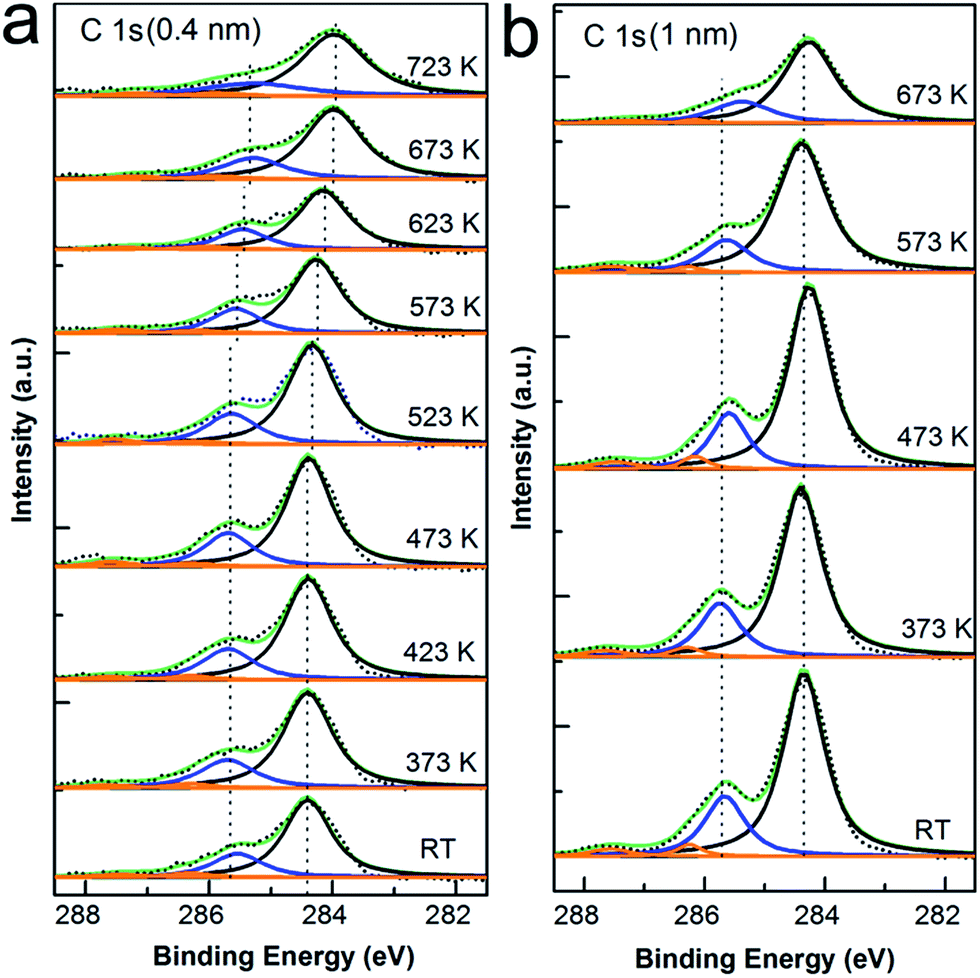 | ||
| Fig. 3 C 1s XP spectra for varying coverages of CoPc adsorbed on Cu(111), followed by subsequent annealing, (a) C 1s XP core level spectra for 1 ML and (b) 3 ML CoPc prepared on the Cu substrate. Black dots: raw data, black curve: the component related to C atoms within the benzene ring, blue curve: the component from the pyrrolic C atoms, orange curves: components from the satellite shake-up. | ||
As seen in Fig. 3, the evolution of C 1s core level spectra is reported for both thin and thick layer of CoPc deposited onto Cu surface as a function of annealing temperature. Detailed analysis of C 1s core level spectra first shows that the shape of C 1s peaks clearly tells the characteristic features for phthalocyanine C 1s spectra.40,48,53,54 At monolayer coverage, the C 1s spectrum is composed by two clearly resolved components with sharp peak, and the intense features are related to the C atoms within the benzene rings (284.4 eV) and pyrrole (285.7 eV). In the case of 3 ML coverage, there is also a pronounced shoulder attributed to the relative shakeup satellites, which locate at 1.9 eV higher binding energy compared to the main peaks and are well consistent with other reports of metal phthalocyanine.23,44,55 Secondly, the peak shape was preserved well for all spectra during annealing either at monolayer or multilayer. Similar to the case of N 1s spectra, there is a peak shift of 0.4 eV when annealed at 673 K for monolayer and less than 0.1 eV for multilayer with respect to the spectrum as deposited at RT, which again shall be attributed to the polarization screening effect from metal substrate due to desorption of macrocycle induced by thermal annealing. In the end, as concluded from both N 1s and C 1s spectra in Fig. 2 and 3, there is no visible indication for chemical interaction between the benzene ring or pyrrole inside the CoPc macrocycle and the Cu(111) surface, again supporting the interface interaction mainly localized between the cobalt ion and copper substrate.
In order to reveal the mechanism underlying the experimental observations and get more convincing insights for the atom exchange process occurring at the CoPc/Cu(111) interface, we have also performed DFT calculations regarding on the stability of metal phthalocyanine adsorbed on Cu(111). The binding energy of MPc (M = Cu or Co) on the Cu(111) surface was calculated utilizing the following formula,
| Ebinding(MPc@Cu(111)) = E(MPc@Cu(111)) − E(MPc) − E(Cu(111)) | (1) |
| Esubstituting = [E(CuPc) + E(Co)] − [E(CoPc) + E(Cu) = [E(CuPc) − E(Cu)] − [E(CoPc) − E(Co)] | (2) |
| Ealloying = E(Cu@CoCu(111)) − E(Co@Cu(111)) | (3) |
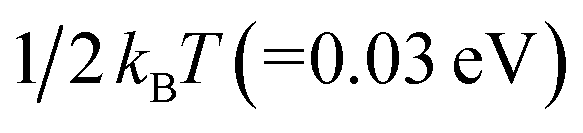 , such embedded thermal vibrations or motions of CoPc macrocycles would provide additional energy to touch off the interface ion-substitution reaction for some of the adsorbed CoPc molecules on Cu(111). In a word, the combination of thermal energy supplied by sublimation and an additional energy released by the subsequent formation of CoCu alloy afterwards, contributes to the ion substitution of Co in CoPc on Cu(111) at RT. Meanwhile, post annealing significantly enhance the ion substation reaction and completely transformed CoPc into CuPc. In another perspective, it can also be understood that the alloying formation of CoCu(111) will definitely prevent the reversed process of CuPc@CoCu(111) back to CoPc@Cu(111). As a summarize and also for a direct picture, the most possible reaction path occurring at CoPc/Cu(111) interface was depicted in Fig. 5, as concluded from DFT calculations.
, such embedded thermal vibrations or motions of CoPc macrocycles would provide additional energy to touch off the interface ion-substitution reaction for some of the adsorbed CoPc molecules on Cu(111). In a word, the combination of thermal energy supplied by sublimation and an additional energy released by the subsequent formation of CoCu alloy afterwards, contributes to the ion substitution of Co in CoPc on Cu(111) at RT. Meanwhile, post annealing significantly enhance the ion substation reaction and completely transformed CoPc into CuPc. In another perspective, it can also be understood that the alloying formation of CoCu(111) will definitely prevent the reversed process of CuPc@CoCu(111) back to CoPc@Cu(111). As a summarize and also for a direct picture, the most possible reaction path occurring at CoPc/Cu(111) interface was depicted in Fig. 5, as concluded from DFT calculations.
| MPc@Cu(111) | M in MPc | Co in CoCu alloy | |||
|---|---|---|---|---|---|
| CuPc | CoPc | Cu | Co | ||
| Ebinding (eV) | −0.157 | −0.380 | −6.070 | −8.101 | −1.128 |
| ΔE (eV) | 0.223 | 2.031 | |||
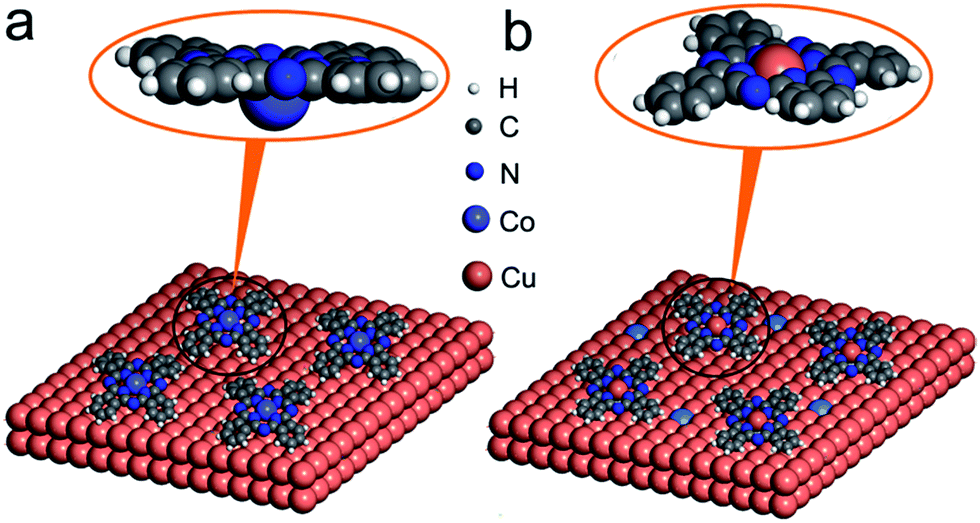 | ||
| Fig. 4 Configurations of MPc adsorbed on Cu(111) surface. (a) Deformed structure of CoPc was discovered once adsorbed on the Cu(111) substrate, where apparently the Co ion was attracted towards substrate from the molecular plane, (b) deformed structure of CuPc on Cu(111). It is visible that molecular ligands are deformed due to the molecular-substrate interaction. | ||
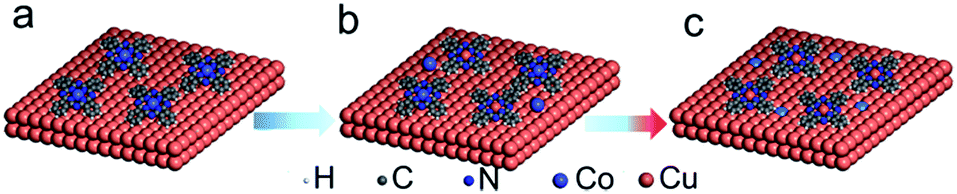 | ||
| Fig. 5 A possible reaction path occurring at the CoPc/Cu(111) interface. (a) As-deposited CoPc on the Cu(111) substrate, (b) chemical reaction between Cu atom and CoPc happened at RT and mild temperature, (c) forming Cu/Co alloy in pretty high temperature. | ||
As reported by Doyle et al., interface atom exchange between nickel porphyrin compounds and Cu adatoms on a Cu(111) substrate was undoubtedly demonstrated especially at RT, however, the principle for such substitution has not been clearly addressed. As a consequence, herein, we have consistently explained the experimental observation of cobalt metal state upon adsorption of CoPc on a Cu(111) surface, which is originated from the ion substitution of Co in CoPc by Cu adatoms at interface. Importantly, the mechanism behind substitution has been properly assigned to the driven force provided either by high-temperature sublimation or post annealing and the following formation of interface alloy, as revealed by detailed DFT calculations.
In short, we have experimentally explored the chemical states of Co 2p, N 1s and C 1s in with consecution and revealed the interface transmetalation process by DFT calculations, initially induced by the combined contribution from strong molecular-substrate interaction, external thermodynamic energy and interface alloying formation. Spontaneously, one will then easily get curious about the change of valence band structure regarding on new electronic states induced by the interface substitution and molecular-substrate interaction. Besides, a detailed knowledge of the valence band structure is fairly beneficial for the understanding of interfacial electronic structures. Consequently, UPS was recorded as well and analysed elaborately to further exploit the on-surface transmetalation. As plotted in Fig. 6, the valence band of 1 ML CoPc adsorbed on the copper substrate was stacked in sequence as a function of post annealing temperature, as well as the change of work function. While Fig. 6(a) shows the overall change of the valence band structure, a detailed investigation of the valence range from Fermi level (Ef) to about 1.5 eV below Ef was performed in Fig. 6(b). First, a peak (labeled as A) located at 1.40 eV below Ef is well resolved, which can be attributed to the HOMO of the organic adsorption. Since the HOMO level of CoPc is similar to that of CuPc adsorbed on Cu(111), as shown at the bottom of Fig. 6(b) for direct comparison and consistent with other theoretical reports,52 it is implausible to discriminate the contribution from CoPc or CuPc below 573 K when both of them exist. Notably, peak A gets weaken after further annealing which might be related to desorption of metallized macrocycle, as also discovered by XPS. Moreover, another peak (labelled as B in Fig. 6(b)) located at 0.90 eV below Fermi level is recognized for all the spectra of CoPc on Cu(111) at varying annealing temperatures, and this feature is most probably connected to the appearance of Co–Cu compound once Co atom is released from CoPc macrocycle and subsequently interacts with the Cu substrate, as pointed out by our DFT calculations. Further convincing insight can also be gained from a comparison to the valence band structure of pristine CuPc deposited on the Cu(111) substrate as shown at the bottom in Fig. 6(b), where no feature located at 0.9 eV below Ef can be resolved, in consistence with the previous report of CuPc adsorbed on Cu foil.58
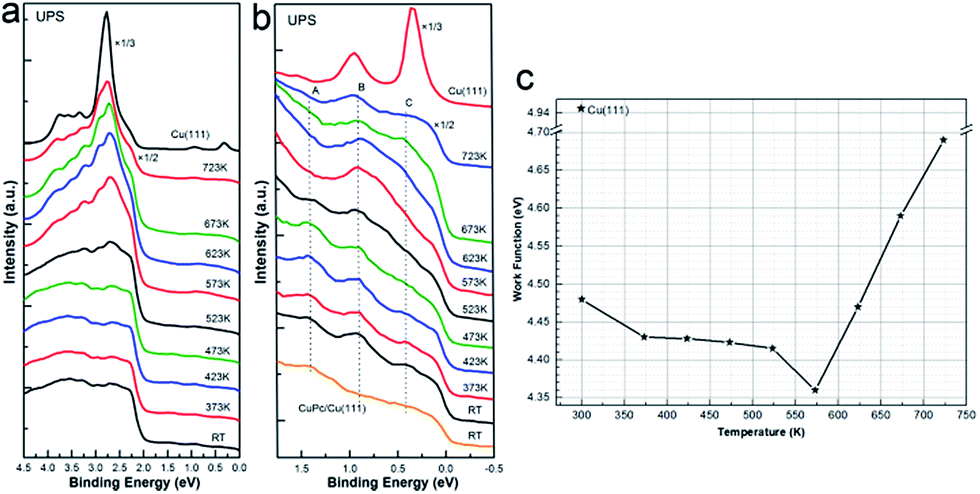 | ||
| Fig. 6 Valence band structure measured at hv = 21.22 eV for 1 ML of CoPc on Cu(111) deposited at RT, followed by step-by-step annealing to the indicated temperature. As a guide to eyes, the spectrum of the clean Cu(111) surface was also shown on top. (a) The evolution of UPS as a function of annealing temperature, (b) the zoom-in spectra of panel (a) from Fermi level (Ef) to 1.7 eV below Ef. (c) The change of working function (WF) at interface during annealing, the WF of clean Cu(111) is also indicated as reference. | ||
As summarized in Fig. 6(c), work function of the clean Cu(111) is determined to be 4.95 eV, while the organic adsorption of CoPc on top dramatically diminish the work function, which is reported to be related with the redistribution of the metal electron cloud or the formation of interface dipole due to the presence of the organic molecules, causing a reduction of the metal work function,59–61 while in our case herein, it can also be related to the appearance of HOMO levels of CoPc on Cu(111), since the occupied HOMO orbitals will definitely affect the electron redistribution at interface. Interestingly, the decreasing of interface work function is enhanced by the thermal annealing until 573 K, where the transmetalation of CoPc into CuPc gets fully completed, indicating the newly formed CuPc/Cu(111) system has a lower work function than the as-prepared CoPc/Cu(111) complex. However, the work function starts to recover after 573 K, which is most probably due to the fact that the organic macrocycle begins to desorb with further annealing as confirmed by XPS.
Experimental and calculations
Photoemission spectroscopy experiments
All the measurements were performed under ultrahigh vacuum (UHV) conditions with the base pressure better than 5 × 10−10 mbar. X-ray photoemission measurements were carried out in the lab using a monochromatic Al-Kα radiation (hν = 1486.6 neV), while the ultraviolet photoelectron spectroscopy (UPS) was done with a helium lamp (He I, hv = 21.2 eV). Photoelectrons were collected at normal emission to the sample surface with the specs electron analyser (Phoibos 100). The photon energies were calibrated against the Au 4f7/2 line and the metal Fermi level, respectively. The surface of the Cu(111) single crystal (Mateck, surface roughness less than 0.01 μm with orientation accuracy <0.1°) has been cleaned by repeated cycles of Ar+ sputtering at 1k–1.5k eV, followed by thermal annealing to around 770 K. The surface cleanliness has been checked by XPS afterwards until no carbon or oxygen contamination detected. The CoPc molecule (Sigma-Aldrich) was degassed in vacuum at 600 K for several hours prior deposition. Thin film of cobalt metal was prepared with a home-made e-beam evaporator. Molecules were deposited from a quartz crucible with thermal couple connected onto the substrate kept at room temperature (RT). The deposition rate and film thickness were calculated from the XPS intensity attenuation of Cu substrate assuming layer-by-layer growth. Herein, one monolayer (ML) is defined as the maximum coverage of molecules adsorbed in direct contact with the substrate as calibrated by annealing a multilayer to 427 K, which is around 4 Å. The intensity of spectra is normalized to the peak height of the most intense component to highlight the changes in the line shape, which were analyzed afterwards using XPSPEAK.DFT calculations
A hexagonal box is used to simulate the unit cell of the adsorption complex, with a dimension of 17.89 Å × 17.89 Å × 19.17 Å including three layers of Cu and 15 Å vacuum. The MPc macrocycles are placed into the vacuum and the absorption location of MPc on Cu(111) surface is determined by the force field simulation method implemented as absorption locator tool in Materials Studio. The MPc@Cu(111) system generated from the force field simulation is used as a input for the accurate total energy calculation.The total energy calculation is carried out using Projector Augmented Wave (PAW) method62 based on the Density Functional Theory (DFT)63,64 as implemented in Vienna Ab initio Simulation Package (VASP).65–67 The exchange–correlation effect is evaluated by the generalized gradient approximation as parameterized by Perdew, Burke and Ernzerhof (PBE).68 C(2s22p2), N(2s22p3), Co(3d84s1), Cu(3d104p1), and H(1s1) are treated as valence electrons. A plane wave basis set with a cutoff energy of 600 eV is used. The convergence criterion for electronic self-consistent relaxation is set to 10–5 eV. The Cu atoms in the substrate are fixed while the atoms in the absorbate macrocycles are fully relaxed with a force convergence criteria of 0.01 eV Å−1. The total energies of free standing MPc macrocycles are calculated by placing them into an orthorhombic cell with a size of 17 × 18 × 13 Å3. Same energy cutoff and convergence criteria with those for total energy calculation of MPc@Cu(111) composite are used.
Conclusions
Although numerous investigations have been done for transition-metal phthalocyanine adsorbed on metal substrates before, essential details of the electronic structure at interfaces are still unclear, to some extent. In the present work, transmetalation occurring at CoPc/Cu(111) interface has been demonstrated by XPS, UPS and well investigated with DFT theory. Whilst the Co(II) oxidation state in CoPc macrocycle is reduced and the Co(0) state assigned to free cobalt metal atom is increasing with external thermal annealing, as concluded from detailed analysis of Co 2p3/2 XP spectra, the substitution of Co ion in the center of CoPc by the surface Cu atom was confirmed. Supported by DFT calculations, strong interaction between CoPc and the copper substrate is proposed; moreover, the mechanism underneath the observed on-surface transmetalation from CoPc to CuPc is revealed, which is jointly contributed by the strong molecular-substrate interaction, external thermodynamic energy and the Co–Cu alloying at surface. Furthermore, UPS study also proves the formation of Co–Cu compound at interface following the release of Co from CoPc. While on-surface engineering of metal–organic nanostructures has been an appealing topic to both fundamental research and practical applications nowadays, our report of on-surface transmetalation from CoPc to CuPc may predict a controllable approach in solid-state environment to engineer functional metal–organic nanostructures for rich applications in molecular electronics and the related devices.Acknowledgements
We thank Dr Yingying Wei and Dr B. V. Tran for fruitful discussions. This work was financially supported by the National Natural Science Foundation of China (91545101 and 11464037), The Hundred Talents Program of Chinese Academy of Sciences, and Shanghai Pujiang Program.Notes and references
- Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401–6408 CrossRef CAS PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- B. R. Takulapalli, G. M. Laws, P. A. Liddell, J. Andréasson, Z. Erno, D. Gust and T. J. Thornton, J. Am. Chem. Soc., 2008, 130, 2226–2233 CrossRef CAS PubMed.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. Nolte, J. A. Elemans and S. Speller, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS PubMed.
- F. Sedona, M. Di Marino, D. Forrer, A. Vittadini, M. Casarin, A. Cossaro, L. Floreano, A. Verdini and M. Sambi, Nat. Mater., 2012, 11, 970–977 CrossRef CAS PubMed.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- C. Z. Zhang, R. Hao, H. Yin, F. Liu and Y. L. Hou, Nanoscale, 2012, 4, 7326–7329 RSC.
- P. F. Siles, T. Hahn, G. Salvan, M. Knupfer, F. Zhu, D. R. T. Zahn and O. G. Schmidt, Nanoscale, 2016, 8, 8607–8617 RSC.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671 CrossRef CAS PubMed.
- J. M. Gottfried, in On-Surface Synthesis, ed. A. Gourdon, Springer, Switzerland, 2016, ch. 7, p. 131 Search PubMed.
- B. W. Caplins, D. E. Suich and A. J. J. Shearer, Phys. Chem. Lett., 2014, 5, 1679–1684 CrossRef CAS PubMed.
- S. Ahmadi, M. N. Shariati, S. Yu and M. Göthelid, J. Chem. Phys., 2012, 137, 084705 CrossRef PubMed.
- H. Peisert, J. Uihlein, F. Petraki and T. Chassé, J. Electron Spectrosc. Relat. Phenom., 2015, 204, 49–60 CrossRef CAS.
- A. Zhao, Q. Li, L. Chen, H. Xiang, W. Wang, S. Pan, B. Wang, X. Xiao, J. Yang, J. G. Hou and Q. Zhu, Science, 2005, 309, 1542–1544 CrossRef CAS PubMed.
- S. Stepanow, A. Mugarza, G. Ceballos, P. Moras and J. C. Cezar, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 014405 CrossRef.
- J. Schaffert, M. C. Cottin, A. Sonntag, H. Karacuban, C. A. Bobisch, N. Lorente, J. Gauyacq and R. Möller, Nat. Mater., 2013, 12, 223–227 CrossRef CAS PubMed.
- F. Song, J. W. Wells, K. Handrup, Z. S. Li, S. N. Bao, K. Schulte, M. Ahola-Tuomi, L. C. Mayor, J. C. Swarbrick, E. W. Perkins, L. Gammelgaard and P. Hofmann, Nat. Nanotechnol., 2009, 4, 373–376 CrossRef CAS PubMed.
- T. Kroll, V. Y. Aristov, O. V. Molodtsova, Y. A. Ossipyan, D. V. Vyalikh, B. Büchner and M. Knupfer, J. Phys. Chem. A, 2009, 113, 8917–8922 CrossRef CAS PubMed.
- E. Annese, J. Fujii, I. Vobornik and G. Rossi, J. Phys. Chem. C, 2011, 115, 17409–17416 CAS.
- J. Uihlein, M. Polek, M. Glaser, H. Adler, R. Ovsyannikov, M. Bauer, M. Ivanovic, A. B. Preobrajenski, A. V. Generalov, T. Chassé and H. Peisert, J. Phys. Chem. C, 2015, 119, 15240–15247 CAS.
- F. Petraki, H. Peisert, F. Latteyer, U. Aygül, A. Vollmer and T. Chassé, J. Phys. Chem. C, 2011, 115, 21334–21340 CAS.
- F. Petraki, H. Peisert, I. Biswas and T. Chassé, J. Phys. Chem. C, 2010, 114, 17638–17643 CAS.
- K. Diller, A. C. Papageorgiou, F. Klappenberger, F. Allegretti, J. V. Barth and W. Auwärter, Chem. Soc. Rev., 2016, 45, 1629–1656 RSC.
- P. Hambright and P. B. Chock, J. Am. Chem. Soc., 1974, 96, 3123–3131 CrossRef CAS PubMed.
- C. M. Doyle, J. P. Cunniffe, S. A. Krasnikov, A. B. Preobrajenski, Z. Li, N. N. Sergeeva, M. O. Senge and A. A. Cafolla, Chem. Commun., 2014, 50, 3447–3449 RSC.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- J. Chastian, R. C. King and J. F. Moulder, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, MN: Physical Electronics, Eden Prairie, 1995 Search PubMed.
- K. Diller, F. Klappenberger, F. Allegretti, A. C. Papageorgiou, S. Fischer, A. Wiengarten, S. Joshi, K. Seufert, D. Écija, W. Auwärter and J. V. Barth, J. Chem. Phys., 2013, 138, 154710 CrossRef CAS PubMed.
- H. Peisert, D. Kolacyak and T. Chassé, J. Phys. Chem. C, 2009, 113, 19244–19250 CAS.
- T. C. Chiang, G. Kaindl and T. Mandel, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 695 CrossRef CAS.
- M. Schmid, A. Kaftan, H. Steinrück and J. M. Gottfried, Surf. Sci., 2012, 606, 945–949 CrossRef CAS.
- J. Uihlein, H. Peisert, M. Glaser, M. Polek, H. Adler, F. Petraki, R. Ovsyannikov, M. Bauer and T. Chassé, J. Chem. Phys., 2013, 138, 081101 CrossRef PubMed.
- J. D. Baran, J. A. Larsson, R. A. J. Woolley, Y. Cong, P. J. Moriarty, A. A. Cafolla, K. Schulte and V. R. Dhanak, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 075413 CrossRef.
- M. G. Betti, P. Gargiani, R. Frisenda, R. Biagi, A. Cossaro, A. Verdini, L. Floreano and C. Mariani, J. Phys. Chem. C, 2010, 114, 21638–21644 CAS.
- M. Toader, P. Shukrynau, M. Knupfer, D. R. T. Zahn and M. Hietschold, Langmuir, 2012, 28, 13325 CrossRef CAS PubMed; M. Toader, M. Knupfer, D. R. T. Zahn and M. Hietschold, Surf. Sci., 2011, 605, 1510–1515 CrossRef.
- A. Mugarza, R. Robles, C. Krull, R. Korytár, N. Lorente and P. Gambardella, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155437 CrossRef.
- E. Salomon, P. Amsalem, N. Marom, M. Vondracek, L. Kronik, N. Koch and T. Ango, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 075407 CrossRef.
- L. Massimi, M. Angelucci, P. Gargiani, M. G. Betti, S. Montoro and C. Mariani, J. Chem. Phys., 2014, 140, 244704 CrossRef CAS PubMed; G. Mette, D. Sutter, Y. Gurdal, S. Schnidrig, B. Probst, M. Iannuzzi, J. Hutter, R. Alberto and J. Osterwalder, Nanoscale, 2016, 8, 7958–7968 RSC.
- Y. Bai, F. Buchner, M. T. Wendahl, I. Kellner, A. Bayer, H. P. Steinruck, H. Marbach and J. M. Gottfried, J. Phys. Chem. C, 2008, 112, 6087–6092 Search PubMed; Y. Bai, M. Sekita, M. Schmid, T. Bischof, H.-P. Steinrück and J. M. Gottfried, Phys. Chem. Chem. Phys., 2010, 12, 4336–4344 RSC.
- K. Eguchi, T. Nakagawa, Y. Takagi and T. Yokoyama, J. Phys. Chem. C, 2015, 119, 9805–9811 CAS.
- A. Calabrese, L. Floreano, A. Verdini, C. Mariani and M. G. Betti, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115446 CrossRef.
- M. Abadía, R. González-Moreno, A. Sarasola, G. Otero-Irurueta, A. Verdini, L. Floreano, A. Garcia-Lekue and C. Rogero, J. Phys. Chem. C, 2014, 118, 29704–29712 Search PubMed.
- Y. Zhang, S. Wang, A. Demasi, I. Reid, L. F. J. Piper, A. Y. Matsuura, J. E. Downes and K. E. Smith, J. Mater. Chem., 2008, 18, 1792–1978 RSC.
- M. Scardamaglia, C. Struzzi, S. Lizzit, M. Dalmiglio, P. Lacovig, A. Baraldi, C. Mariani and M. G. Betti, Langmuir, 2013, 29, 10440–10447 CrossRef CAS PubMed.
- S. Yu, S. Ahmadi, C. Sun, P. T. Z. Adibi, W. Chow, A. Pietzsch and M. M. Göthelid, J. Chem. Phys, 2012, 136, 154703 CrossRef PubMed.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H. Steinrück, J. Phys. Chem. C., 2007, 111, 3090–3098 CAS.
- Y. Bai, F. Buchner, I. Kellner, M. Schmid, F. Vollnhals, H. Steinrueck, H. Marbach and J. M. Gottfried, New J. Phys., 2009, 11, 125004 CrossRef.
- R. Liu, J. Duay, T. Lane and S. Bok Lee, Phys. Chem. Chem. Phys., 2010, 12, 4309–4316 RSC.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- G. Di Santo, C. Castellarin-Cudia, M. Fanetti, B. Taleatu, P. Borghetti, L. Sangaletti, L. Floreano, E. Magnano, F. Bondino and A. Goldoni, J. Phys. Chem. C, 2011, 115, 4155–4162 CAS.
- F. Klappenberger, A. Weber-Bargioni, W. Auwärter, M. Marschall, A. Schiffrin and J. V. Barth, J. Chem. Phys., 2008, 129, 214702 CrossRef CAS PubMed.
- H. Peisert, M. Knupfer and J. Fink, Surf. Sci., 2002, 515, 491–498 CrossRef CAS.
- A. Ruocco, F. Evangelista, R. Gotter, A. Attili and G. Stefani, J. Phys. Chem. C, 2008, 112, 2016–2025 CAS.
- S. Ahmadi, B. Agnarsson, I. Bidermane, B. M. Wojek, Q. Noel, C. Sun and M. Gothelid, J. Chem. Phys., 2014, 140, 174702 CrossRef PubMed.
- S. J. Grabowski, J. Phys. Org. Chem., 2004, 47, 18–31 CrossRef.
- T. Allmers and M. Donath, Surf. Sci., 2011, 605, 1875–1924 CrossRef CAS.
- S. Sinha and M. Mukherjee, Appl. Surf. Sci., 2015, 353, 540–547 CrossRef CAS.
- H. Peisert, M. Knupfer and J. Fink, Appl. Phys. Lett., 2002, 81, 2400 CrossRef CAS.
- X. Crispin, V. Geskin, A. Crispin, J. Cornil, R. Lazzaroni, W. R. Salaneck and J. Brédas, J. Am. Chem. Soc., 2002, 124, 8131–8141 CrossRef CAS PubMed.
- P. S. Bagus, V. Staemmler and C. Woll, Phys. Rev. Lett., 2002, 89, 096104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS; G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00636e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |