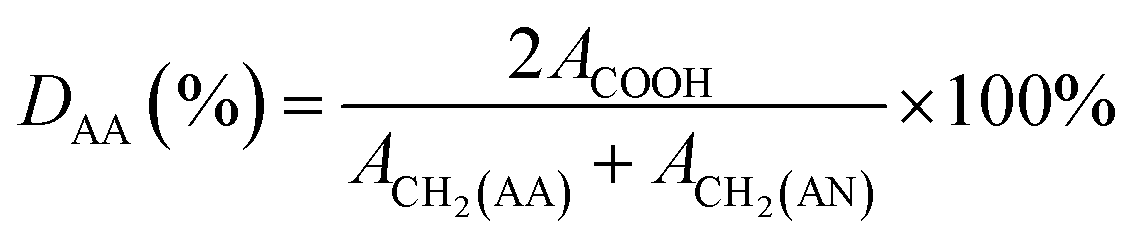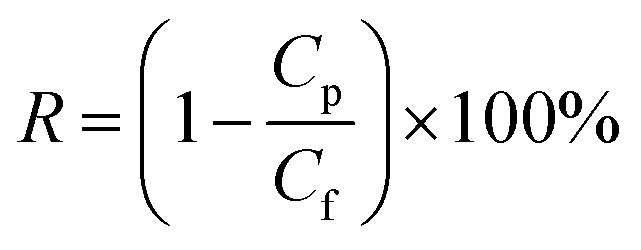DOI:
10.1039/C7RA00621G
(Paper)
RSC Adv., 2017,
7, 18001-18013
A durable thin-film nanofibrous composite nanofiltration membrane prepared by interfacial polymerization on a double-layer nanofibrous scaffold
Received
15th January 2017
, Accepted 17th March 2017
First published on 24th March 2017
Abstract
A novel kind of thin-film nanofibrous composite (TFNC) nanofiltration membrane consisting of a polypiperazine amide (PPA) barrier layer, an ultrathin electrospun poly(acrylonitrile-co-acrylic acid) (PAN–AA) transitional mid-layer and an electrospun polyacrylonitrile (PAN) nanofibrous supporting layer, was successfully fabricated by interfacial polymerization with piperazine (PIP) and trimesoyl chloride (TMC) onto the PAN–AA/PAN double-layer substrate. The PAN–AA nanofibrous mid-layer played two important roles between the PPA barrier layer and the PAN nanofibrous supporting layer. It could be swollen in the alkaline aqueous monomer (PIP) solution to form an intermediate hydrogel film, which acted as the transitional mid-layer to cover the majority of the large surface pores of the electrospun PAN nanofibrous substrate. On the other hand, the hydrophilic PAN–AA hydrogel film could capture and reserve abundant PIP monomer to facilitate interfacial polymerization with TMC to form an endurable ultrathin PPA barrier layer, resulting in an integrated composite membrane confirmed by the mechanical properties. The resultant TFNC membranes demonstrated a high rejection rate (98.2%) and high permeate flux (64.4 L m−2 h−1) for MgSO4 aqueous solution (2.0 g L−1), and also exhibited excellent structural stability due to the strong interactions between the barrier layer and the nanofibrous support that were enhanced by the transitional PAN–AA mid-layer.
1. Introduction
In recent decades, thin film composite (TFC) membranes have been heavily researched for developing nanofiltration (NF) membranes, which have an asymmetric structure composed of a thin functional barrier layer on a porous substrate.1,2 It is considered as a key advantage that each layer in a TFC membrane can be optimized. The thin dense barrier layer is usually obtained through dip-coating,3 layer-by-layer self-assembly4,5 or interfacial polymerization,6,7 by which the filtration performance of TFNC membranes is mainly determined. Meanwhile, the conventional reinforced porous matrix is a dense asymmetric membrane made by phase inversion methods which could provide enough mechanical strength to support a thin barrier layer.8 Recently, an alternative highly porous substrate made of electrospun nanofibrous mat has received much attention as the support layer to fabricate TFC membrane due to its high permeability that derived from its high porosity (up to over 80%), interconnectivity, micro scale interstitial space, and a large surface-to-volume ratio.9,10 This kind of TFC membranes was named as thin film nanofibrous composite (TFNC) membranes, which have been proven to be an effective media for microfiltration (MF),11 ultrafiltration (UF)12–14 and NF,8,15–17 as the permeability of the composite membrane was enhanced while maintaining good solute rejection capability.
However, high permeability of the nanofibrous substrates from its high porous structure will bring new challenges for the preparation of the top barrier layer onto the nanofibrous substrate. For example, it is difficult to avoid the permeation of coating solution into the inside of the nanofibrous scaffold resulting in a much thicker barrier layer during the conventional surface coating process. Especially for the barrier layer prepared by interfacial polymerization method, the low viscous aqueous phase monomer solution held in nanofibrous substrate could be easily drained away from the surface during the interfacial polymerization process, readily leading to a defective and ultrathin barrier layer. In addition, the diameters of most polymer fibers generated by electrospinning are in the range of 100–1000 nm, the mean pore sizes of electrospun nanofibrous membranes (ENMs) are relatively large (more than 300 nm) according to the relationship between membrane pore size and fiber diameter in an electrospun non-woven structure, i.e., the mean pore size of nanofibrous membranes is about 3 ± 1 times the mean fiber diameter.18,19 Therefore, the ultrathin interfacial polymerized barrier layer supported by ENMs with the relatively large pore size may cause new problems about the stability and durability of the TFNC membrane. To overcome these shortcomings, various approaches have been attempted such as soaking in a coagulant bath,20,21 treating the nanofibrous substrates by hot-pressing,22 introducing a dopamine intermediate layer7,15 or coating a regenerated cellulose nanowhiskers layer on the nanofibrous substrate.13,17,23,24 The introduction of intermediate transitional layer not only can reduce the surface pore size of nanofibrous substrates, but also can introduce functional groups such as hydroxyl groups, carboxyl groups,23 and amino groups15 leading to a more hydrophilic surface for further modification. Herein, the intermediate transitional layer can fill the gaps between functional barrier layer and nanofibrous substrate and build up the interconnection on both sides, and the integrity of TFNC membranes will be significantly enhanced.
In the present study, a double-layer nanofibrous substrate containing an ultrathin poly(acrylonitrile-co-acrylic acid) (PAN–AA) nanofibrous transitional layer and a thick PAN nanofibrous support layer was used to prepare the TFNC membranes. The more hydrophilic PAN–AA nanofibers could be swollen in aqueous amine solution and merged into an ultrathin hydrogel film which could cover most of the large pores (0.3–1.0 μm) on the surface of PAN nanofibrous substrate and enhanced the membrane surface hydrophilicity.25 The swollen PAN–AA transitional layer not only can form interconnection with PAN nanofibrous substrate by hydrogen bond between the nitrile group of PAN and the carboxylic acid group of PAN–AA, but also can capture the aqueous amine monomer for the following interfacial polymerization process, i.e. the flow away of aqueous monomer in the process of conventional interfacial polymerization could be avoided effectively, and further improved the interfacial bonding between the polyamide barrier layer and the PAN nanofibrous supporting layer. The effect of the transitional PAN–AA mid-layer on the fabrication of the interfacial polyamide barrier layer onto the nanofibrous support was investigated in details. Various parameters were optimized for the preparation of an endurable TFNC nanofiltration membrane with high filtration performance.
2. Experiment
2.1 Materials
PAN (Mw = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from J&K Scientific Ltd., which was dried in vacuum oven at 50 °C for 24 h before use. Piperazine (PIP), trimesoyl chloride (TMC), n-hexane, sodium carbonate (Na2CO3), sodium hydroxide (NaOH), magnesium sulfate (MgSO4), sodium sulfate (Na2SO4), magnesium chloride (MgCl2), sodium chloride (NaCl) and potassium chloride (KCl) were purchased from Sinopharm Chemical Reagent Co., Ltd., without further purification before use. Acrylonitrile (AN), acrylic acid (AA), azobisisobutyronitrile (AIBN), methanol, ethanol, N,N′-dimethylformamide (DMF) and dimethylsulfoxide (DMSO) was kindly supplied by Shanghai Chemical Reagent Plant.
000 g mol−1) was purchased from J&K Scientific Ltd., which was dried in vacuum oven at 50 °C for 24 h before use. Piperazine (PIP), trimesoyl chloride (TMC), n-hexane, sodium carbonate (Na2CO3), sodium hydroxide (NaOH), magnesium sulfate (MgSO4), sodium sulfate (Na2SO4), magnesium chloride (MgCl2), sodium chloride (NaCl) and potassium chloride (KCl) were purchased from Sinopharm Chemical Reagent Co., Ltd., without further purification before use. Acrylonitrile (AN), acrylic acid (AA), azobisisobutyronitrile (AIBN), methanol, ethanol, N,N′-dimethylformamide (DMF) and dimethylsulfoxide (DMSO) was kindly supplied by Shanghai Chemical Reagent Plant.
2.2 Synthesis of poly(acrylonitrile-co-acrylic acid)
Polyacrylonitrile-co-acrylic acid (PAN–AA) with a viscosity-averaged molecular weight (Mη) of 2.4 × 105 g mol−1 was synthesized by radical polymerization according to the method reported by Takaomi Kobayashi et al.26 AN was dried over molecular sieves and distilled under atmospheric pressure at 78 °C to remove the inhibitor. AA was distilled under reduced pressure (16 mmHg, 50 °C). AIBN was recrystallized in supersaturated ethanol solution. All other reagent grade chemicals were used without further purification. Radical polymerization was carried out in DMSO solution as follows. In a reaction vessel with 500 mL capacity, 30.4 g (566 mmol) of purified AN, 7.51 g (104 mmol) of AA, 110.5 g of DMSO and 0.22 g of AIBN were introduced. Polymerization was carried out at 60 °C for 6 h in nitrogen flow. The reaction was terminated and the mixture was poured into a large quantity of water to precipitate the crude copolymer. After washed with hot deionized water and methanol alternatively several times, the copolymer was then dried under vacuum at 40 °C for at least 24 h. The copolymerization was confirmed by FTIR and 1H-NMR (d6-DMSO, 400 MHz) measurements, as the absorbance peaks of FTIR νmax/cm−1: 3470 (–OH), 1732 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1627 (–COOH) and chemical shifts of 1H-NMR δH/ppm: 12.87 (–COOH of AA), 2.05 (2H, –CH2– of AN), 1.85 (2H, –CH2– of AA). The result were in accordance with the reports in literature.27 The degree value of the acrylic acid (DAA) was about 9.95%, which was calculated by following equation:
O), 1627 (–COOH) and chemical shifts of 1H-NMR δH/ppm: 12.87 (–COOH of AA), 2.05 (2H, –CH2– of AN), 1.85 (2H, –CH2– of AA). The result were in accordance with the reports in literature.27 The degree value of the acrylic acid (DAA) was about 9.95%, which was calculated by following equation:| |
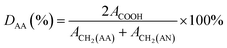 | (1) |
where ACOOH is the peak area of protons from carboxyl group (–COOH) attached to AA, ACH2(AA) is the peak area of protons from methylene (–CH2–) attached to AA, and ACH2(AN) is the peak area of protons from methylene (–CH2–) attached to AN.27 The peak area of protons from carboxyl group (ACOOH) was 1.0, and the peak area of protons from methylene (–CH2–) was 20.11 in total. From the polymer structure, peak area of protons from methylene attached AA (ACH2(AA)) was relative 2 times of the ACOOH. And the intrinsic viscosity of copolymer was determined by Ubbelodhe-type viscometer in DMF at 30 °C. Molecular weight of poly(acrylonitrile-co-acrylic acid) was calculated by the following Mark–Houwink equation:| | |
[η] = 2.78 × 10−4M0.76
| (2) |
2.3 Fabrication of PAN single-layer and PAN–AA/PAN double-layer nanofibrous substrates
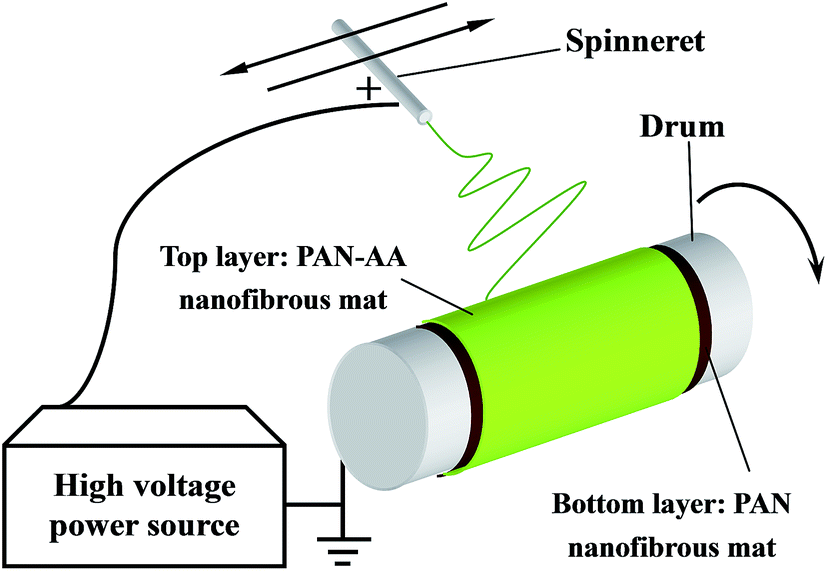
Dried PAN and PAN–AA powder was dissolved in DMF with gentle stirring at ∼55 °C in water bath for at least 12 h until it became a homogeneous solution respectively. The above polymer solutions were subject to a customer-build electrospinning setup described in our previous work.28 The fabrication process of PAN–AA/PAN double-layer nanofibrous membranes were showed in Scheme 1. Firstly, 10.0 mL PAN solution (10 wt%) was electrospun on a grounded rotating metal drum covered with aluminum foil (the applied electric voltage was 20 kV and the solution feed rate was 16.6 μL min−1) until the thickness of the PAN nanofibrous substrate reached to around 40 μm. Then an ultrathin layer of PAN–AA nanofibers was electrospun on the PAN nanofibrous substrate for a period of time (the applied electric voltage was 24 kV and the solution feed rate was 10.0 μL min−1), and the thickness of PAN–AA layer was adjusted by controlling the depositing time. The spinneret with diameter of 0.7 mm was made a translational oscillatory motion perpendicular to the drum rotation direction (the oscillation distance was about 30 cm and the distance between the spinneret and the grounded drum was 15 cm) driven by a step motor to ensure the production of uniform electrospun membrane with sufficient area (i.e. 30 × 31.4 cm2) for measurements. The surrounding temperature was ∼40 °C and the environmental humidity was controlled at ∼40%. The as-prepared PAN single-layer and PAN–AA/PAN double-layer nanofibrous membranes were dried at 50 °C in vacuum for at least 24 h after electrospinning for further use, and the resultant samples were denoted as PAN–AAx/PAN, where x stands for the depositing time of PAN–AA nanofibers.
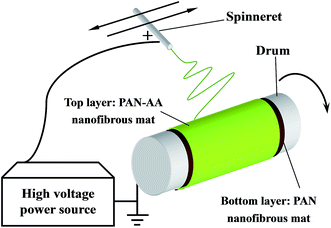 |
| | Scheme 1 Schematic diagram illustrating the fabrication of PAN–AA/PAN double-layer nanofibrous substrate. | |
2.4 Fabrication of TFNC nanofiltration (NF) membranes on different substrates by interfacial polymerization
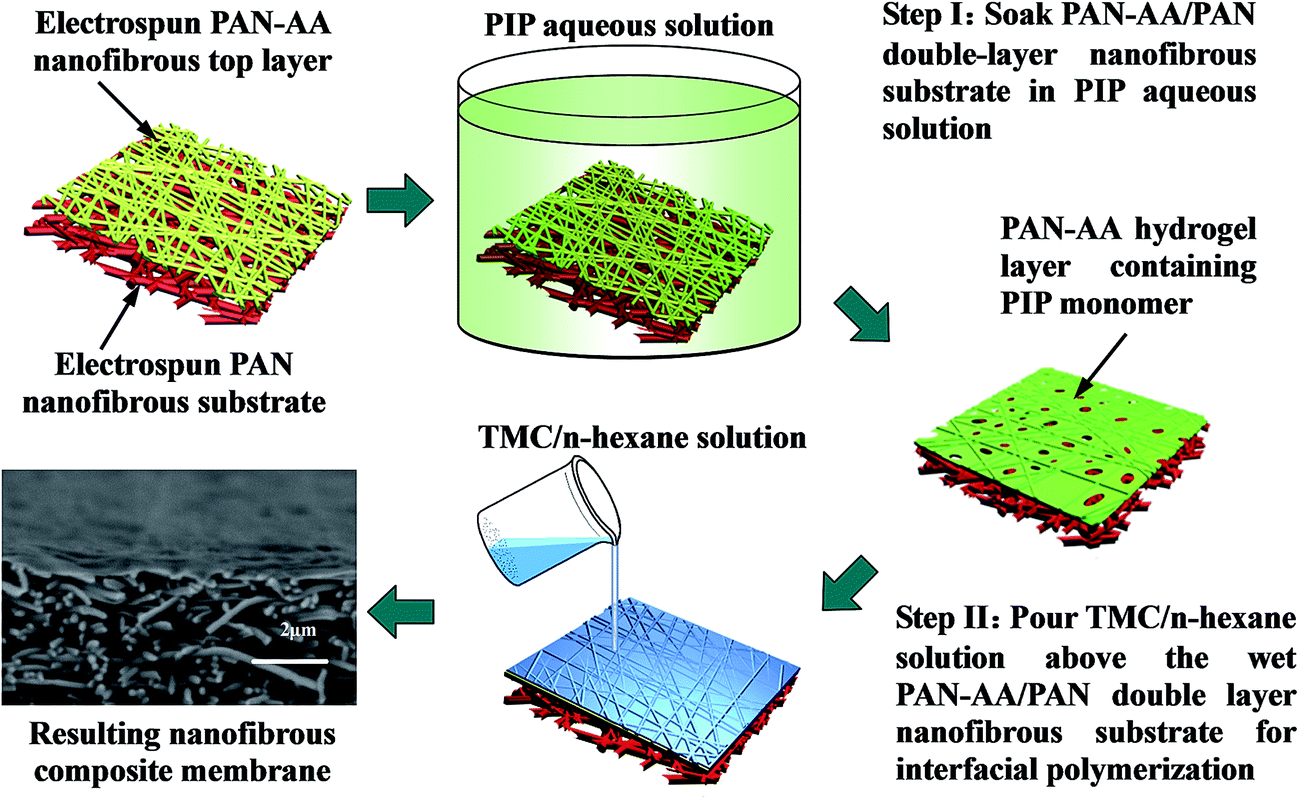
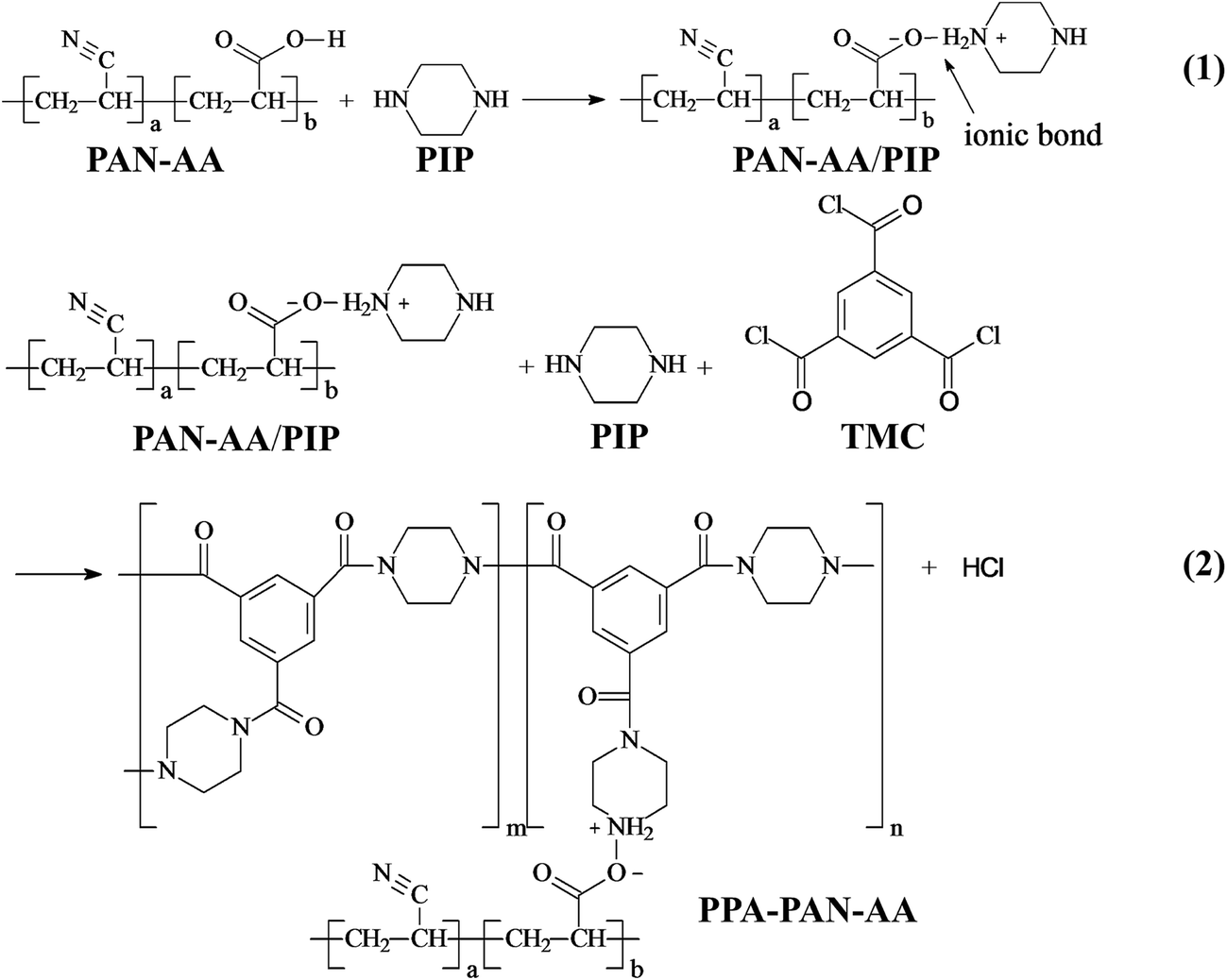
The fabrication procedure of TFNC NF membrane with a PPA barrier layer by interfacial polymerization technique onto PAN–AA/PAN double-layer nanofibrous substrate was shown in Scheme 2. For comparison, same procedure was performed for the preparation of PPA/PAN TFNC membrane with PAN single-layer nanofibrous substrate. For the process of interfacial polymerization, the aqueous phase solution was prepared by dissolving PIP (0.2 wt%), Na2CO3 (0.2 wt%) and NaOH (0.1 wt%) in deionized water, and the organic phase was prepared by dissolving TMC (0.05–0.20 wt%) into n-hexane. Firstly, the PAN or PAN–AA/PAN nanofibrous substrates were soaked and saturated in the PIP aqueous solution for 1 h to absorb aqueous monomer. Wherein, carboxyl groups of PAN–AA nanofibers on the top layer would react with PIP to form carboxyl amine intermediate (PAN–AA/PIP) as formula (1) shown in Fig. 1. Then, the substrate surface was rolled with a glass rod to remove extra aqueous solution. The wetted substrate was clamped with a four-edge Teflon holder, and the n-hexane solution of TMC was poured onto the reactive surface of the wetted substrate gently. The chemical structures of the monomers (PIP and TMC) used during the interfacial polymerization process were presented in formula (2) of Fig. 1. The amine functional groups in PAN–AA/PIP and PIP would react with acryl chloride moieties in TMC, and resulted in the amide linkages to form polypiperazine-amide (PPA). The unreacted acryl chloride groups were hydrolyzed into carboxyl groups after contacted with water.29 After a certain period of reacting time (60 s), the excess organic solution was removed from the surface and the membranes were held in an oven for 0.5 h at 60 °C so that a PPA barrier layer was formed on the nanofibrous substrate. Finally, the obtained TFNC membranes were successively washed and stored in deionized water for use, and the resultant samples were denoted as PPAy–PAN–AA/PAN and PAy/PAN TFNC membranes, where y stand for the TMC concentration.
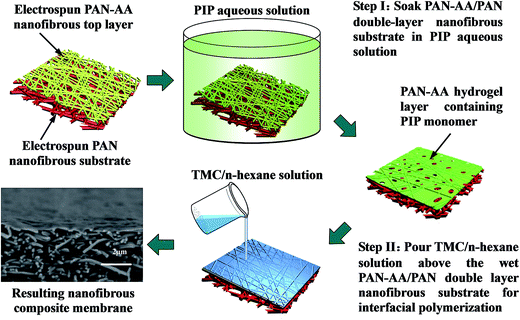 |
| | Scheme 2 Illustration for the fabrication procedure of PPA–PAN–AA/PAN nanofibrous composite membrane. | |
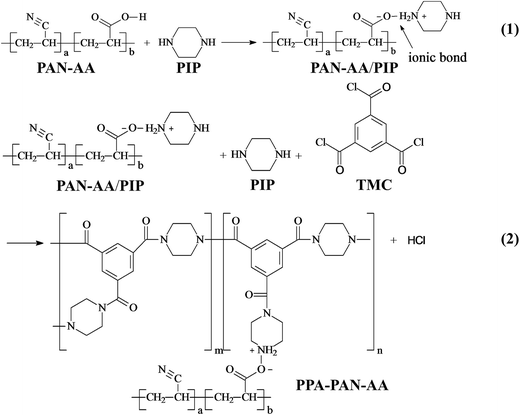 |
| | Fig. 1 Reaction (1): piperazine (PIP) and carboxyl group on PAN–AA carboxyl form carboxyl amine salt while soaking PAN–AA nanofibrous substrate in PIP aqueous solution; reaction (2): trimesoyl chloride (TMC) react with free PIP and carboxyl amine salt on PAN–AA nanofibrous substrate form polypiperazine amide during interfacial polymerization process. | |
2.5 Characterizations of the resultant membranes
The morphology of the electrospun PAN and PAN–AA nanofibers, PAN–AA/PAN double-layer substrate, PPA/PAN and PPA–PAN–AA/PAN nanofibrous composite membrane were examined by field emission scanning electron microscopy (FESEM) system (Hitachi, S4800). All specimens received 30 s of gold coating to minimize the charging effect. For cross-sectional views, all samples were prepared by fracturing the sample in liquid nitrogen. The average fiber diameter of PAN and PAN–AA nanofibers and the thickness of PPA barrier layer were determined from the FE-SEM image by using the ImageJ analysis program (http://rsb.info.nih.gov/ij/) after calibration with standards. All data were expressed as mean ± standard deviation (SD).
The chemical structures of PAN nanofibrous substrates, PAN–AA nanofibrous substrates, PAN–AA/PIP swollen mid-layer, PPA/PAN and PPA–PAN–AA/PAN composite membranes were investigated by FTIR spectroscopy using a FTIR spectrometer (Nicolet 8700, Thermo Scientific, USA) with resolution of 4 cm−1 with an OMNIC Sampler in attenuated total reflectance (ATR) mode.
The mechanical properties of PAN and PAN–AA/PAN nanofibrous substrates, PPA/PAN and PPA–PAN–AA/PAN TFNC membranes were measured by using a tensile testing machine (Model WDW3020, Changchun Kexin, China). All of measurements were carried out at room temperature. For the tensile test, the gauge length was 45 mm and the narrow width at center was 10 mm. The thickness of specimen was ∼40 μm. The chosen cross-head speed was 10 mm min−1.
The zeta potential for PPA barrier layers of PPA–PAN–AA/PA and PPA/PAN nanofibrous composite membranes were determined based on streaming potential method with an homemade electrokinetic analyzer which is customized according to the structure that described in the literature.30 The streaming potential was measured by forcing the electrolyte solutions (KCl, 0.001 mol L−1, pH = 6.0) with operation pressure at 0.010 MPa at 25 ± 0.5 °C. The resulting electrical potential difference (ΔE) as the pressure difference across the membrane (ΔP) changed was measured through a digital multimeter (UT33D, Uni-Trend, China) using reversible Ag/AgCl electrodes, which were placed on both sides of the thin slit channel. Then an equivalent zeta potential (ξ) can be deduced from these measurements according to the Helmholtz–Smoluchowski equation.
2.6 Nanofiltration performance evaluation
A custom-built flat-sheet laboratory-scale cross-flow nanofiltration (NF) system with 5 flat-sheet permeation cells in parallel (effective area in each: 24.0 cm2) was used in total recycle mode to evaluate the water permeability and salt rejection of the PPA/PAN and PPA–PAN–AA/PAN composite membranes.31 All membranes were pressured at 0.5 MPa for 30 min with pure water before the permeation tests to stabilize the membrane. After that, the water flux and salt rejection performance tests were conducted with MgSO4 aqueous solution (2.0 g L−1) under certain operating conditions (25 °C, 0.7 MPa); the water flux was determined by direct measurement of the permeate flow in terms of liter per square meter per hour (L m−2 h−1). The salt rejection rate was calculated using the following equation:| |
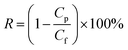 | (3) |
where Cp and Cf are the conductivity values of permeate and feed solutions, respectively, which were monitored by using a conductivity detector (FE30, Mettler Toledo, Germany).9 The NF performance of these membranes was further tested by different feed solutions (2.0 g L−1), containing sodium sulfate (Na2SO4), magnesium chloride (MgCl2), and sodium chloride (NaCl), respectively.
3. Results and discussion
3.1 Preparation of PAN and PAN–AA/PAN nanofibrous substrates
For the preparation of hydrophilic transitional layer, an ultrathin PAN–AA nanofibrous mat was electrospun on the thick PAN nanofibrous supporting substrate (the thickness is about 40 μm). Fig. 2A showed the typical SEM image of PAN nanofibrous mat, as can be seen, the average diameter of PAN nanofibers was about 350 nm. The representative SEM images of the PAN–AA electrospun morphology with different PAN–AA concentrations were shown in Fig. 2B–D. As can be seen from Fig. 2B, some beaded nanofibers appeared since the capillary breakup of the jet by surface tension from low PAN–AA concentration of 6 wt% could lead to the formation of beads during electrospinning process. With the increase of PAN–AA concentration (8 wt%), the PAN–AA nanofibers exhibited a smooth and uniform surface with average diameter of 120 nm but without any beads (Fig. 2C). When the concentration increased to 10 wt%, the average diameter of PAN–AA nanofibers increased to 250 nm (Fig. 2D). In general, the finer the PAN–AA nanofibers were, the smoother the surface of PAN–AA nanofibrous layer will be. For achieving a smoother surface, the PAN–AA solution with concentration of 8 wt% was chosen to fabricate the ultrathin PAN–AA nanofibrous transitional layer on the PAN nanofibrous support. The thickness of the PAN–AA nanofibrous top layer could be easily adjusted by the depositing time of electrospinning PAN–AA.
 |
| | Fig. 2 SEM images of electrospun PAN nanofibers (A) and electrospun PAN–AA nanofibers with different concentration (B) 6 wt%, (C) 8 wt%, and (D) 10 wt%. | |
Normally, the hydrophilic PAN–AA was readily to be swollen in basic buffer solution due to the presence of carboxyl acid groups. Therefore, PAN–AA nanofibrous layer could be swollen into a hydrogel film onto the PAN nanofibrous support when the as-prepared double layer immersed into the aqueous PIP monomer solution for a certain time, in which the pH value was higher than 10.0. It can be imagined that the swollen PAN–AA hydrogel film can not only cover and minimize the most pores of PAN nanofibrous support, but also its abundant carboxyl acid functional groups are favorable to capture and reserve adequate PIP monomers for the following interfacial polymerization process. The thickness of PAN–AA nanofibrous layer will be optimized for achieving a desirable minimum thickness of PPA barrier layer. The surface morphologies of the electrospun PAN–AA nanofibers onto PAN nanofibrous substrate with different depositing time were showed in Fig. 3A–C. It is easy to identify the PAN–AA nanofibers with PAN nanofibrous substrate. Because the average diameter of bottom PAN nanofibers is ∼350 nm, which is obviously coarser than that of top PAN–AA nanofibers (∼120 nm). It could be observed that the thickness and packing density of PAN–AA nanofibers increased with the increase of depositing time of PAN–AA nanofibers. Meanwhile, the PAN nanofibrous substrates were completely covered with the fine PAN–AA nanofibers after the electrospinning time of 30 minutes, the mean surface pore size of PAN nanofibrous substrate decreased remarkably from ∼1 μm to ∼360 nm due to the PAN–AA nanofibrous layer.
 |
| | Fig. 3 SEM images of PAN–AA nanofibers deposited on the PAN nanofibrous substrate with different depositing time (first line) and the corresponding surface morphology (second line) after immersed into the PIP aqueous solution for 1 h (A, D) 10 min, (B, E) 20 min, and (C, F) 30 min. | |
The double-layer PAN–AAx/PAN nanofibrous substrates were immersed into the PIP (0.2 wt%) aqueous solution for 1 h, which is long enough for the fully swelling of PAN–AA nanofibers to form an ultrathin hydrogel film. Fig. 3D–F showed the surface morphologies of the swollen PAN–AA layer with different thickness. It is clearly observed that the top PAN–AA nanofibrous layer was gradually swollen and imperceptibly merged into an ultrathin film on the PAN nanofibrous surface when the depositing time of the PAN–AA nanofibers was prolonged to 30 min, which covered most of the large pores on the surface of PAN nanofibrous substrate. The pure water flux of the PAN–AA30/PAN nanofibrous substrate before and after swollen in PIP solution were tested by a homemade cross-flow filtration system with an operation pressure of 0.2 MPa. Compared to the pure water flux (6496 L m−2 h−1) of the PAN–AA30/PAN substrate before swollen, the flux value after swollen was decreased to ∼3094 L m−2 h−1, which was still much higher than that of the traditional commercial ultrafiltration support for the interfacial polymerization process with cross-flow filtration at 0.2 MPa (20–30 L m−2 h−1, PAN400 UF membrane from Sepro Company, USA). Therefore, the double-layer PAN–AA30/PAN nanofibrous mats were selected as the substrate for the subsequent interfacial polymerization process for the preparation of PPA barrier layer.
3.2 Preparation and characterization of TFNC membranes based on double-layer nanofibrous substrates
The PAN–AA30/PAN double-layer nanofibrous substrate soaked in aqueous PIP solution would form a requisite surface to support the interfacial polymerization process for the fabrication of PPA barrier layer. Fig. 4 showed the typical surface and cross-section SEM images of the PPA–PAN–AA/PAN composite membrane prepared by the reaction between the swollen PAN–AA/PAN substrates with abundant PIP monomer and the TMC organic solution with different concentration (0.025–0.20 wt%). As can be seen, the surface morphology of the nanofibrous substrates were clearly observed from the top views, the nanofibers were partially embedded in the top barrier layers, which indicated a tight combination of the thin barrier layer and the support. And the formed PPA barrier layers impregnated into the PAN–AA transitional layers were completely covered onto the bottom PAN nanofibrous substrate. Meanwhile, with the increase of TMC concentration, the average thickness of the top barrier layer was increased from ∼120 nm to ∼270 nm (roughly labeled in right column of Fig. 4), which include the thickness of swollen PAN–AA layer, and the surface roughness were gradually increased as shown left column of Fig. 4.
 |
| | Fig. 4 SEM images of surface (left column) and cross-section (right column) images of the PPA–PAN–AA/PAN TFNC membranes prepared by interfacial polymerization between PIP (0.2 wt%) and TMC with different concentration (A, B) 0.025 wt%, (C, D) 0.05 wt%, (E, F) 0.10 wt% and (G, H) 0.20 wt% (reacting time 60 s, based on PAN–AA30 nanofibrous substrate soaked in 0.2 wt% PIP aqueous solution for 1 h). | |
The average thickness of the top PPA layers of the PPA/PAN TFNC membranes (interfacial polymerization directly based on PAN nanofibrous substrate without PAN–AA transitional layer) ranged from ∼160 nm to ∼200 nm when the TMC concentration changed from 0.05 wt% to 0.20 wt%, as shown in Fig. 5, which was thinner than that of PPA–PAN–AA/PAN TFNC membrane (prepared by the interfacial polymerization based on the swollen PAN–AA30/PAN substrate with the same TMC concentration, i.e., 0.05 wt%: ∼190 nm, 0.20 wt%: ∼270 nm) due to the swollen PAN–AA ultrathin film was imbedded in PPA barrier layer. But the filtration performance of the PPA/PAN TFNC membranes was much worse than that of the PPA–PAN–AA/PAN TFNC membranes since the swollen PAN–AA ultrathin film could provide abundant water channels for the permeate flow,24 which will be discussed in the following section. Meanwhile, there existed obvious wrinkles on the surface of PPA barrier layer in PPA/PAN TFNC membranes, which might be caused by the shrinkage of PPA layer during the thermal treatment, as shown in Fig. 5A and C. In contrast, much smoother surface of PPA barrier layer on PAN–AA/PAN TFNC membranes were obtained, which was attributed to the swollen PAN–AA ultrathin film could cover most of the surface pores of PAN nanofibrous mat and then decreased the surface roughness that derived from the intrinsic PAN nanofibrous structures.
 |
| | Fig. 5 SEM images of surface (left column) and cross-section (right column) images of the PPA/PAN nanofiltration composite membrane prepared by interfacial polymerization between PIP (0.2 wt%) and TMC with different concentration (A, B) 0.05 wt% and (C, D) 0.20 wt% within 60 s based on PAN nanofibrous substrate. | |
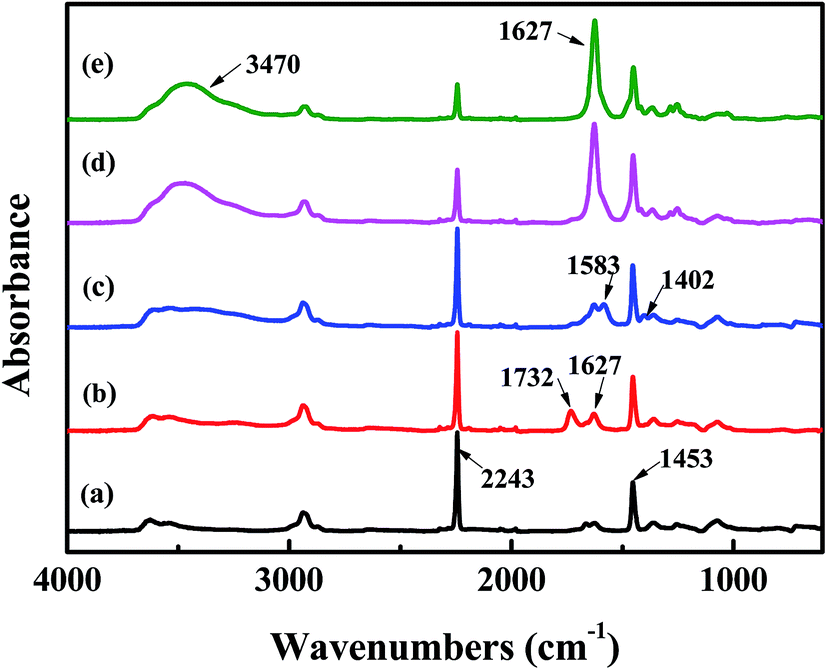
The surface chemical composition of each layer related to TFNC membranes was provided by FTIR spectroscopy, i.e., PAN nanofibrous substrate, PAN–AA nanofibrous mid-layer, PAN–AA/PIP swollen layer (PAN–AA nanofibrous mid-layer swollen in the PIP aqueous solution), PPA barrier layer of the PPA–PAN–AA/PAN and the PPA/PAN TFNC membranes (Fig. 6). As shown in Fig. 6a, the characteristic absorption peak at 2243 cm−1 and 1732 cm−1 were attributed to cyan groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) in PAN and methylene groups mainly come from the initiator remained in terminal group of PAN. In Fig. 6b, those characteristic absorption peaks appeared at 1627 cm−1 and 1732 cm−1 corresponded to the vibrations of carbonyl in the non-ionized carboxyl group from the PAN–AA nanofibrous middle layer. After swollen in the PIP solution, the carboxyl acid groups of PAN–AA nanofibers would dissociate into carboxylate ions in this alkaline surrounding, these carboxylate ions from PAN–AA nanofibers would react with the secondary amino group of PIP and form the carboxyl amine salt (–COO−NH2+) as formula (1) in Fig. 1. Thus, a characteristic absorption peak appeared at 1583 cm−1 as shown in Fig. 6c, which could be attributed to the ionic bond of carboxyl amine salt. Meanwhile, a peak at 1402 cm−1 related to the antisymmetric vibrations of the carboxylate anion could also prove the existence of the carboxyl amine salt.32,33 As can be seen from Fig. 6d and e, a new adsorption peak at 1627 cm−1 and a broad peak at about 3470 cm−1 were assigned to the amide link (OC–N) and the stretching vibrations of amino group (N–H) from the formed PPA top layer of PPA–PAN–AA/PAN and PPA/PAN TFNC membranes.27 It could be seen that there is no significant difference between the FTIR spectra of PPA–PAN–AA/PAN TFNC membrane and that of PPA/PAN TFNC membrane as the surface PPA functional layer was prepared by the same interfacial polymerization process as formula (2) in Fig. 1.
N) in PAN and methylene groups mainly come from the initiator remained in terminal group of PAN. In Fig. 6b, those characteristic absorption peaks appeared at 1627 cm−1 and 1732 cm−1 corresponded to the vibrations of carbonyl in the non-ionized carboxyl group from the PAN–AA nanofibrous middle layer. After swollen in the PIP solution, the carboxyl acid groups of PAN–AA nanofibers would dissociate into carboxylate ions in this alkaline surrounding, these carboxylate ions from PAN–AA nanofibers would react with the secondary amino group of PIP and form the carboxyl amine salt (–COO−NH2+) as formula (1) in Fig. 1. Thus, a characteristic absorption peak appeared at 1583 cm−1 as shown in Fig. 6c, which could be attributed to the ionic bond of carboxyl amine salt. Meanwhile, a peak at 1402 cm−1 related to the antisymmetric vibrations of the carboxylate anion could also prove the existence of the carboxyl amine salt.32,33 As can be seen from Fig. 6d and e, a new adsorption peak at 1627 cm−1 and a broad peak at about 3470 cm−1 were assigned to the amide link (OC–N) and the stretching vibrations of amino group (N–H) from the formed PPA top layer of PPA–PAN–AA/PAN and PPA/PAN TFNC membranes.27 It could be seen that there is no significant difference between the FTIR spectra of PPA–PAN–AA/PAN TFNC membrane and that of PPA/PAN TFNC membrane as the surface PPA functional layer was prepared by the same interfacial polymerization process as formula (2) in Fig. 1.
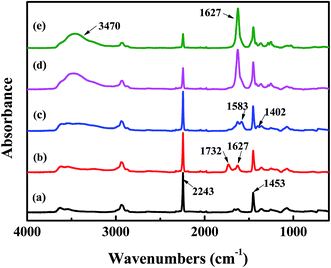 |
| | Fig. 6 FTIR spectra of (a) PAN nanofibrous substrates, (b) PAN–AA nanofibrous layer, (c) PAN–AA/PIP swollen layer, (d) PPA–PAN–AA/PAN TFNC membrane and (e) PPA/PAN TFNC membrane. | |
3.3 Mechanical properties of nanofibrous substrates and TFNC membranes
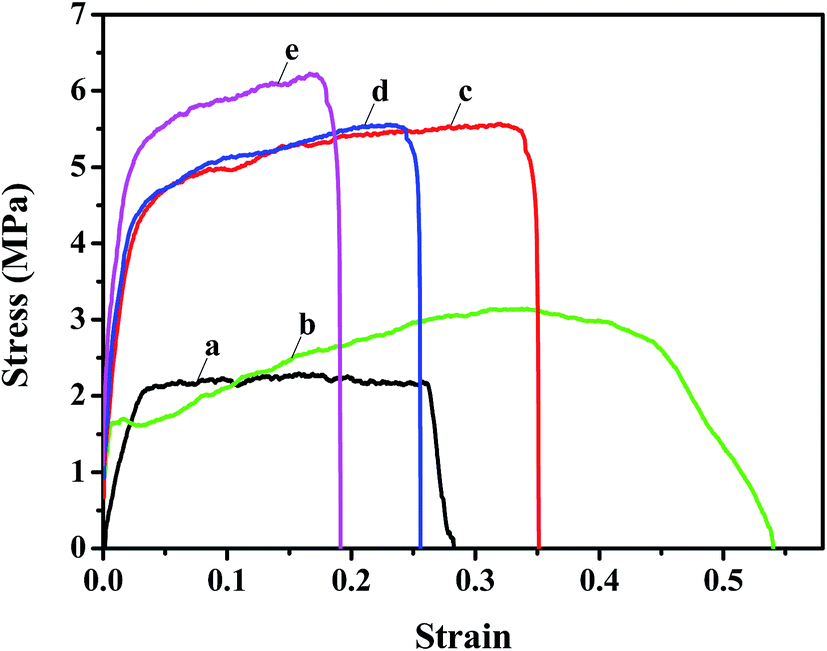
The mechanical properties of the nanofibrous substrates and the corresponding TFNC membranes were investigated by tensile test. The strain–stress curves clearly showed that the mechanical properties of PAN nanofibrous mats (Fig. 7a) were remarkably enhanced from 2.2 MPa to 5.6 MPa after depositing the PAN–AA nanofibers for 30 minutes (Fig. 7c). By immersing into the PIP aqueous solution for 1 h, the PAN–AA nanofibrous layer was swollen into a piece of PAN–AA microporous hydrogel film, which would adhere to the bottom PAN nanofibrous substrate and enhance the structural integrity of the PAN–AA/PAN double-layer substrate. Thus, the swollen PAN–AA30/PAN substrates could keep its high tensile strength (5.6 MPa) but with a lower elongation at break as shown in Fig. 7d, which might be attributed to the generation of an ionic bond of carboxyl amine salt (–COO−NH2+) between the carboxyl groups of PAN–AA nanofibers and the secondary amine groups of PIP. As a reactive substrate, the PAN–AA mid-layer could capture much more amine monomer through ionic bonds on the surface of the swollen PAN–AA/PAN substrates than that of the PAN single-layer substrate. Additionally, the number of amine monomer involved in interfacial polymerization will be increased and a denser PPA barrier layer with fewer defects can be prepared. Meanwhile, the ionic bond cross-linkage between the PPA barrier layer and the PAN–AA/PAN substrate could enhance the interfacial compatibility and structural integrity of the TFNC membranes.25 Therefore, the structurally integrated PPA–PAN–AA/PAN TFNC membrane based on the PAN–AA/PAN double-layer substrate exhibited relatively higher tensile breaking strength of 6.2 MPa (Fig. 7e) than that of the PPA/PAN TFNC membrane based on the PAN single-layer substrate (3.1 MPa, Fig. 7b).
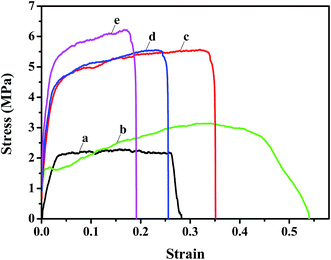 |
| | Fig. 7 Stress–strain curve of the TFNC membranes and the nanofibrous basic membranes (a) PAN nanofibrous substrate, (b) PPA/PAN TFNC membrane, (c) PAN–AA30/PAN nanofibrous substrate, (d) PAN–AA30/PAN swollen substrate, and (e) PPA–PAN–AA/PAN TFNC membrane. | |
3.4 Nanofiltration performance of TFNC membranes
The separation performance of nanofiltration (NF) membranes could be influenced by several variables in the process of interfacial polymerization. To demonstrate the advantage of using an electrospun double-layer PAN–AA/PAN nanofibrous substrate over a single-layer PAN nanofibrous substrate, the same interfacial polymerization conditions (i.e. 0.2 wt% PIP aqueous solution, interfacial polymerization reacted for 60 s, heat treatment at 60 °C for 0.5 h) were used to fabricate the PPA barrier layer. In addition, the concentration of TMC in hexane could control the average thickness of PPA barrier layer as shown in Fig. 4 and 5. The nanofiltration performance of the resultant TFNC membranes was evaluated in terms of permeate flux and salt rejection by using MgSO4 feed solution (2 g L−1) under the cross-flow filtration at 0.7 MPa.
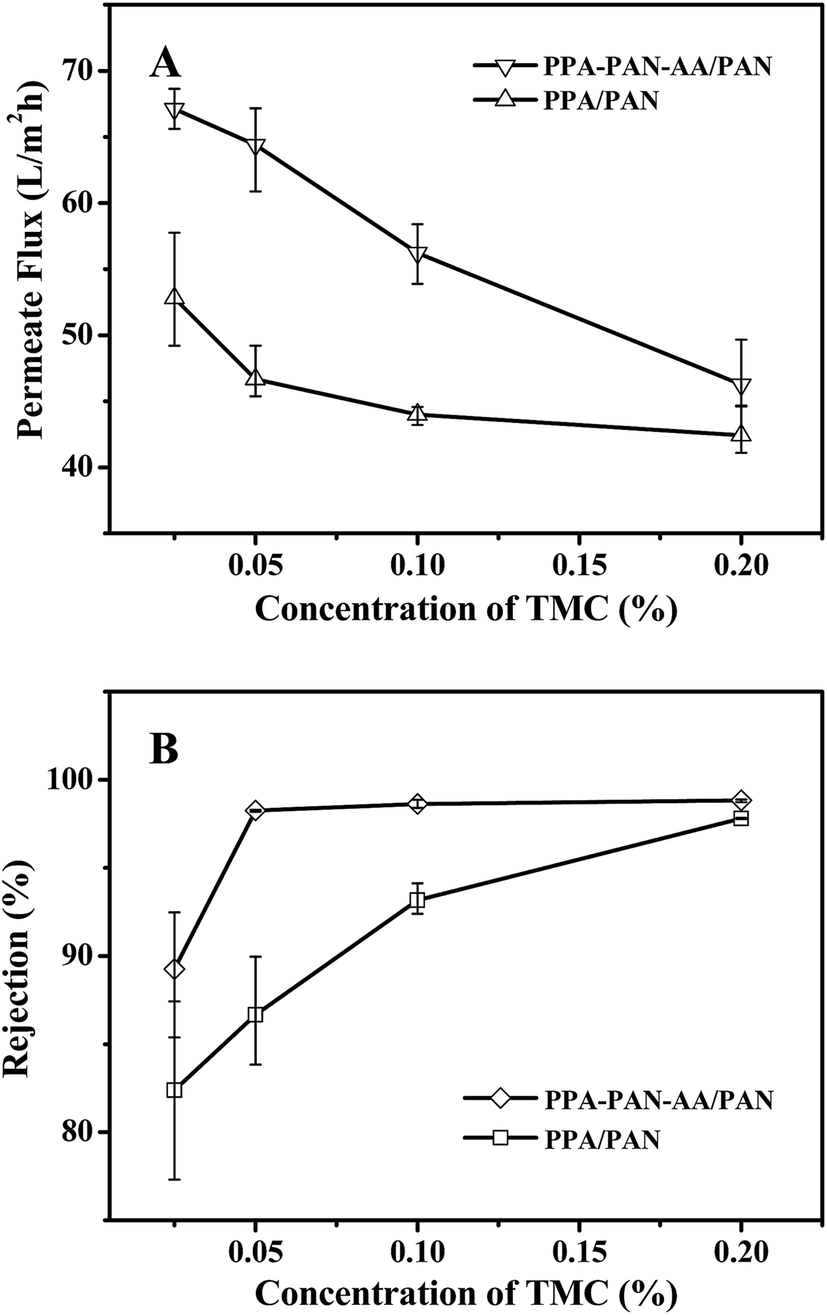
When the PIP concentration was fixed at 0.2 wt%, the NF performance exhibited a systematic change with the variation of TMC concentration in hexane (i.e. the rejection increased while the permeate flux decreased with the increment of TMC concentration from 0.025 to 0.20 wt%). As shown in Fig. 8, there was obvious difference in filtration performance of the TFNC membranes based on PAN–AA/PAN and PAN nanofibrous substrates. The rejection ratio for MgSO4 of the PPA/PAN TFNC membrane was much lower than that of PPA–PAN–AA/PAN TFNC membrane until the TMC concentration was increased to 0.20 wt%, and the rejection ratio of PPA/PAN TFNC membrane could only reach to 97.8%. However, the rejection ratio of PPA–PAN–AA/PAN TFNC membrane could maintain at around 98.7% when the TMC concentration was higher than 0.05 wt%. The different rejections against MgSO4 of the TFNC membranes based on PAN and PAN–AA nanofibrous substrates could be attributed to the compactness of PPA barrier layer and the membrane surface charge density.24 The PIP content enriched within PAN–AA hydrogel layer should be higher than the PIP content deposited on the PAN nanofiber surface, which would influence the structural and filtration performance of the resultant PPA barrier layer. On the other hand, the PPA/PAN TFNC membrane was negatively charged (zeta potential: −1.3 mV) as the PPA barrier layer possessed negative charged carboxylic groups, which could be hydrolyzed from the excess acyl chloride group on TMC during the interfacial polymerization process.17 The PPA–PAN–AA/PAN TFNC membranes (zeta potential: −4.5 mV) could provide more negative charged carboxylic groups by introducing the PAN–AA nanofibers on PAN nanofibrous mats. Although some of carboxylic groups reacted with the secondary amino groups after swollen in PIP solution, the PAN–AA mid-layer could still contain some residual carboxyl groups, which has been confirmed by the FTIR analysis as shown in Fig. 6. The higher negative charge density on the membrane surface can strongly influence the ion–exchange equilibrium and impede the passage of anions in the feed solution according to the Donan ion repulsion effect. Thus, the PPA–PAN–AA/PAN TFNC membrane could obtain better rejection than the PPA/PAN TFNC membrane. Furthermore, the permeate flux of PPA/PAN TFNC membrane was also lower than that of PPA–PAN–AA/PAN TFNC membrane. In details, when the TMC concentration increased from 0.025 wt% to 0.20 wt%, the permeate flux of PPA–PAN–AA/PAN TFNC membrane decreased from 67.1 L m−2 h−1 to 46.3 L m−2 h−1, while that of PPA/PAN TFNC membranes decreased from 52.8 L m−2 h−1 to 42.4 L m−2 h−1. The relatively higher flux of PPA–PAN–AA/PAN TFNC membrane may be attributed to that the formed PPA barrier layer was impregnated into the PAN–AA transitional hydrogel layer which could be acted as the water channels in PPA barrier layer.34
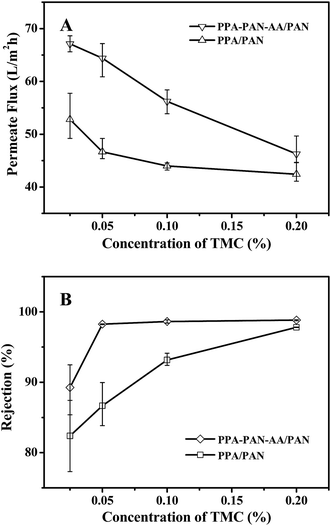 |
| | Fig. 8 Effect of TMC concentration on permeate flux (A) and rejection rate (B) for the TFNC membranes prepared by interfacial polymerization based on different nanofibrous substrates (feed with 2 g L−1 MgSO4 aqueous solution). | |
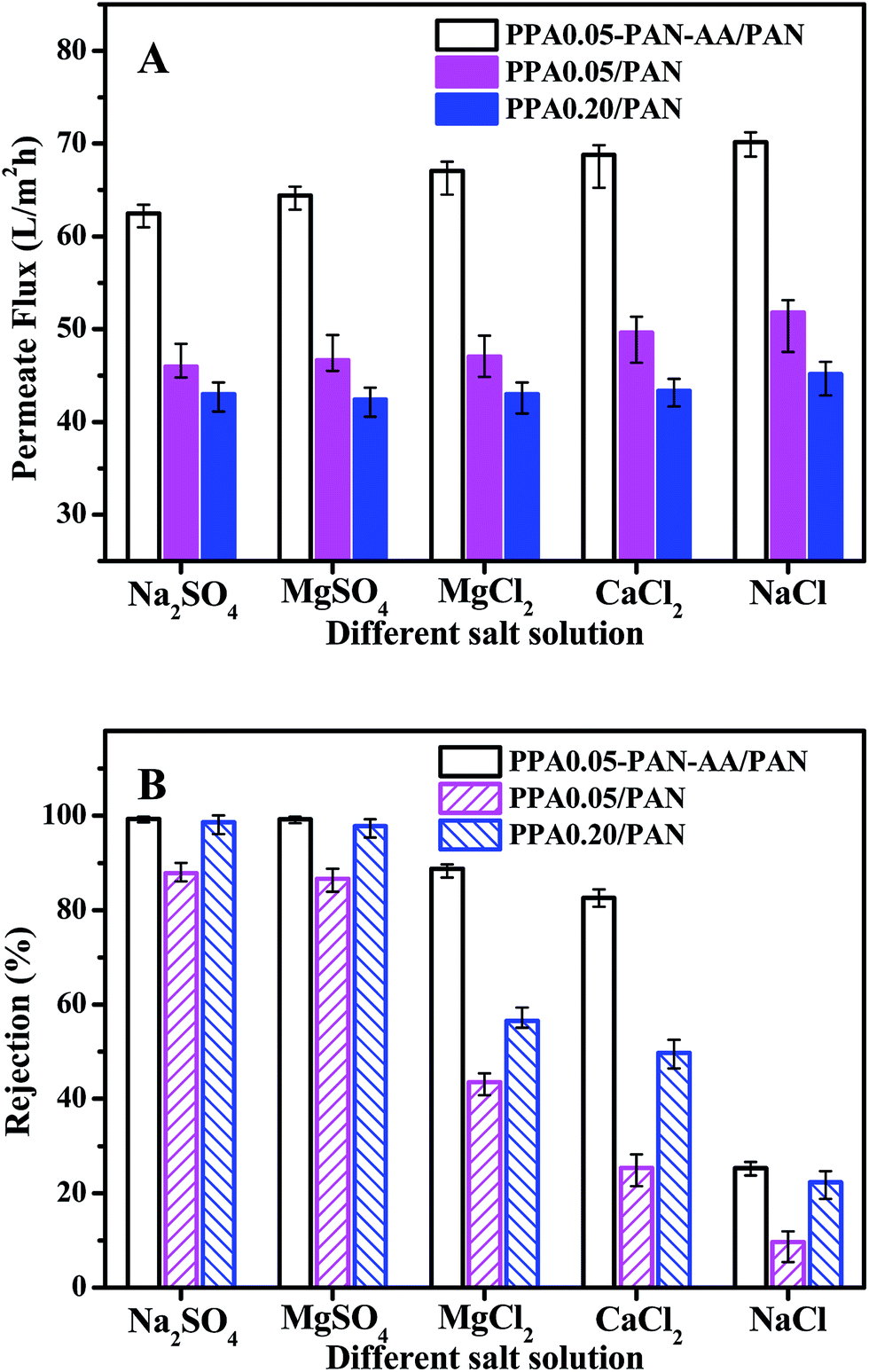
To further evaluate the nanofiltration performance of PPA–PAN–AA/PAN TFNC membranes, several kinds of salt aqueous solutions were used as the feed solutions for the cross-flow filtration to measure the nanofiltration property of PPA0.05–PAN–AA/PAN, PPA0.05/PAN and PPA0.20/PAN TFNC membranes with different divalent cations and anions. As shown in Fig. 9, in the case of the solute containing monovalent cation (Na+) and divalent anion (i.e. sulfate ions, SO42−), both the PPA0.05–PAN–AA/PAN and PPA0.20/PAN TFNC membrane exhibited a high rejection rate (>98.5%) for sodium sulfate (Na2SO4) and kept almost the same permeate flux as values for the filtration of MgSO4 solution. While, with respect to the solute with a constitution of divalent cation and monovalent anion, the PPA0.05–PAN–AA/PAN TFNC membrane showed much better rejection rate for MgCl2 (88.8%) and CaCl2 (82.6%) compared to those of the PPA0.05/PAN and PPA0.20/PAN TFNC membranes (the rejection for MgCl2 of PPA0.05/PA and PPA0.20/PAN was 43.6% and 56.5%, respectively, and the value for CaCl2 of PPA0.05/PA and PPA0.20/PAN was 23.4% and 49.8%, respectively). Even for the NaCl solution, the PPA0.05–PAN–AA/PA TFNC membrane still possessed a rejection of 25.3%. Consequently, the rejections for divalent anions were obviously higher than that of monovalent anions, and the cations with bigger atomic number exhibited lower rejection ratio (i.e. the rejection order of the PPA0.05–PAN–AA/PA TFNC membranes for different kinds of salt solution was as following: Na2SO4 ≈ MgSO4 > MgCl2 > CaCl2 > NaCl), which is the typical characteristics of the negatively charged membranes, further confirming the surface property that derived from the zeta potential. Meanwhile, the permeate flux of PPA0.05–PAN–AA/PAN TFNC membranes was 35.4% to 58.2% higher than that of PPA/PAN TFNC membranes by replacing the PAN single-layer nanofibrous substrate with PAN–AA/PAN double-layer nanofibrous substrate. Additionally, the nanofiltration properties of the PPA0.05–PAN–AA/PAN TFNC membrane were also very comparable with those data from other membranes prepared by interfacial polymerization method reported in literatures (as shown in Table 1).35–38
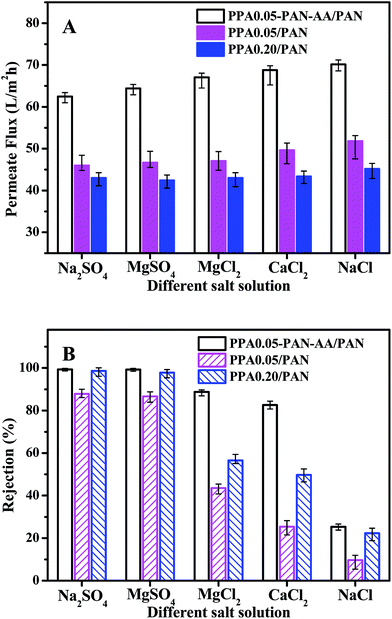 |
| | Fig. 9 The permeate flux (A) and rejection rate (B) for different salt solutions with TFNC membranes prepared by interfacial polymerization based on different nanofibrous substrates (operating pressure at 0.7 MPa, the concentration of all feed solutions are 2 g L−1). | |
Table 1 Permeate flux and salt rejection ratio for the different types of NF membranes
| Barrier layer/substrate |
Salt solution |
Applied pressure (MPa) |
Flux (L m−2 h−1 bar) |
Rejection (%) |
| NF270 |
2000 ppm MgSO4 |
0.7 |
8.07 |
97.2 |
| PA–PEI/PS35 |
75 ppm MgSO4 |
0.8 |
4.38 |
85.1 |
| PA–PEI/PS35 |
75 ppm MgCl2 |
0.8 |
4.75 |
93.2 |
| PVAm–TMC/PS36 |
500 ppm Na2SO4 |
0.6 |
8.53 |
94.8 |
| PEI–TMC/PES37 |
500 ppm MgSO4 |
0.8 |
3.06 |
94.4 |
| PA–MWCNT/PSF38 |
2000 ppm Na2SO4 |
0.6 |
5.3 |
96.6 |
| PA–MWCNT/PSF38 |
2000 ppm MgSO4 |
0.6 |
6.9 |
93.7 |
| PPA0.05/PAN (this work) |
2000 ppm MgSO4 |
0.7 |
6.24 |
86.7 |
| PPA0.20/PAN (this work) |
2000 ppm MgSO4 |
0.7 |
6.58 |
97.8 |
| PPA0.05–PAN–AA/PAN (this work) |
2000 ppm Na2SO4 |
0.7 |
9.18 |
99.3 |
| PPA0.05–PAN–AA/PAN (this work) |
2000 ppm MgSO4 |
0.7 |
9.31 |
98.7 |
3.5 Stabilities of TFNC membranes
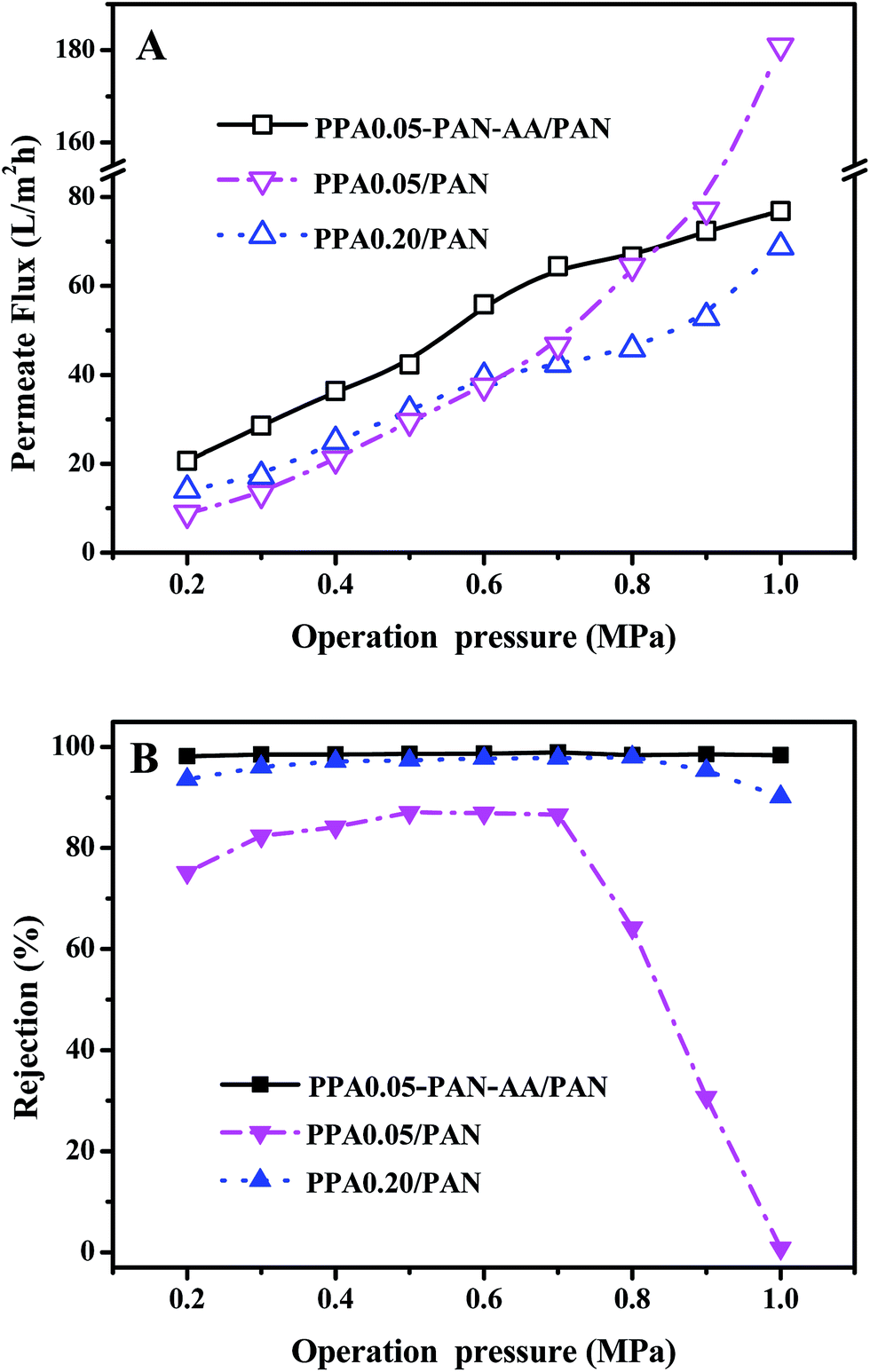
The stability of TFNC membranes is very important for the practical application. The nanofiltration performances of TFNC membranes at different operation pressure were tested to evaluate the stability of the TFNC membranes. As shown in Fig. 10, three kinds of typical TFNC membranes, i.e., PPA0.05–PAN–AA/PAN, PPA0.05/PAN and PPA0.20/PAN TFNC membranes were used in this test. The permeate flux of these TFNC membranes were increased synchronously with the increment of the operation pressure from 0.2 to 1.0 MPa (Fig. 10A). When the operation pressure was higher than 0.7 MPa, the permeate flux of TFNC membranes based on PAN nanofibrous substrates increased much faster than that of PPA0.05–PAN–AA/PAN TFNC membrane, but the rejection of PPA0.05/PAN TFNC membrane decreased sharply from 86.6 to 0.0% due to the breakage of PPA barrier layer, and the rejection of PPA0.20/PAN TFNC membrane was gradually decreased when the applied pressure was over 0.8 MPa. Under the same operating pressure, the rejection of PPA0.05–PAN–AA/PAN TFNC membrane could still maintain a high value more than 98.0% and the permeate flux increased to 76.8 L m−2 h−1 at 1.0 MPa, which was ascribed to the PAN–AA intermediate layer that swollen to hydrogel film in the PIP aqueous solution, and then reduced the surface pore size of PAN nanofibrous substrate and increased the stressed area to provide enough strength for supporting the ultra-thin PPA barrier layer. Furthermore, the PAN–AA swollen layer could also form a loose structure to provide abundant water channels for the permeate flow in the PPA barrier layer.
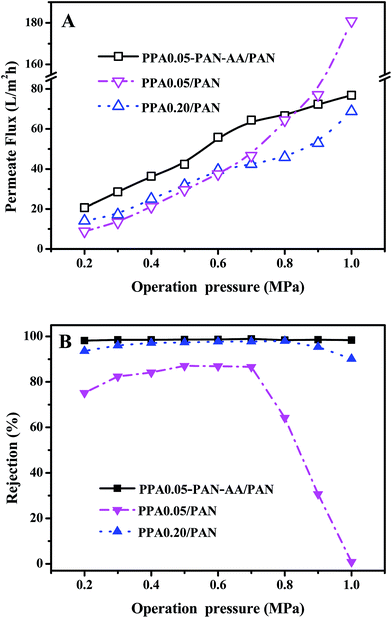 |
| | Fig. 10 Effect of the operating pressure on (A) permeate flux and (B) rejection for the TFNC membranes prepared by interfacial polymerization with different TMC concentration based on different nanofibrous substrates (feed with 2 g L−1 MgSO4 aqueous solution). | |
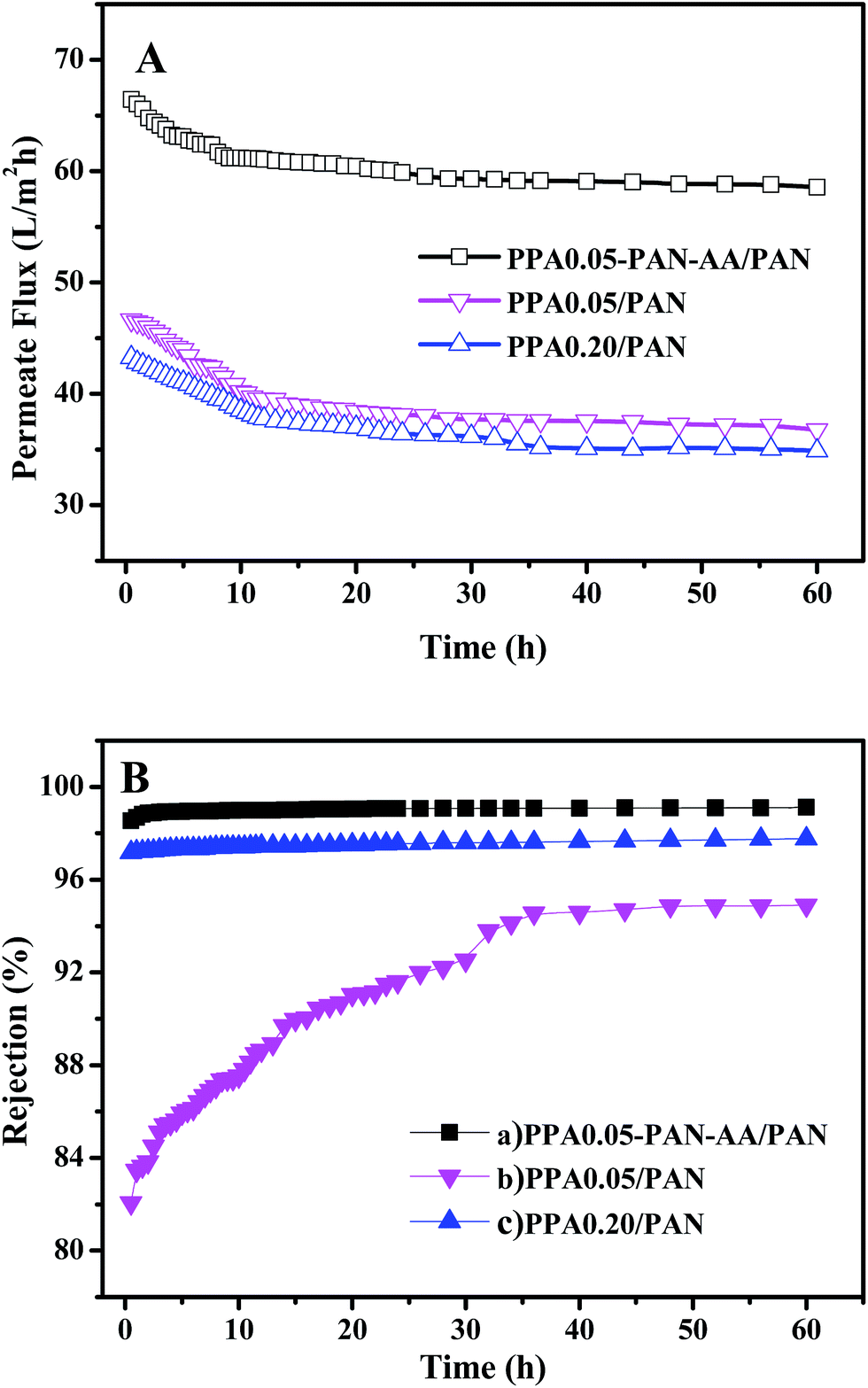
A long period filtration test of these TFNC membranes were also performed for 60 h with MgSO4 aqueous solution (2 g L−1) at a limit operation pressure of 0.7 MPa, which could evaluate the anti-fouling property and the long-term stability of these membranes. The permeate fluid was collected after every one hour and the results were shown in Fig. 11. After operating at 0.7 MPa for 2 hours, the filtration performance of PPA0.05–PAN–AA/PAN TFNC membrane became more stable, which could provide higher average permeate flux (60.9 L m−2 h−1) and higher average rejection for MgSO4 (99.0%). Meanwhile, the average rejection of PPA0.05/PAN TFNC membrane was 89.7%, and the permeate flux was 40.0 L m−2 h−1. The PPA0.20/PAN TFNC membrane could obtain an approximate rejection for MgSO4 (∼97.5%) but provide much lower permeate flux (37.9 L m−2 h−1). After 60 h operation, the rejection of PPA0.05–PAN–AA/PAN TFNC membrane maintained the excellent rejection ratio above 98.0%, and that of PPA0.20/PAN TFNC membrane was still under 98.0%, but the rejection of PPA0.05/PAN TFNC membranes increased from 84.5 to 94.9% as the PPA0.05/PAN TFNC membrane was gradually compacted by water pressure. On the other hand, the permeate flux of PPA0.05–PAN–AA/PAN TFNC membrane decreased from 64.4 to 58.6 L m−2 h−1, while that of PPA0.05/PAN and PPA0.20/PAN membranes decreased from 45.6 to 36.7 L m−2 h−1 and from 42.1 to 34.9 L m−2 h−1, respectively. This result revealed that the PPA0.05–PAN–AA/PAN TFNC membrane showed excellent anti-fouling property and long-term stability throughout the test process, even the permeate flux existed a slight fluctuation (less than 9.0%), which was much better than that of the PPA0.05/PAN TFNC membrane (permeate flux decreased 19.4%) and the PPA0.20/PAN TFNC membranes (permeate flux decreased 17.2%). Compared with the PPA0.05/PAN TFNC membrane, not only the average rejection of the PPA0.05–PAN–AA/PAN TFNC membrane enhanced more than 9.4%, but also the average permeate flux was remarkably improved by 34.4%. Therefore, the filtration performance of the PPA0.05/PAN TFNC membrane was strongly enhanced by introducing the PAN–AA intermediate layer under the same interfacial polymerization conditions (PIP 1.0%, TMC 0.05 wt%, 60 s reacting time, heat treatment at 60 °C for 0.5 h) and the same filtration process (cross-flow filtration, 2 g L−1 MgSO4 solution, operating pressure at 0.7 MPa).
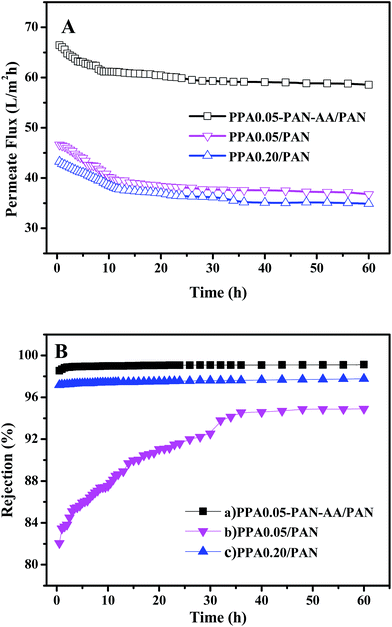 |
| | Fig. 11 Effect of long period operation on (A) permeate flux and (B) rejection for the TFNC membranes prepared by interfacial polymerization with different TMC concentration based on different nanofibrous substrates (operating pressure at 0.7 MPa, feed with 2 g L−1 MgSO4 aqueous solution). | |
4. Conclusion
The TFNC nanofiltration membranes were prepared by interfacial polymerization based on PAN single nanofibrous layer or PAN–AA/PAN double-layer nanofibrous substrate. The structural integrity and mechanical properties of the PAN–AA/PAN nanofibrous substrates and the resultant PPA–PAN–AA/PAN TFNC membranes were significantly enhanced due to the introduction of transitional PAN–AA nanofibrous mid-layer that increased the interconnection between PPA barrier layer and PAN nanofibrous substrate. The PAN–AA nanofibrous layer could be swollen into an ultrathin hydrogel film which would cover most of the surface pores of PAN nanofibrous support but with a high permeability. The PAN–AA hydrogel transitional mid-layer could capture much more aqueous monomers (PIP) which would be beneficial to the stability of PPA barrier layer. Consequently, the well-designed PPA0.05–PAN–AA/PAN TFNC membranes showed much better nanofiltration performance (i.e. the permeate flux was 64.4 L m−2 h−1 and the rejection rate was 98.2%) than that of PPA0.05/PAN TFNC membranes for MgSO4 (2.0 g L−1) solution under 0.7 MPa. Meanwhile, the rejection order of the PPA0.05–PAN–AA/PA TFNC membranes for different kinds of salt solution was Na2SO4 ≈ MgSO4 > MgCl2 > CaCl2 > NaCl. Furthermore, the PPA0.5–PAN–AA/PAN TFNC membrane exhibited excellent anti-fouling property and long-term stability after a test period of 60 h (the permeate flux decreased less than 9.0% and the rejection kept above 98%). Therefore, it may lead to a further improvement of the composite membrane performance by introducing an ultrathin intermediate layer between the functional barrier layer and the porous basic support.
Acknowledgements
This work was supported by National Natural Science Foundation of China (51273042, 21174028), Program for New Century Excellent Talents in University (NCET-13-0725) and Program of Shanghai Science and Technology Innovation International Exchange and Cooperation (15230724700).
References
- N. Misdan, W. J. Lau, C. S. Ong, A. F. Ismail and T. Matsuura, Study on the thin film composite poly(piperazine-amide) nanofiltration membranes made of different polymeric substrates: effect of operating conditions, Korean J. Chem. Eng., 2015, 32, 753–760 CrossRef CAS.
- W. J. Lau, A. F. Ismail, P. S. Goh, N. Hilal and B. S. Ooi, Characterization methods of thin film composite nanofiltration membranes, Sep. Purif. Rev., 2015, 44, 135–156 CrossRef CAS.
- L. Y. Jiang, Y. Wang, T.-S. Chung, X. Y. Qiao and J.-Y. Lai, Polyimides membranes for pervaporation and biofuels separation, Prog. Polym. Sci., 2009, 34, 1135–1160 CrossRef CAS.
- H. Guo, Y. Ma, P. Sun, S. Cui, Z. Qin and Y. Liang, Self-cleaning and antifouling nanofiltration membranes – superhydrophilic multilayered polyelectrolyte/CSH composite films towards rejection of dyes, RSC Adv., 2015, 5, 63429–63438 RSC.
- J. R. Kovacs, C. Liu and P. T. Hammond, Spray layer-by-layer assembled clay composite thin films as selective layers in reverse osmosis membranes, ACS Appl. Mater. Interfaces, 2015, 7, 13375–13383 CAS.
- S. Zhu, S. Zhao, Z. Wang, X. Tian, M. Shi, J. Wang and S. Wang, Improved performance of polyamide thin-film composite nanofiltration membrane by using polyethersulfone/polyaniline membrane as the substrate, J. Membr. Sci., 2015, 493, 263–274 CrossRef CAS.
- L. Shen, Y. Yang, J. Zhao and X. Wang, High-performance nanofiltration membrane prepared by dopamine-assisted interfacial polymerization on PES nanofibrous scaffolds, Desalin. Water Treat., 2016, 57, 9549–9557 CrossRef CAS.
- L. Yung, H. Ma, X. Wang, K. Yoon, R. Wang, B. S. Hsiao and B. Chu, Fabrication of thin-film nanofibrous composite membranes by interfacial polymerization using ionic liquids as additives, J. Membr. Sci., 2010, 365, 52–58 CrossRef CAS.
- K. Yoon, B. S. Hsiao and B. Chu, High flux nanofiltration membranes based on interfacially polymerized polyamide barrier layer on polyacrylonitrile nanofibrous scaffolds, J. Membr. Sci., 2009, 326, 484–492 CrossRef CAS.
- L. A. Hoover, J. D. Schiffman and M. Elimelech, Nanofibers in thin-film composite membrane support layers: enabling expanded application of forward and pressure retarded osmosis, Desalination, 2013, 308, 73–81 CrossRef CAS.
- Y. Liu, M. Park, B. Ding, J. Kim, M. El-Newehy, S. S. Al-Deyab and H. Y. Kim, Facile electrospun polyacrylonitrile/poly(acrylic acid) nanofibrous membranes for high efficiency particulate air filtration, Fibers Polym., 2015, 16, 629–633 CrossRef CAS.
- H. You, X. Li, Y. Yang, B. Wang, Z. Li, X. Wang, M. Zhu and B. S. Hsiao, High flux low pressure thin film nanocomposite ultrafiltration membranes based on nanofibrous substrates, Sep. Purif. Technol., 2013, 108, 143–151 CrossRef CAS.
- Z. Wang, H. Ma, B. S. Hsiao and B. Chu, Nanofibrous ultrafiltration membranes containing cross-linked poly(ethylene glycol) and cellulose nanofiber composite barrier layer, Polymer, 2014, 55, 366–372 CrossRef CAS.
- L. Shen, X. Li, Y. Yang and X. Wang, High flux PEO doped chitosan ultrafiltration composite membrane based on PAN nanofibrous substrate, Mater. Res. Innovations, 2014, 18, S4808–S4811 CrossRef.
- J. Zhao, Y. Su, X. He, X. Zhao, Y. Li, R. Zhang and Z. Jiang, Dopamine composite nanofiltration membranes prepared by self-polymerization and interfacial polymerization, J. Membr. Sci., 2014, 465, 41–48 CrossRef CAS.
- S. J. Park, R. K. Cheedrala, M. S. Diallo, C. Kim, I. S. Kim and W. A. Goddard, Nanofiltration membranes based on polyvinylidene fluoride nanofibrous scaffolds and crosslinked polyethyleneimine
networks, J. Nanopart. Res., 2012, 14, 884–898 CrossRef.
- X. Wang, T. M. Yeh, Z. Wang, R. Yang, R. Wang, H. Ma, B. S. Hsiao and B. Chu, Nanofiltration membranes prepared by interfacial polymerization on thin-film nanofibrous composite scaffold, Polymer, 2014, 55, 1358–1366 CrossRef CAS.
- H. Ma, C. Burger, B. S. Hsiao and B. Chu, Ultra-fine cellulose nanofibers: new nano-scale materials for water purification, J. Mater. Chem., 2011, 21, 7507–7510 RSC.
- Y. Liu, R. Wang, H. Ma, B. S. Hsiao and B. Chu, High-flux microfiltration filters based on electrospun polyvinylalcohol nanofibrous membranes, Polymer, 2013, 54, 548–556 CrossRef CAS.
- Z. Zhao, J. Zheng, M. Wang, H. Zhang and C. C. Han, High performance ultrafiltration membrane based on modified chitosan coating and electrospun nanofibrous PVDF scaffolds, J. Membr. Sci., 2012, 394–395, 209–217 CrossRef CAS.
- K. Yoon, K. Kim, X. Wang, D. Fang, B. S. Hsiao and B. Chu, High flux ultrafiltration membranes based on electrospun nanofibrous PAN scaffolds and chitosan coating, Polymer, 2006, 47, 2434–2441 CrossRef CAS.
- S. Kaur, R. Barhate, S. Sundarrajan, T. Matsuura and S. Ramakrishna, Hot pressing of electrospun membrane composite and its influence on separation performance on thin film composite nanofiltration membrane, Desalination, 2011, 279, 201–209 CrossRef CAS.
- H. Ma, C. Burger, B. S. Hsiao and B. Chu, Fabrication and characterization of cellulose nanofiber based thin-film nanofibrous composite membranes, J. Membr. Sci., 2014, 454, 272–282 CrossRef CAS.
- X. Wang, D. Fang, B. Hsiao and B. Chu, Nanofiltration membranes based on thin-film nanofibrous composites, J. Membr. Sci., 2014, 469, 188–197 CrossRef CAS.
- J. Peng, Y. Su, W. Chen, X. Zhao, Z. Jiang, Y. Dong, Y. Zhang, J. Liu and C. Xingzhong, Polyamide nanofiltration membrane with high separation performance prepared by EDC/NHS mediated interfacial polymerization, J. Membr. Sci., 2013, 427, 92–100 CrossRef CAS.
- T. Kobayashi, H. Y. Wang and N. Fujii, Molecular Imprinting of Theophylline in Acrylonitrile-acrylic Acid Copolymer Membrane, Chem. Lett., 1995, 10, 927–928 CrossRef.
- B. Fang, Q. Ling, W. Zhao, Y. Ma, P. Bai, Q. Wei, H. Li and C. Zhao, Modification of polyethersulfone membrane by grafting bovine serum albumin on the surface of polyethersulfone/poly(acrylonitrile-co-acrylic acid) blended membrane, J. Membr. Sci., 2009, 329, 46–55 CrossRef CAS.
- X. Wang, K. Zhang, M. Zhu, H. Yu, Z. Zhou, Y. Chen and B. S. Hsiao, Continuous polymer nanofiber yarns prepared by self-bundling electrospinning method, Polymer, 2008, 49, 2755–2761 CrossRef CAS.
- Y. Mo, A. Tiraferri, N. Y. Yip, A. Adout, X. Huang and M. Elimelech, Improved antifouling properties of polyamide nanofiltration membranes by reducing the density of surface carboxyl groups, Environ. Sci. Technol., 2012, 46, 13253–13261 CrossRef CAS PubMed.
- Y. Tang, B. Tang and P. Wu, Preparation of a positively charged nanofiltration membrane based on hydrophilic-hydrophobic transformation of a poly(ionic liquid), J. Mater. Chem. A, 2015, 3, 12367–12376 CAS.
- J. Tanninen, M. Mänttäri and M. Nyström, Effect of salt mixture concentration on fractionation with NF membranes, J. Membr. Sci., 2006, 283, 57–64 CrossRef CAS.
- A. R. Rudman, R. I. Kalyuzhnaya, N. A. Vengerova, B. S. Eltsefon, Y. F. Razvodovskii and A. B. Zezin, Structure and properties
of polyelectrolyte complexes of polyacrylic acid and polymeric heterocyclic amines, Polym. Sci. U.S.S.R., 1983, 25, 2796–2806 CrossRef.
- H. W. Sun, G. H. Chen, R. H. Huang and C. J. Gao, A novel composite nanofiltration (NF) membrane prepared from glycolchitin/poly(acrylonitrile) (PAN) by epichlorohydrin cross-linking, J. Membr. Sci., 2007, 297, 51–58 CrossRef CAS.
- X. Wang, T.-M. Yeh, Z. Wang, R. Yang, R. Wang, H. Ma, B. S. Hsiao and B. Chu, Nanofiltration membranes prepared by interfacial polymerization on thin-film nanofibrous composite scaffold, Polymer, 2014, 55, 1358–1366 CrossRef CAS.
- Y. Zhou, Z. Dai, D. Zhai and C. Gao, Surface modification of polypiperazine-amide membrane by self-assembled method for dye wastewater treatment, Chin. J. Chem. Eng., 2015, 23, 912–918 CrossRef CAS.
- M. H. Liu, Y. P. Zheng, S. Shuai, Q. Zhou, S. C. Yu and C. J. Gao, Thin-film composite membrane formed by interfacial polymerization of polyvinylamine (PVAm) and trimesoyl chloride (TMC) for nanofiltration, Desalination, 2012, 288, 98–107 CrossRef CAS.
- D. Wu, Y. Huang, S. Yu, D. Lawless and X. Feng, Thin film composite nanofiltration membranes assembled layer-by-layer via interfacial polymerization from polyethylenimine and trimesoyl chloride, J. Membr. Sci., 2014, 472, 141–153 CrossRef CAS.
- S. M. Xue, Z. L. Xu, Y. J. Tang and C. H. Ji, Polypiperazine-amide nanofiltration membrane modified by different functionalized multiwalled carbon nanotubes (MWCNTs), ACS Appl. Mater. Interfaces, 2016, 8, 19135–19144 CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Benjamin S. Hsiao*c
*a and
Benjamin S. Hsiao*c