
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the effective factors affecting the conformational preferences and the early and late transition states of the unimolecular retro-ene decomposition reactions of ethyl cyanate, ethyl thiocyanate and ethyl selenocyanate†
Hooshang Atabakia,
Davood Nori-Shargh *b and
Mohamad Momen-Heravia
*b and
Mohamad Momen-Heravia
aDepartment of Chemistry, College of Science, Islamic Azad University, Mashhad Branch, Mashhad, Iran
bDepartment of Chemistry, College of Science, Islamic Azad University, Arak Branch, Arak, Iran. E-mail: D-norishargh@iau-arak.ac.ir; nori_ir@yahoo.com
First published on 26th April 2017
Abstract
The structural and conformational properties of ethyl cyanate (1), ethyl thiocyanate (2), and ethyl selenocyanate (3) and also their corresponding unimolecular retro-ene decomposition and isomerization reactions were investigated by means of G3(MP2) and MP2/6-311++G** methods and natural bond orbital interpretations. We assessed the role and contributions of the hyperconjugative interactions on the conformational preferences of compounds 1–3 by the deletion of the orbitals overlapping from the Fock matrices of the gauche- and anti-conformations, where the results obtained showed that the deletion of these hyperconjugative interactions from the Fock matrices leads to an increase in the anti-conformations preferences, revealing the significant impacts of the hyperconjugative interactions on the gauche-conformations preferences going from compound 1 to compound 3. The hyperconjugative interactions, Pauli exchange-type repulsions (PETR), and electrostatic model associated with the dipole–dipole interactions were in favor of the gauche-conformation of compound 1. Contrary to the conclusions published in the literature, there is no fact that justifies the anti-conformation preference in compound 1. Accordingly, we concluded that there are two rotamers (i.e., gauche- and anti-conformations) with literally the same populations for compound 1. The unimolecular retro-ene decomposition reactions of compounds 1–3 were more feasible than their corresponding cyanate → isocyanate isomerization reactions. The Pauli exchange-type repulsions have determining impacts on the retro-ene decomposition reactions of these compounds. The correlations between the activation Gibbs free energies and the advancements of the transition states (δBav) of the retro-ene decomposition reactions of compounds 1–3 according to the Hammond–Leffler postulate were also analyzed. Interestingly, the variations of the bond lengths in the transition state structures of the retro-ene decomposition reactions of compounds 1–3 were in accordance with the Hammond–Leffler postulate.
Introduction
Organyl cyanates and their thio- and seleno-analogs (as ambient functional groups) are widely used in the pharmaceutical industry, medicine, organic synthesis, etc.1–3 It is worth noting that the thiocyanate anion could be converted to the hypothiocyanite ion (OSCN−) in the human body, which is an antibiotic. The lactoperoxidase system is effective against staphylococci, Escherichia coli, and pseudomonads.4–6 It uses hydrogen peroxide to catalyze oxidation of the thiocyanate anion to the antibiotic to function in the airway lumen. The antileshmanial potential of selenocyanates has also been assayed and established.7Since cyanates and their thio- and seleno-analogs play important roles in human safety and organic syntheses, the exploration of effective factors on the structural and conformational behaviors of their organyl derivatives could be of interest to synthetic and theoretical chemists. Due to the instability of covalent (organyl) cyanates,8–14 there is little published information about their structures.15–18
Sakaizumi and co-workers performed microwave spectroscopy to investigate the conformational behavior of ethyl cyanate (1) and, based on their results, the anti-conformation of compound 1 was found to be more stable than its gauche-conformation.18 They claimed that they could not calculate the energy difference between the anti- and gauche-conformations of ethyl cyanate due to the weakness of the lines from the gauche-conformation. In addition, due to the insufficient number of isotope substituents, no geometrical data could be derived.18
In 1998, Leszczynski and co-workers performed ab initio calculations at the MP2 level of theory with a triple-ζ basis set augmented with polarization and diffusion functions to investigate the structure and conformational stability of ethyl cyanate (1).19 In addition, they investigated the relative stabilities of the rotational conformers of compound 1 at the MP4 level using MP2 optimized reference geometries. Based on the results obtained, they pointed out that the anti-conformation of compound 1 was predicted to be slightly more stable (0.14 kcal mol−1) than its gauche-conformation.19
In 2003, Pasinszki and co-workers performed mid-infrared spectroscopy and quantum mechanical calculations to study the structure, conformation, and isomerization of gaseous ethyl cyanate (1),20 and their results indicated the presence of two conformations of ethyl cyanate in gas phase, the gauche- and the anti-conformations. Also, they mentioned that the anti-conformation of ethyl cyanate is more stable than its gauche-conformation by about 0.16 kcal mol−1 (as calculated at the CCSD(T)/6-311+G(2d,2p)//B3LYP/6-311+G(2d,2p) level of theory), which is similar to the result obtained by Leszczynski and co-workers.19 It is worth noting that Pasinszki and co-workers20 mentioned that the conversion of ethyl thiocyanate to its isothiocyanate isomer in the gas phase could not be take place via a unimolecular process at ambient temperature [B3LYP/6-311+G(2d,2p): ΔG≠(ethyl cyanate→ethyl isocyanate) = 52.31 kcal mol−1], which is in the line with the results obtained by Faustov and co-workers21 for methyl cyanate [G2(MP2, SVP): ΔG≠(methyl cyanate→methyl isocyanate) = 62.6 kcal mol−1].
In addition to ethyl cyanate (1), the conformational behavior of ethyl thiocyanate (2) has been the subject of several infrared, Raman, and microwave investigations.22–27 In 1964, Hirschmann and co-workers performed infrared spectroscopy to study the conformational properties of the liquid and solid states of ethyl thiocyanate (2) and they concluded that the molecule exists in the gauche- and anti-conformations.22 They calculated an enthalpy difference of 0.49 ± 0.03 kcal mol−1 between the more stable anti- and higher energy gauche-conformations.
Contrary to the conclusion obtained by Hirschmann and co-workers concerning the anti-conformation preference in ethyl thiocyanate (2), the microwave spectrum analysis of this compound (by Bjørseth and Marstokk) revealed only one rotational isomer (the gauche-conformation).23 The discrepancy between the results of the vibrational spectroscopy23 and the microwave investigations of ethyl thiocyanate (2)23 prompted Ellestad and Torgrimsen to reevaluate the vibrational spectrum of this compound and their results showed only the gauche-conformation.24
In 1984, Durig and co-workers reanalyzed the vibrational spectrum of gaseous and solid ethyl thiocyanate (2) and also the Raman spectra of its liquid and solid sates.25 From the variable-temperature Raman study of the liquid state of ethyl thiocyanate (2), they concluded that the gauche-conformation of ethyl thiocyanate (2) is more stable than its anti-conformation (an enthalpy difference of 1.68 ± 0.07 kcal mol−1).25 In 1986, Braathen and Gatial analyzed the infrared spectrum of ethyl thiocyanate (2) isolated in argon and nitrogen matrices at 12 K and they found that the gauche-conformation was about 1.0 ± 0.07 kcal mol−1 more stable than its anti-conformation.26
To the best of our knowledge, there is only one report published outlining the rotational spectral data from the structural parameters of ethyl selenocyanate (3).27 Evaluations of the experimental data published about the conformational properties of ethyl cyanate (1), ethyl thiocyanate (2), and ethyl selenocyanate (3) imply an anti-conformation preference in compound 1 and the gauche-conformation preference in compounds 2 and 3, but there is no published information about the origin of the conformational behaviors of compounds 1–3.
The purpose of the present study was to explore the roles and contributions of the effective factors on the conformational behaviors of compounds 1–3 and also their retro-ene decomposition reactions, which take place earlier than their corresponding cyanate → isocyanate isomerization reactions.19,20 In this work, we examined the role and contributions of the generalized manifestation of the anomeric effect,28–48 the steric exchanges [total steric exchange energies, TSEE, which is considered to represent the Pauli exchange-type repulsions between filled orbitals (or the quasi-classical “Lennard-Jones repulsion”) between hard-shell sphere atoms],49–53 the electrostatic model associated with the dipole–dipole interactions, and also the attractive electrostatic interactions between two adjacent atoms on the conformational preferences and unimolecular retro-ene decomposition reactions of compounds 1–3. Importantly, the potential energy surfaces of the retro-ene decomposition reactions of compounds 1–3 in accordance with the Hammond–Leffler postulate were analyzed and the correlations between the early and late transition state structures, the advancements of the transition states (the average bond orders values, δBav), and the steric effects associated with the Pauli exchange-type repulsions were explored.
In order to explore the contributions of the hyperconjugative interactions on the anomeric relationships in compounds 1–3, we deleted the  hyperconjugative interactions [which are changed by the rotations around the X–CH2 bonds, where X = O (1), S (2), Se (3)] from the Fock matrices of the gauche- and anti-conformations. Then, by rediagonalization and comparison of the current Fock matrices with their original forms (Scheme 1), we evaluated the contributions of the hyperconjugation interactions mentioned above on the anomeric relationships in compounds 1–3. It is worth noting that the procedure mentioned above is an efficient approach and can be performed to evaluate the role and contributions of some specific hyperconjugative interactions on the conformational properties of chemical compounds.28,29,47
hyperconjugative interactions [which are changed by the rotations around the X–CH2 bonds, where X = O (1), S (2), Se (3)] from the Fock matrices of the gauche- and anti-conformations. Then, by rediagonalization and comparison of the current Fock matrices with their original forms (Scheme 1), we evaluated the contributions of the hyperconjugation interactions mentioned above on the anomeric relationships in compounds 1–3. It is worth noting that the procedure mentioned above is an efficient approach and can be performed to evaluate the role and contributions of some specific hyperconjugative interactions on the conformational properties of chemical compounds.28,29,47
 | ||
| Scheme 1 Schematic representation of the gauche- and anti-conformations of compounds 1–3 [X = O (1), S (2), Se (3)] and their corresponding unimolecular retro-ene decomposition reactions. | ||
Computational details
The modified version of Gaussian-2 (G2), which uses second-order Møller–Plesset perturbation theory (MP2) calculations54–56 instead of MP4 for the basis set extension corrections, and also the MP2 method with the 6-311++G** basis set57–65 on all atoms (MP2/6-311++G**) were performed to optimize the structural parameters of the gauche- and anti-conformations and the transition state structures of the unimolecular retro-ene decomposition and cyanate → isocyanate isomerization reactions of compounds 1–3 and also to calculate their corresponding electronic energies and thermodynamic functions together with the GAMESS US package of programs.66,67The natural bond orbital (NBO) interpretation was performed to examine quantitatively the impacts of the plausible hyperconjugative interactions and the Pauli exchange-type repulsions on the conformational and structural properties and also the potential energy surfaces of the retro-ene decomposition of compounds 1–3. In addition, the bonding and antibonding orbital occupancies and energies, the total natural resonance theory (NRT) bond orders (natural bond orders, nbo) of the gauche- and anti-conformations, and also the transition state structures of the retro-ene decomposition reactions of compounds 1–3 were analyzed by means of natural bond orbital interpretation with the NBO 5.G program.68
The stabilization energies (second-order perturbative estimations of donor–acceptor electronic interactions) in the NBO basis are inversely proportional to the energy differences between the donor (i) and acceptor (j) orbitals, Δεij, and directly proportional to the magnitudes of the orbital overlap integrals (Sij):69–71
Stabilization or resonance energy ∝ 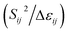
Accordingly, the stabilization or resonance energy (second-order perturbational energies, E2) associated with i → j electron delocalization can be explicitly estimated by the following equation: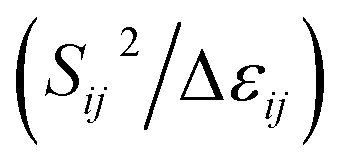
 | (1) |
The impacts of the hyperconjugative interactions (which change with the rotation around the C4–X3 bonds:  electron delocalizations) on the conformational behaviors of compounds 1–3 were assessed by deletions of these electron delocalizations from the Fock matrices of their gauche- and anti-conformations (Fig. 1 and 2). It is worth noting that the natural bond orbital interpretation is a capable and sufficient theoretical approach to assess quantitatively the impacts of the hyperconjugative interactions associated with the electron delocalizations, the electrostatic interactions, and the steric effects on the dynamic behaviors and reactivity of chemical compounds.74
electron delocalizations) on the conformational behaviors of compounds 1–3 were assessed by deletions of these electron delocalizations from the Fock matrices of their gauche- and anti-conformations (Fig. 1 and 2). It is worth noting that the natural bond orbital interpretation is a capable and sufficient theoretical approach to assess quantitatively the impacts of the hyperconjugative interactions associated with the electron delocalizations, the electrostatic interactions, and the steric effects on the dynamic behaviors and reactivity of chemical compounds.74
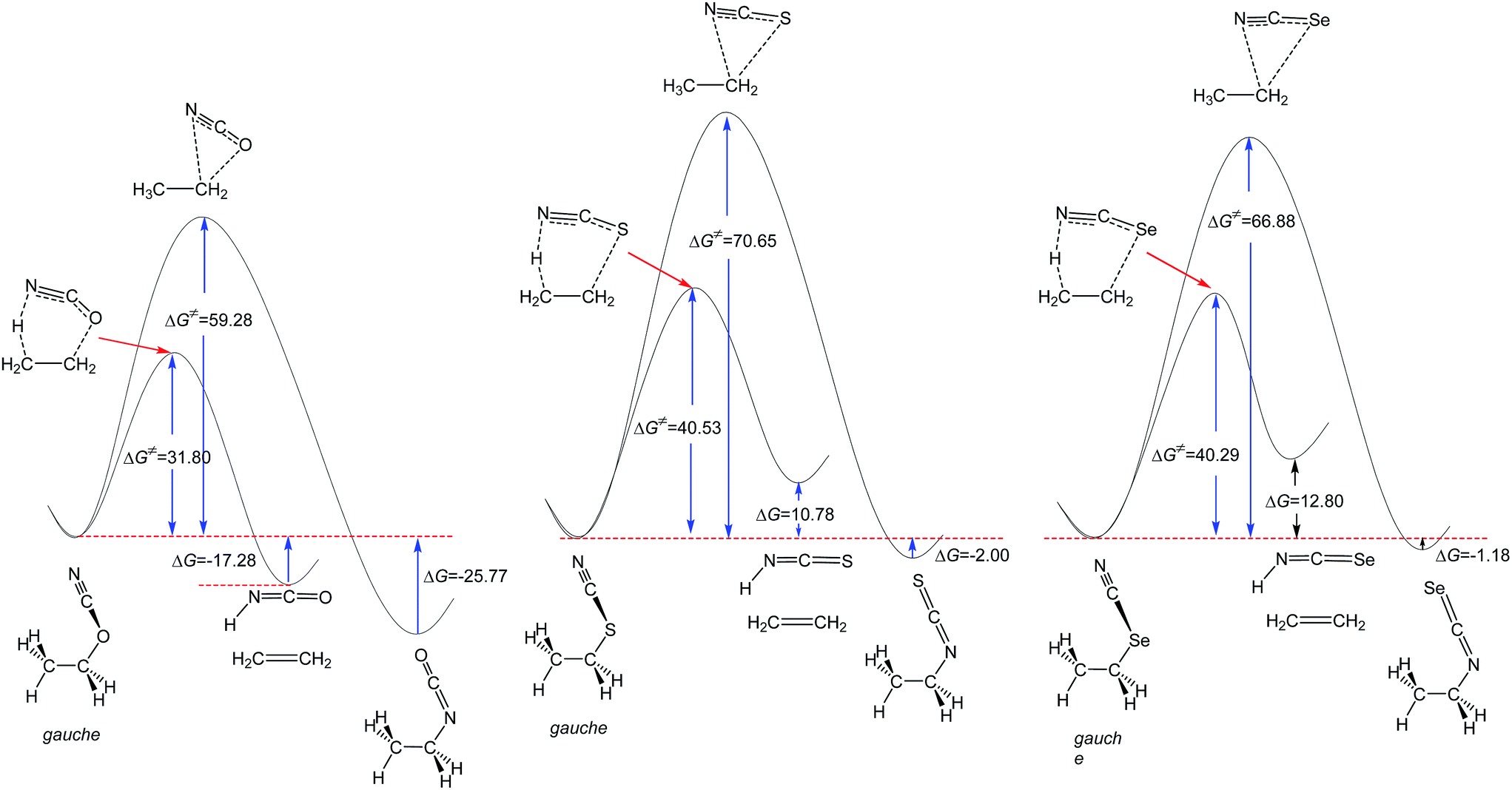 | ||
| Fig. 1 MP2/6-311++G** calculated comparative potential energy profiles of the unimolecular isomerization and retro-ene decomposition reactions of compounds 1–3. ΔG and ΔG‡ values are in kcal mol−1. | ||
 | ||
| Fig. 2 MP2/6-311++G** calculated comparative potential energy profiles of the retro-ene decomposition reactions of compounds 1–3 in accordance with the Hammond–Leffler postulate. ΔG, ΔG‡, and ΔTSEE‡ values are in kcal mol−1. Δμ‡ values are in Debye. | ||
Results and discussion
1. Conformational preference and retro-ene decomposition reactions in compounds 1–3
The MP2/6-311++G** and G3MP2 calculated zero-point energies (ZPE), corrected zero-point energies (Eo = Eel + ZPE), enthalpy, entropy, Gibbs free energy, and their differences (i.e., ΔZPE, ΔEo, ΔH, ΔS and ΔG) for the gauche- and anti-conformations of compounds 1–3 and also their corresponding retro-ene decomposition reactions are summarized in Table 1 and ESI-1.† According to the results of this work and the previously published data, there is no significant difference between the energies of the gauche- and anti-conformations of compound 1. Since the energy difference between the gauche- and anti-conformations of compound 1 is negligible; we may expect the presence of two rotamers (i.e., gauche- and anti-conformations) at ambient temperature with literally the same populations for compound 1. In order to justify this claim, we examined the contributions of the stabilization energies associated with the hyperconjugative interactions, the electrostatic model associated with the dipole–dipole interactions, and the steric effects on the conformational behaviors of compound 1. Surprisingly, the factors mentioned above were in favor of the gauche-conformation of compound 1, contradicting the preference of the anti-conformation of compound 1 compared to its gauche-conformation. Therefore, the conclusion published in the literature concerning the anti-conformation preference in compound 1 could be questioned.18| Geometry | MP2/6-311++G** | G3MP2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔZPE | ΔEo | ΔH | ΔS | ΔG | ΔZPE | ΔEo | ΔH | ΔS | ΔG | |
| a From MP2, ref. 19.b From MP4, ref. 19.c From CCSD(T)/6-311+G(2d,2p)//QCISD/6-311+G(d,p), ref. 20.d From infrared spectroscopy, ref. 26.e From Raman spectroscopy, ref. 25. | ||||||||||
| 1-gauche | 0.00 | 0.00 | 0.00 | 0.000 | 0.00 | 0.00b | 0.00b | 0.00b | 0.000b | 0.00b |
| 1-anti | −0.14 | 0.09 | 0.20 | 1.192 | −0.15 | 0.09b | 0.05b | 0.12b | 0.371b | 0.01b |
| (0.05)a | (−0.16)c | |||||||||
| (−0.14)b | ||||||||||
| (−0.11)c | ||||||||||
| 1-TS | −4.017 | 31.21 | 30.79 | −3.373 | 31.80a | −4.733 | 31.27 | 31.10 | −1.951 | 31.68b |
| 2-gauche | 0.08 | 0.00 | 0.00 | 0.000a | 0.00a | 0.02b | 0.00b | 0.00b | 0.000b | 0.00b |
| 2-anti | 0.00 | 1.12 | 1.25 | 1.871a | 0.69a | 0.00b | 0.86b | 0.93b | 0.672b | 0.73b |
| (1.06)a | ||||||||||
| (1.02)b | ||||||||||
| 1.00 ± 0.07d | ||||||||||
| 1.68 ± 0.07e | ||||||||||
| 2-TS | −4.145 | 39.86 | 39.39 | −3.829 | 40.53a | −4.580 | 40.538 | 40.40 | −1.307 | 40.79b |
| 3-gauche | 0.07a | 0.00a | 0.00a | 0.000a | 0.00a | 0.02b | 0.00b | 0.00b | 0.000b | 0.00b |
| 3-anti | 0.00 | 1.19a | 1.30a | 1.480a | 0.86a | 0.00b | 0.90b | 0.96b | 0.787b | 0.72b |
| (1.07)a | ||||||||||
| (1.36)b | ||||||||||
| 3-TS | −4.046 | 39.58 | 39.09 | −4.064 | 40.29a | −4.474 | 40.99 | 40.82 | −1.310 | 41.21b |
The MP2/6-311++G** results showed that the gauche-conformation preference increases significantly from compound 1 to compound 2, while it increase only slightly from compound 2 to compound 3. Seemingly, compared with the experimental data published about the conformational preferences in compounds 2 and 3,22–27 the MP2/6-311++G** method gives more reliable results concerning their conformational preferences than the results obtained at the G3(MP2) level (Table 1).
The results of this work (Fig. 1) and the previously published data indicate that the alkyl cyanate → alkyl isocyanate unimolecular isomerization reactions could not take place at ambient temperature.20,21 In this work, we examined the potential energy surfaces of the cyanate → isocyanate unimolecular isomerization reactions of compounds 1–3 (Fig. 1) and also their corresponding retro-ene decomposition reactions (Fig. 2) at the MP2/6-311++G** level of theory. Based on our findings and also the published theoretical data in the literature,20,21 we found that the retro-ene decomposition reactions of compounds 1–3 proceed through pathways with lower activation barriers (with more stable transition states) and produce less stable products compared to their corresponding cyanate → isocyanate unimolecular isomerization reactions. Accordingly, the retro-ene decomposition reactions of compounds 1–3 could be kinetically controlled at lower temperatures. At higher temperatures, the cyanate → isocyanate unimolecular isomerization reactions of compounds 1–3 may proceed through pathways with greater activation barriers (with less stable transition states) and the major products are the thermodynamically more stable systems.
Fig. 1 shows that the barrier heights of the unimolecular cyanate → isocyanate unimolecular isomerization reactions increase significantly from compound 1 to compound 2, but decrease from compound 2 to compound 3. It is worth noting that the unimolecular cyanate → isocyanate unimolecular isomerization reaction of compound 1 is evidently endothermic, but the corresponding endothermicity decreases drastically from compound 1 to compound 2, while it does not change significantly from compound 2 to compound 3.
The MP2/6-311++G** results showed that the retro-ene decomposition reaction barrier height increases drastically from compound 1 to compound 2, but it does not change significantly going from compound 2 to compound 3 (Fig. 2). It is worth noting that the retro-ene decomposition reaction of compound 1 is exothermic. On the contrary, the retro-ene decomposition reactions of compounds 2 and 3 are endothermic and the endothermicity increases from compound 2 to compound 3 (Table 1 and ESI-1†).
2. Assessing the impacts of the hyperconjugative interactions on the conformational preferences and retro-ene decomposition reactions of compounds 1–3
The contributions of the generalized anomeric effects associated with the hyperconjugative interactions (HCGAE) on the conformational preferences in compounds 1–3 were analyzed and the corresponding results are given in Table 2. The NBO-MP2/6-311++G** analysis showed that the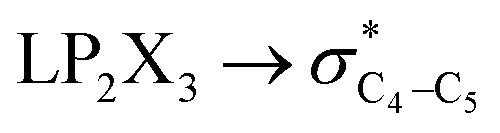 [X = O (1), S (2), Se (3)] and
[X = O (1), S (2), Se (3)] and  electron delocalizations have the greatest impact on the magnitudes of the total hyperconjugative anomeric effects (HCAEtotal) of compounds 1–3. Seemingly, the HCAEtotal values in compounds 1–3 are controlled by the
electron delocalizations have the greatest impact on the magnitudes of the total hyperconjugative anomeric effects (HCAEtotal) of compounds 1–3. Seemingly, the HCAEtotal values in compounds 1–3 are controlled by the  electron delocalizations. Based on the results obtained, the HCGAEtotal (HCGAEanti − HCGAEgauche) values associated with the
electron delocalizations. Based on the results obtained, the HCGAEtotal (HCGAEanti − HCGAEgauche) values associated with the  hyperconjugative interactions are in favor of the gauche-conformations of compounds 1–3 compared with their corresponding anti-conformations. It is worth noting that the HCGAEtotal values decrease slightly from compound 1 to compound 3, revealing that the generalized anomeric effects associated with the hyperconjugative interactions are in favor of the gauche-conformations in compounds 1–3 but are not solely responsible for the variations of the gauche-conformations stabilities compared with their corresponding anti-conformations going from compound 1 to compound 3. Therefore, the roles and contributions of other factors (e.g., the electrostatic model and steric effects) should be accounted for (Table 2).
hyperconjugative interactions are in favor of the gauche-conformations of compounds 1–3 compared with their corresponding anti-conformations. It is worth noting that the HCGAEtotal values decrease slightly from compound 1 to compound 3, revealing that the generalized anomeric effects associated with the hyperconjugative interactions are in favor of the gauche-conformations in compounds 1–3 but are not solely responsible for the variations of the gauche-conformations stabilities compared with their corresponding anti-conformations going from compound 1 to compound 3. Therefore, the roles and contributions of other factors (e.g., the electrostatic model and steric effects) should be accounted for (Table 2).
| Hyperconjugative interactions | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| Gauche | Anti | Gauche | Anti | Gauche | Anti | |
| HCGAE(C4–X3 strengthening) | ||||||
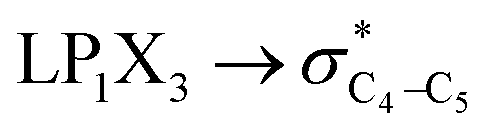 |
0.79 | — | 0.91 | — | 0.68 | — |
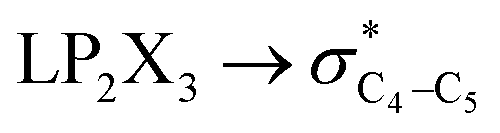 |
4.50 | 1.00 | 4.01 | — | 3.13 | — |
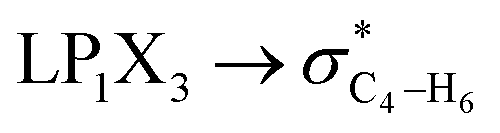 |
5.08 | 5.45 | 5.31 | 5.24 | 4.05 | 3.92 |
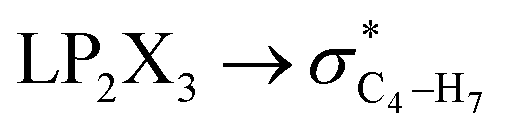 |
2.94 | 5.45 | 0.86 | 5.24 | 0.00 | 3.92 |
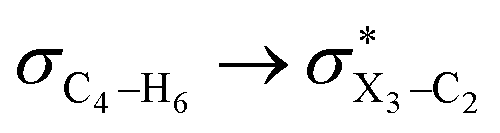 |
0.00 | 2.48 | 0.00 | 0.75 | 0.00 | 0.52 |
 |
3.64 | — | 1.86 | — | 1.54 | — |
| ∑HCGAE(C4–X3 strengthening) | 16.95 | 14.38 | 12.95 | 11.23 | 9.4 | 8.36 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| HCGAE(C4–X3 weakening) | ||||||
 |
2.84 | 2.49 | 3.06 | 3.12 | 2.43 | 2.45 |
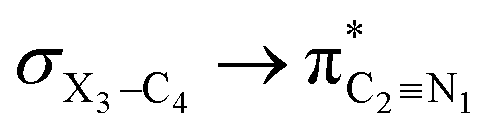 |
12.34 | 12.75 | 7.59 | 7.55 | 6.76 | 6.63 |
 |
6.50 | 5.66 | 6.71 | 5.39 | 7.24 | 5.95 |
| ∑HCGAE(C4–X3 weakening) | 21.68 | 20.9 | 17.36 | 16.06 | 16.43 | 15.03 |
| ∑HCGAE | 38.63 | 35.28 | 30.31 | 27.29 | 25.83 | 23.39 |
| HCGAEtotal | −3.35 | −3.02 | −2.44 | |||
|
||||||
| Fij | ||||||
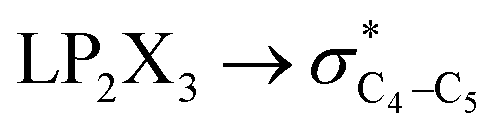 |
0.066 | 0.035 | 0.058 | — | 0.051 | — |
 |
0.121 | 0.122 | 0.079 | 0.079 | 0.073 | 0.072 |
In order to estimate quantitatively the impacts of the hyperconjugative interactions on the conformational preferences in compounds 1–3, we deleted the hyperconjugative interactions (in which the overlapping of their corresponding orbitals change with the rotations around the X3–C4 bonds: 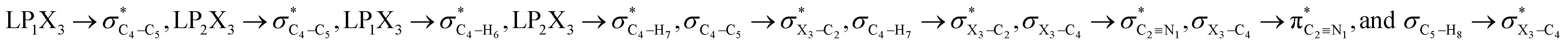 electron delocalizations) from the Fock matrices of the gauche- and anti-conformations. Then, with rediagonalization and comparison of the current Fock matrices with their original forms, we found that the anti-conformations of compounds 1–3 became more stable than their corresponding chiral gauche-conformations, revealing that the anomeric relationships in compounds 1–3 may have a hyperconjugative interactions origin (Table 3).
electron delocalizations) from the Fock matrices of the gauche- and anti-conformations. Then, with rediagonalization and comparison of the current Fock matrices with their original forms, we found that the anti-conformations of compounds 1–3 became more stable than their corresponding chiral gauche-conformations, revealing that the anomeric relationships in compounds 1–3 may have a hyperconjugative interactions origin (Table 3).
 electron delocalizations and their corresponding energy changes (in a.u.) in the gauche- and anti-conformations of compounds 1–3
electron delocalizations and their corresponding energy changes (in a.u.) in the gauche- and anti-conformations of compounds 1–3
| Geometries | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| Gauche | Anti | Gauche | Anti | Gauche | Anti | |
| a Values are in kcal mol−1. | ||||||
| Total SCF energies | −245.8535160 | −245.854611210 | −568.521273696 | −568.521085748 | −2570.781555431 | −2570.781318328 |
| Energies of deletions | −245.801561 | −245.807529571 | −568.479413418 | −568.483811670 | −2570.745744656 | −2570.749046882 |
| Energy changes (EC) | 0.051955(32.60)a | 0.047082(29.54)a | 0.041860(26.27)a | 0.037274(23.39)a | 0.035811(22.47)a | 0.032271(20.25)a |
| Δ(ECgauche − ECanti) | 3.06a | 2.88a | 2.22a | |||
Since the X3–C4 [X = O (1), S (2), Se (3)] bonds break in the transition state structures of the retro-ene decomposition reactions, we may expect that the retro-ene decomposition reaction barrier heights may decrease going from compound 1 to compound 3. The results obtained in this work, however, do not confirm this expectation. Based on the results obtained at the MP2/6-311++G** level of theory, the retro-ene decomposition reaction barrier height increases drastically from compound 1 to compound 2, but does not change significantly going from compound 2 to compound 3 (Table 1, ESI-1 and ESI-2†).
3. Orbital amplitudes (electron densities) associated with the hyperconjugative interactions
The impacts of the hyperconjugative generalized anomeric effect (HCGAE) on the retro-ene decomposition reactions barrier heights of compounds 1–3 were investigated. Seemingly, the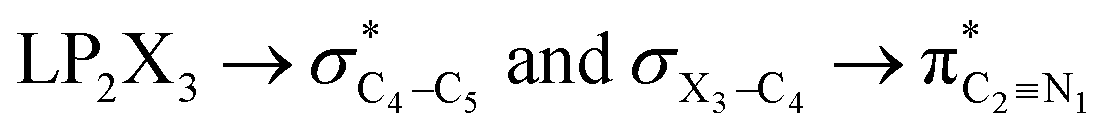 electron delocalizations have the greatest impact on the easiness of the breaking of the X3–C4 bonds, thus we analyzed their corresponding donor–acceptor orbital amplitude profiles (or electron densities) (Fig. 3 and 4). Fig. 3 shows that the mixings of the lobes of the donor LP2X3 nonbonding and acceptor
electron delocalizations have the greatest impact on the easiness of the breaking of the X3–C4 bonds, thus we analyzed their corresponding donor–acceptor orbital amplitude profiles (or electron densities) (Fig. 3 and 4). Fig. 3 shows that the mixings of the lobes of the donor LP2X3 nonbonding and acceptor  antibonding orbitals decrease from the gauche-conformations of compound 1 to compound 3, revealing that the variations of the off-diagonal elements control the variations of the mixing of the doubly occupied orbitals of LP2M3 [X = O (1), S (2), Se (3)] with the adjacent unoccupied orbitals of the CH2–CH3 bonds
antibonding orbitals decrease from the gauche-conformations of compound 1 to compound 3, revealing that the variations of the off-diagonal elements control the variations of the mixing of the doubly occupied orbitals of LP2M3 [X = O (1), S (2), Se (3)] with the adjacent unoccupied orbitals of the CH2–CH3 bonds  . A same trend was observed for mixing of the lobes of the donor σX3–C4 bonding and acceptor
. A same trend was observed for mixing of the lobes of the donor σX3–C4 bonding and acceptor  antibonding orbitals, which is in accordance with the Mulliken (or Wolfberg–Helmholtz) approximation.47,48 Based on the Mulliken (or Wolfberg–Helmholtz) approximation, the desirable orbital overlapping between the donor and acceptor orbitals are reachable by substantial adjustments of their off-diagonal terms.
antibonding orbitals, which is in accordance with the Mulliken (or Wolfberg–Helmholtz) approximation.47,48 Based on the Mulliken (or Wolfberg–Helmholtz) approximation, the desirable orbital overlapping between the donor and acceptor orbitals are reachable by substantial adjustments of their off-diagonal terms.
 | ||
Fig. 3 Calculated profiles of the orbital amplitudes (electron densities) for the 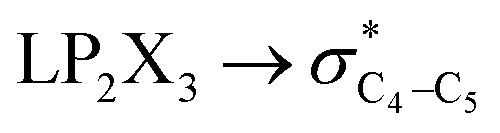 [X = O (1), S (2), Se (3)] negative hyperconjugative interactions that shorten the X3–C4 bonds in the gauche-conformations of compounds 1–3. E2 values are in kcal mol−1. [X = O (1), S (2), Se (3)] negative hyperconjugative interactions that shorten the X3–C4 bonds in the gauche-conformations of compounds 1–3. E2 values are in kcal mol−1. | ||
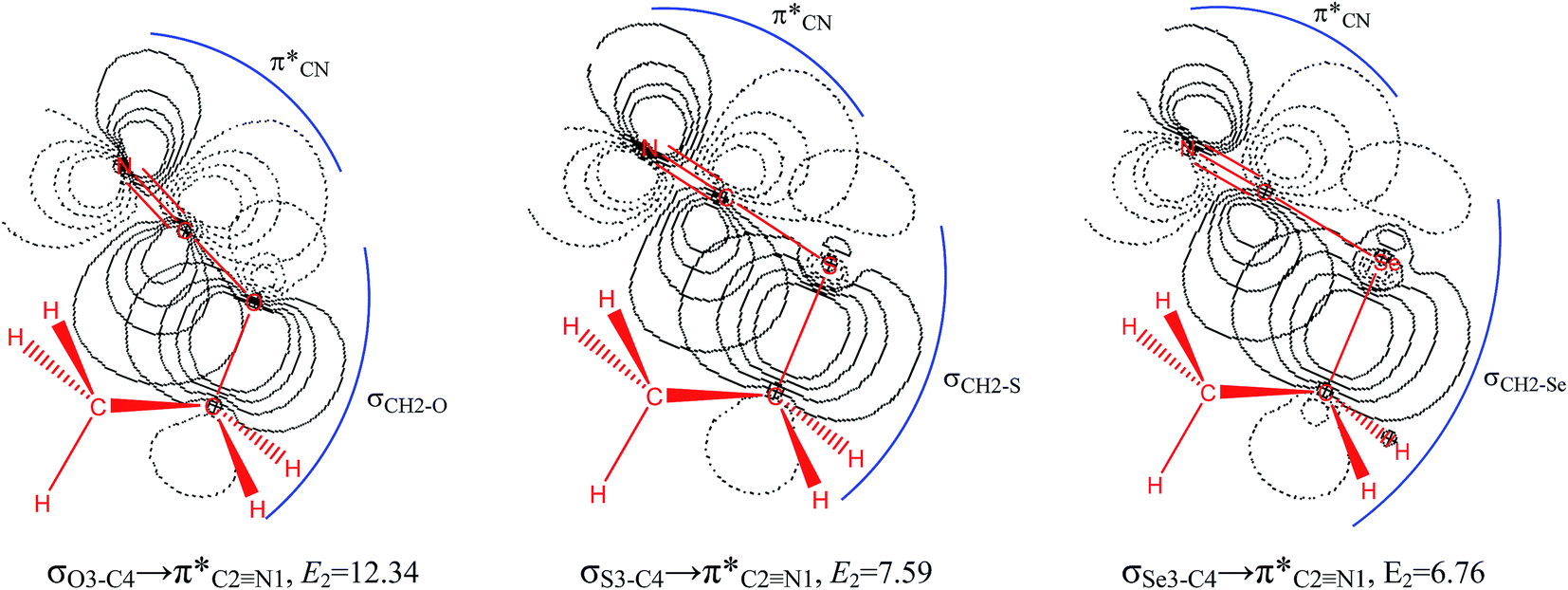 | ||
Fig. 4 Calculated profiles of the orbital amplitudes (electron densities) for the 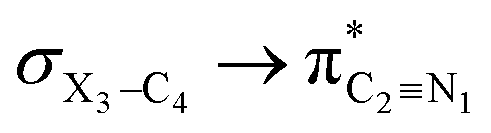 [X = O (1), S (2), Se (3)] hyperconjugative interactions that lengthen the X3–C4 bonds in the gauche-conformations of compounds 1–3. E2 values are in kcal mol−1. [X = O (1), S (2), Se (3)] hyperconjugative interactions that lengthen the X3–C4 bonds in the gauche-conformations of compounds 1–3. E2 values are in kcal mol−1. | ||
4. HCGAEtotal decomposition into the X3–C4 bonds strengthening and weakening components
It is worth noting that HCGAEtotal could be decomposed into two components: the first component has an impact on the decrease of the X3–C4 bond orders [HCGAE(X3–C4 strengthening)], while the second one has an opposite impact [HCGAE(X3–C4 weakening)] (see Table 2). The calculated HCGAE(X3–C4 strengthening) values associated with the electron delocalizations and the HCGAE(X3–C4 weakening) values associated with the
electron delocalizations and the HCGAE(X3–C4 weakening) values associated with the 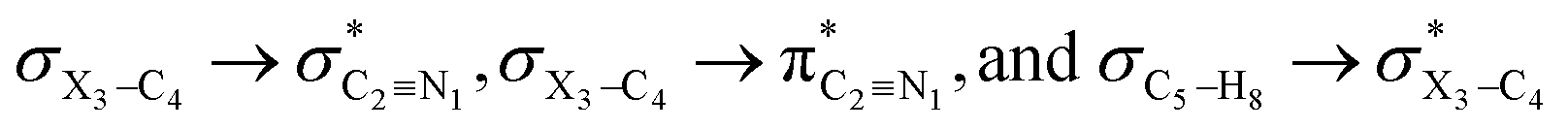 hyperconjugative interactions decrease from compound 1 to compound 3, while their differences (Δ[HCGAE(X3–C4 weakening) − HCGAE(X3–C4 strengthening)]) increase significantly from compound 1 to compound 2, but decrease slightly from compound 2 to compound 3. Surprisingly, the variations of the Δ[HCGAE(X3–C4 weakening) − HCGAE(X3–C4 strengthening)] parameters justifies the variations of the retro-ene decomposition reactions barrier heights going from compound 1 to compound 3 (Table 2 and Fig. 2).
hyperconjugative interactions decrease from compound 1 to compound 3, while their differences (Δ[HCGAE(X3–C4 weakening) − HCGAE(X3–C4 strengthening)]) increase significantly from compound 1 to compound 2, but decrease slightly from compound 2 to compound 3. Surprisingly, the variations of the Δ[HCGAE(X3–C4 weakening) − HCGAE(X3–C4 strengthening)] parameters justifies the variations of the retro-ene decomposition reactions barrier heights going from compound 1 to compound 3 (Table 2 and Fig. 2).
5. Retro-ene decomposition reactions of compounds 1–3 in connection with the Hammond–Leffler postulate
The MP2/6-311++G** results showed that the retro-ene decomposition reaction of compound 1 is exothermic and its transition state structure more closely resembles the gauche-conformation (with an early transition state). On the contrary, the retro-ene decomposition reactions of compounds 2 and 3 are endothermic and their transition state structures more closely resemble their corresponding products (with late transition states). In order to assess the validity of the Hammond–Leffler postulate for the retro-ene reactions of compounds 1–3, we examined the total natural resonance theory (NRT) bond orders (natural bond orders, nbo) of the reactants, the transition state structures, and the products of the retro-ene reactions of compounds 1–3 (Table 4). The examinations of the bond orders of the transition state structures revealed that the bond orders of the retro-ene decomposition reaction of compound 1 more resemble the gauche-conformation (Fig. 5), which is in agreement with the Hammond–Leffler postulate. Interestingly, the bond orders of the six-membered cyclic transition state structures of the retro-ene decomposition reactions of compounds 2 and 3 are closer to their corresponding products.| 1 | 2 | 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Gauche | TS | P | Gauche | TS | P | Gauche | TS | P | |
| WBI | |||||||||
| N1–C2 | 2.9248 | 2.4089 | 2.0061 | 2.9301 | 2.3596 | 2.0929 | 2.9447 | 2.4249 | 2.1856 |
| C2–X3 | 1.0710 | 1.5524 | 1.9861 | 1.0652 | 1.5257 | 1.8968 | 1.0501 | 1.4746 | 1.7951 |
| X3–C4 | 0.9897 | 0.5908 | 0.0000 | 0.9906 | 0.3736 | 0.0000 | 0.9852 | 0.3230 | 0.0000 |
| C4–C5 | 1.0200 | 1.1547 | 2.0154 | 1.0234 | 1.4747 | 2.0154 | 1.0255 | 1.5254 | 2.0154 |
| C5–H9 | 0.9910 | 0.8173 | 0.0000 | 0.9908 | 0.4308 | 0.0000 | 0.9905 | 0.3839 | 0.0000 |
| N1–H9 | 0.0000 | 0.0355 | 0.9731 | 0.0000 | 0.3876 | 0.9662 | 0.0000 | 0.4481 | 0.9606 |
|
|||||||||
| δBi | |||||||||
| δBN1–C2 | 0.5614 | 0.6814 | 0.6848 | ||||||
| δBC2–X3 | 0.5261 | 0.5538 | 0.5698 | ||||||
| δBX3–C4 | 0.4031 | 0.6229 | 0.6721 | ||||||
| δBC4–C5 | 0.1353 | 0.4549 | 0.5050 | ||||||
| δBC5–H9 | 0.1753 | 0.5652 | 0.6124 | ||||||
| δBN1–H9 | 0.0365 | 0.4012 | 0.4665 | ||||||
| δBav | 0.3063 | 0.5466 | 0.5851 | ||||||
| ASy | 0.3733 | 0.0018 | 0.0736 | ||||||
| Sy | 0.6267 | 0.9816 | 0.9264 | ||||||
 | ||
| Fig. 5 MP2/6-311++G** calculated total natural resonance theory (NRT) bond orders (natural bond orders, nbo) of the transition state structures of the retro-ene decomposition reactions of compounds 1–3. | ||
Although the C–X (X = O, S, Se) bond dissociation energy decreases when going from C–O to C–Se bonds, the barrier heights of the decomposition and isomerization reactions of ethyl selenocyanate (3) are significantly greater than those in ethyl cyanate (1) and also are close to the corresponding values for ethyl thiocyanate (2). It is worth noting that six bonds (N1![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) C2, C2X3, X3⋯C4, C4C5, C5⋯H9, H9⋯N1) are involved in the transition state structures of the retro-ene decomposition reactions of compounds 1–3. The C5⋯H9 bond in the transition state structure of the retro-ene decomposition reaction of compound 1 has a significant covalent character, while there is no considerable bond order for the H9⋯N1 bonds. Importantly, the bond orders of the C5⋯H9 bonds decrease going from the transition state structures of compound 1 to compound 3, while the bond orders of the H9⋯N1 bonds increase inversely. Also, the bond orders of the C–C bonds in the transitions state structures increase significantly from the transition state structures of compound 1 to compound 3. It can thus be concluded that the variations of the barrier heights of the retro-ene decomposition reactions of compounds 1–3 could not be controlled only by the X3–C4 (X = O, S, Se) bonds and therefore the contributions of the other bonds involved in the retro-ene decomposition reactions and their corresponding angle strains should be considered.
C2, C2X3, X3⋯C4, C4C5, C5⋯H9, H9⋯N1) are involved in the transition state structures of the retro-ene decomposition reactions of compounds 1–3. The C5⋯H9 bond in the transition state structure of the retro-ene decomposition reaction of compound 1 has a significant covalent character, while there is no considerable bond order for the H9⋯N1 bonds. Importantly, the bond orders of the C5⋯H9 bonds decrease going from the transition state structures of compound 1 to compound 3, while the bond orders of the H9⋯N1 bonds increase inversely. Also, the bond orders of the C–C bonds in the transitions state structures increase significantly from the transition state structures of compound 1 to compound 3. It can thus be concluded that the variations of the barrier heights of the retro-ene decomposition reactions of compounds 1–3 could not be controlled only by the X3–C4 (X = O, S, Se) bonds and therefore the contributions of the other bonds involved in the retro-ene decomposition reactions and their corresponding angle strains should be considered.
It is worth noting that the variations of the bond orders of the transition state structures of the retro-ene reactions of compounds 1–3 are reflected in their structural parameters. Based on the results obtained at the MP2/6-311++G** level of theory, the C-C bond lengths and the H⋯N distances decrease going from the transition state structures of the retro-ene reactions of compound 1 to compound 3 (Fig. 6). As the C–C bond lengths and the H⋯N distances decrease, the C–H bond lengths increase inversely, leading to the formation of the late transition structures that resemble their corresponding products (Fig. 1 and 2). Accordingly, the variations of the bond lengths in the transition state structures of the retro-ene decomposition reactions of compounds 1–3 are in accordance with the Hammond–Leffler postulate (Table ESI-3†).
 | ||
| Fig. 6 MP2/6-311++G** calculated structural parameters (bond lengths, Å) of the transition state structures of the retro-ene decomposition reactions of compounds 1–3. | ||
6. Exploring the impacts of the attractive electrostatic interactions between two adjacent atoms on the retro-ene decomposition reactions of compounds 1–3
In order to assess the impacts of the attractive electrostatic interactions on the retro-ene decomposition reactions of compounds 1–3, we examined the natural atomic charges (NAC) of the X3 and C4 atoms (Scheme 1). The results obtained showed that the natural atomic charge of the O atom is negative, while the S and Se atoms possess positive charges.Effectively, the calculated natural atomic charge differences between the X3 and C4 atoms in the gauche-conformations (Δ[NAC(X3) − NAC(C4)]) increase from compound 1 to compound 3 (Table 5). The smaller Δ[NAC(X3) − NAC(C4)] parameter in compound 1 compared to those in compounds 2 and 3 leads to an early transition state (smaller δBav value). The greater attractive electrostatic interactions between the two adjacent S3–C4 and Se3–C4 atoms in compounds 2 and 3, respectively, (greater Δ[NAC(X3) − NAC(C4)] parameters) lead to the relatively late transition states (greater δBav values) together with the increased overall synchronicity in the retro-ene decomposition reactions of compounds 2 and 3. This fact could also be rationalized by the smaller  electron delocalizations in the gauche-conformations of compounds 2 and 3 compared with that in compound 1. Note that the greater
electron delocalizations in the gauche-conformations of compounds 2 and 3 compared with that in compound 1. Note that the greater  electron delocalization in the gauche-conformation of compound 1 facilitates more O3–C4 bond breaking in the retro-ene decomposition reaction of compound 1 compared with the smaller
electron delocalization in the gauche-conformation of compound 1 facilitates more O3–C4 bond breaking in the retro-ene decomposition reaction of compound 1 compared with the smaller  electron delocalizations in compounds 2 and 3 (Table 2).
electron delocalizations in compounds 2 and 3 (Table 2).
| a From rotational spectrum, ref. 18.b From microwave spectrum, ref. 23.c From microwave spectrum, ref. 27.d From B3LYP/6-311+G(2d,2p), ref. 20.e From QCISD(full)/6-311+G(d,p), ref. 20.f From MP2 with a triple-ξ basis set augmented with polarization and diffusion function, ref. 19. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | 1 | 2 | 3 | ||||||
| Geometry | Gauche | Anti | TS | Gauche | Anti | TS | Gauche | Anti | TS |
| μ | 5.05 | 5.24 | 4.63 | 4.52 | 4.84 | 3.51 | 4.71 | 5.06 | 3.56 |
| (4.72 ± 0.33)a | (4.01 ± 0.12)b | (4.47 ± 0.10)c | |||||||
| (4.92)d (4.60)e | (5.13)d (5.17)e | (3.98)f | (4.27)f | (4.13)f | (4.46)f | ||||
| (4.59)f | (4.68)f | ||||||||
| Δ(μanti − μgauche) | 0.19 | 0.32 | 0.35 | ||||||
| Δ(μTS − μgauche) | −0.42 | −1.01 | −1.15 | ||||||
| TSEE | 132.76 | 135.71 | 157.01 | 123.91 | 121.86 | 183.82 | 133.21 | 126.79 | 188.85 |
| Δ(TSEEanti − TSEEgauche) | 2.95 | −2.05 | −6.42 | ||||||
| Δ(TSEETS − TSEEgauche) | 24.25 | 59.91 | 55.64 | ||||||
|
|||||||||
| NAC | |||||||||
| X3 | −0.60116 | −0.60663 | 0.28657 | 0.28134 | 0.40735 | 0.40180 | |||
| C4 | 0.07274 | 0.07204 | −0.43066 | −0.42671 | −0.48241 | −0.47864 | |||
| N1 | −0.49298 | −0.48980 | −0.36828 | −0.36510 | −0.37473 | −0.37081 | |||
| Δ[NC(X3) − NC(C4)] | 0.67390 | 0.67867 | 0.71723 | 0.70805 | 0.88976 | 0.88044 | |||
7. Assessing the synchronicities and advancements of the transition states (δBav) of the retro-ene decomposition reactions of compounds 1–3
To gain further insights into the nature and mechanisms of the retro-ene decomposition reactions of compounds 1–3 and the applicability of the Hammond–Leffler postulate in these reactions, we examined their synchronicity indices by calculating the bond orders [total natural resonance theory (NRT) bond orders (natural bond orders, nbo)] of the gauche, transition state, and product structures.75–78The synchronous or asynchronous nature of the mechanisms of the retro-ene decomposition reactions of compounds 1–3 were also considered by the synchronicity, Sy, as calculated from eqn (2):
| Sy = 1 − ASy | (2) |
 | (3) |
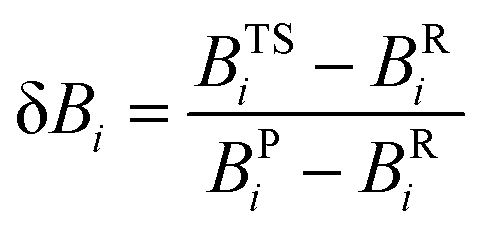 | (4) |
From the calculated δBi values, we can measure the degree of advancement of the transition state structures along the reaction paths. δBav is the average value (eqn (5)) and is defined as:
 | (5) |
The calculated total natural resonance theory (NRT) bond orders (natural bond orders, nbo), δBi, δBav, ASy, and Sy values are given in Table 4. A low synchronicity was found for the retro-ene decomposition reaction of compound 1 compared with those in compound 2 and compound 3. This fact could be justified by the bond orders of the C5⋯H9⋯N1 moiety in the transition state structure of compound 1 (Fig. 6). The examination of the bond orders of the C5⋯H9⋯N1 moiety in the transition state structure of compound 1 revealed that the C5⋯H9 bond had a significantly covalent character, while there was no considerable bond order for the H9⋯N1 bond. This fact may lead to an early transition state structure. Contrary to compound 1, we found high synchronicity indices for the retro-ene decomposition reactions of compounds 2 and 3. Importantly, the decrease of the C5⋯H9 bond orders and also the increase of the H9⋯N1 bond orders in the C5⋯H9⋯N1 moieties of the transition state structures of compounds 2 and 3 compared with that in compound 1 led to their greater synchronicity indices. Despite the low synchronicity found for the retro-ene decomposition reaction of compound 1, the intrinsic reaction coordinate (IRC) analysis asserted the one-step nature of this reaction.
8. Pauli exchange-type repulsions
Natural steric analysis, which states that steric exchange repulsion is the energy difference due to orbital orthogonalization, can be used to express the well-established physical picture of steric repulsions.49–53 All occupied orbitals effects are included in the Pauli exchange-type repulsion (or steric exchange energy) and therefore it typically contains contributions from covalent (intrabond) groups.NBO-MP2/6-311++G** analysis was used to calculate the steric repulsion contributions in the anti- and gauche-conformations and also in the transition state structures of the retro-ene reactions of 1–3. Based on the results obtained, Pauli exchange-type repulsion of the anti-conformation of compound 1 was found to be greater than that in its gauche-conformation, thus favoring the gauche-conformation. Contrary to the trend observed for compound 1, the exchange components of the gauche-conformations of compounds 2 and 3 were greater than those in their anti-conformations, thus favoring their corresponding anti-conformations. Accordingly, the exchange component tends to increase the anti-conformation preference compared to its gauche-conformation when going from compound 1 to compound 3. Based on the results obtained, the calculated total steric exchange energy differences between the anti- and gauche-conformations [Δ(TSEEanti − TSEEgauche)] decrease going from compound 1 to compound 3. Accordingly, the conformational preferences for the exchange component does not justify solely the conformational preferences in compounds 1–3 (Table 5).
It is worth noting that the Pauli exchange component values of the transition state structures of the retro-ene decomposition reactions of compounds 1–3 are greater than those in their corresponding anti- and gauche-conformations. Most importantly, the calculated total exchange energy differences between the transition state structures of the retro-ene decomposition reactions of compounds 1–3 correlate well with their corresponding Gibbs free energy differences, revealing that the variations of the exchange component have a significant impact on the barrier heights of these reactions (Fig. 2).
9. Impact of the electrostatic model associated with the dipole–dipole interactions on the conformational preferences and the retro-ene decomposition reactions of compounds 1–3
In the conformations or configurations of a molecule with larger dipole moments (μ), a larger polarizability may result from the local dipole moments repulsive interactions, therefore, it is acceptable that a conformation or configuration of a molecule with the largest resultant dipole moment (in the gas phase or in the nonpolar media)35 may possess the largest electrostatic energy compared to its structural isomers with smaller diploe moments.The calculated dipole moments for the gauche- and anti-conformations of compounds 1–3 and the transition state structures of their corresponding retro-ene decomposition reactions are given in Table 5. Based on the results obtained, the dipole moments of the gauche-conformations of compounds 1–3 are smaller than those in their corresponding anti-conformations, revealing that the electrostatic model associated with the dipole–dipole interaction is in favor of the gauche-conformations of compounds 1–3. It is worth noting that the electrostatic model associated with the dipole–dipole interactions, the Pauli exchange-type repulsions, and the hyperconjugative interactions are in favor of the gauche-conformation of compound 1. Since the results of this work and previously published data19,20 indicate that there is a small energy difference between the gauche- and anti-conformations of compound 1, we suggest a re-evaluation of the conformational properties of compound 1 by spectroscopic techniques (e.g., gas electron diffraction or microwave spectroscopy).
Importantly, the transition state structures of the retro-ene decomposition reactions of compounds 1–3 possess smaller dipole moments compared to their corresponding gauche-conformations. Based on the calculated dipole moments for the transition state and the gauche-conformations, the Δ[μ(TS) − μ(gauche)] parameters for compounds 1–3 possess negative values and slightly increase from compound 1 to compound 3, revealing that the rationalization of the retro-ene decomposition reactions solely in terms of the electrostatic model associated with the dipole–dipole interaction fails to fully account for compounds 1–3. This fact contradicts the conclusions proposed by some researchers who claimed that “the isomer with larger molecular dipole moment is the less stable one”.35
10. Kinetic parameters of the retro-ene decomposition reactions of compounds 1–3
The kinetic parameters of the retro-ene decomposition reactions of compounds 1–3 were estimated using the classical transition state theory (TST).79,80 The Eyring–Polanyi equation was used to estimate the temperature-dependent rate constant [k(T)] of the retro-ene decomposition reactions of compounds 1–3 (eqn (6)):
 | (6) |
| Ea = ΔH≠(T) + RT | (7) |
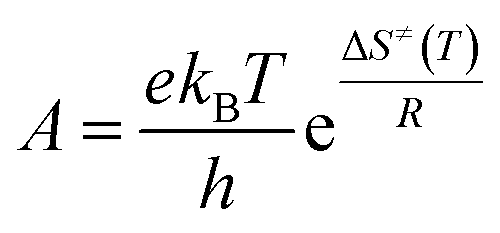 | (8) |
The activation energy (Ea) for the retro-ene decomposition reactions increases drastically from compound 1 to compound 2, but does not change significantly going from compound 2 to compound 3 (Table 6). Interestingly, the results obtained showed that the calculated Arrhenius A factors for the retro-ene decomposition reactions of compounds 1–3 are relatively the same and fall within the range 1011.5 to 1014.5 s−1, which is acceptable for such unimolecular reactions.81
| MP2/6-311++G** | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 300 K | |||||||||
| Ea | A | k | ΔH≠ | ΔS≠ | ΔG≠ | ΔH | ΔS | ΔG | |
| 1-gauche → [TS]≠ → 1-P | 31.38 | 3.07 × 1012 | 2.51 × 10−11 | 30.79 | −3.373 | 31.80 | −6.49 | 35.667 | −17.12 |
| 2-gauche → [TS]≠ → 2-P | 39.98 | 2.44 × 1012 | 9.45 × 10−18 | 39.39 | −3.829 | 40.53 | 20.25 | 34.088 | 10.09 |
| 3-gauche → [TS]≠ → 3-P | 39.67 | 2.17 × 1012 | 1.41 × 10−17 | 39.08 | −4.064 | 40.29 | 33.84 | 11.950 | 19.41 |
The calculated entropies of activation (ΔS≠) for the retro-ene decomposition reactions of compounds 1–3 were used in support of the reaction mechanisms occurring via a concerted six-membered cyclic transition state (Table 6). The calculated activation entropies (ΔS≠) of the retro-ene decomposition of compounds 1–3 lie between −7.00 and 7.00 cal mol−1 K−1, demonstrating that their calculated Arrhenius factors are normal. The ΔS≠ value of the retro-ene decomposition reactions decrease when going from compound 1 to compound 3. By considering the calculated ΔS≠ values of the retro-ene decomposition reactions of compounds 1–3, we can expect their transition state structures to have significantly greater rigidity than their corresponding initial states. Accordingly, the transition state structures of the retro-ene decomposition reactions of compounds 1–3 had much less freedom of vibrations than their corresponding initial states.
Conclusion
The results obtained at the MP2/6-311++G** and G3(MP2) levels of theory and the natural bond orbital (NBO) interpretations provided a reasonable picture from an energetic, structural, bonding, and stereoelectronic point of view for the conformational preferences in compounds 1–3 and their corresponding retro-ene decomposition reactions. The role and contributions of the hyperconjugative interactions on the conformations preferences of compounds 1–3 were assessed by the deletion of the overlapping orbitals [which are changed by the rotations around the M–CH2 bond [M = O (1), M = S (2), M = Se (3)] from the Fock matrices of the gauche- and anti-conformations. Based on the results obtained, the deletion of the hyperconjugative interactions mentioned above from the Fock matrices leads to an increase in the preference for the anti-conformation, demonstrating the significant impacts of the hyperconjugative interactions on the gauche-conformation preferences in compounds 1–3. The electrostatic model associated with the dipole–dipole interactions is in favor of the gauche-conformations of compounds 1–3, but does not justify the potential energy surfaces of the retro-ene decomposition reactions of compounds 1–3.Since the hyperconjugative interactions, the electrostatic model associated with the dipole–dipole interactions, and steric effects are in favor of the gauche-conformation of compound 1, no fact justifies the anti-conformation preference in compound 1. Accordingly, we claim that there is some doubt concerning the interpretations of the rotational spectra of compound 1. As experimental data (infrared spectroscopy)20 indicated the presence of two forms (gauche- and anti-conformations), we suggest a re-evaluation of the conformational and structural properties of compound 1 by gas electron diffraction or microwave spectroscopy to be carried out with more consciousness.
In accordance with the results of this work and the previously published theoretical data,20,21 compounds 1–3 were found to be stable in the gas phase and their corresponding cyanate → isocyanate unimolecular isomerization reactions could not take place at ambient temperature. Based on the results of this work, the retro-ene decomposition reactions of compounds 1–3 will occur earlier than their corresponding unimolecular cyanate → isocyanate unimolecular isomerization reactions. Importantly, the exchange component has a determining role on the retro-ene decomposition reactions of these compounds. The calculated potential energy surfaces and the advancements of the transition states (δBav) of the retro-ene decomposition reactions of compounds 1–3 are in agreement with the Hammond–Leffler postulate. The greater overall synchronicities of the retro-ene decomposition reactions of compounds 2 and 3 compared to that of compound 1 results from the greater attractive electrostatic interactions between the two adjacent S3–C4 and Se3–C4 atoms in compounds 2 and 3, respectively, in comparison to the smaller one (i.e., O3–C4) in compound 1.
Since the X3–C4 bonds break in the transition state structures of the retro-ene decomposition reactions of compounds 1–3, we decomposed the hyperconjugative generalized anomeric effects (HCGAEtotal) (which have impacts on the strengthening or weakening of the X3–C4 bonds) into two components. Most interestingly, the variations of Δ[HCGAE(X3–C4 weakening) − HCGAE(X3–C4 strengthening)] parameters correlated well with the variations of the retro-ene decomposition reactions barrier heights when going from compound 1 to compound 3. Based on this finding, this procedure could be an applicable and suitable approach to investigate the feasibility of various chemical reactions (especially unimolecular reactions).
References
- J. Gonda, M. Martinkova, J. Raschmanova and E. Balentova, Tetrahedron: Asymmetry, 2006, 17, 1875 CrossRef CAS.
- J. Burmeister, Coord. Chem. Rev., 1990, 105, 77 CrossRef CAS.
- T. Castanheiro, J. Suffert, M. Donnard and M. Gulea, Chem. Soc. Rev., 2016, 45, 494 RSC.
- C. Johansen, P. Falholt and L. Gram, Appl. Environ. Microbiol., 1997, 63, 3724 CAS.
- L. Bjorck, C. Rosen, V. Marshall and B. Reiter, Appl. Microbiol., 1975, 30, 199 CAS.
- B. Reiter, V. M. Marshall, L. Bjorck and C. G. Rosen, Infect. Immun., 1976, 13, 800 CAS.
- D. Plano, Y. Baquedano, D. Moreno-Mateos, M. Font, A. Jiménez-Ruiz, J. A. Palop and C. Sanmartín, Eur. J. Med. Chem., 2011, 46, 3315 CrossRef CAS PubMed.
- E. Riedle and H. Mayr, Chem.–Eur. J., 2008, 14, 3866 CrossRef PubMed.
- D. A. Ben-Efraim, in The Chemistry of Cyanates and Their Thio Derivatives, ed. S. Patai, Wiley, Chichester, 1977 Search PubMed.
- S. S. Al-Juaid, A. A. K. Al-Nasr, G. A. Ayoko, C. Eaborn and P. Hitchcock, J. Organomet. Chem., 1995, 488, 155 CrossRef CAS.
- C. Eaborn, P. D. Lickiss, G. Markuina-Chidsey and E. Y. Thorli, J. Chem. Soc., Chem. Commun., 1982, 1326 RSC.
- D. Martin, Tetrahedron Lett., 1964, 39, 2829 CrossRef.
- A. Holm and E. Huge-Jensen, Acta Chem. Scand., Ser. B, 1974, 28, 705 CrossRef CAS.
- H. Fischer, S. Zeuner, K. Ackermann and U. Schubert, J. Organomet. Chem., 1984, 263, 201 CrossRef CAS.
- P. Reich and D. Martin, Chem. Ber., 1965, 98, 2063 CrossRef CAS.
- N. Groving and A. Holm, Acta Chem. Scand., 1965, 19, 443 CrossRef CAS.
- K. A. Jensen, A. Holm and C. Wentrup, Acta Chem. Scand., 1966, 20, 2107 CrossRef CAS.
- T. Sakaizumi, H. Mure, O. Ohashi and I. Yamaguchi, J. Mol. Spectrosc., 1989, 138, 375 CrossRef CAS.
- J. Urban, A. Nowek, R. Venkatraman, P. Babinec and J. Leszczynski, Struct. Chem., 1998, 9, 161 CrossRef CAS.
- T. Pasinszki, B. Havasi and A. Kovács, J. Phys. Chem. A, 2003, 107, 1720 CrossRef CAS.
- V. I. Faustov, E. G. Baskir and A. A. Biryukov, Russ. Chem. Bull., Int. Ed., 2003, 52, 2328 CrossRef CAS.
- R. P. Hirschmann, R. N. Kniseley and V. A. Fassel, Spectrochim. Acta, 1964, 20, 809 CrossRef CAS.
- A. Bjørseth and K. M. Marstokk, J. Mol. Struct., 1972, 11, 15 CrossRef.
- O. H. Ellestad and T. Torgrimsen, J. Mol. Struct., 1972, 12, 79 CrossRef CAS.
- J. R. Durig, J. F. Sullivan and H. L. Heusel, J. Phys. Chem., 1984, 88, 374 CrossRef CAS.
- G. O. Braathen and A. Gatial, Spectrochim. Acta, Part A, 1986, 42, 615 CrossRef.
- T. Sakaizumi and T. Itakura, J. Mol. Spectrosc., 1994, 163, 1 CrossRef CAS.
- P. A. Petillo and L. E. Lerner, in The Anomeric Effect and Associated Steroelectronic Effects, ed. G. R. J. Thacher, American Chemical Society, ACS Symposium Series No. 539, Washington, DC, 1993 Search PubMed.
- A. Behrouz and D. Nori-Shargh, Aust. J. Chem., 2016, 70, 61 CrossRef.
- J. Kirby, The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, Springer Verlag, New York, NY, 1983 Search PubMed.
- P. Deslongchamps, Stereoelectronic Effects in Organic Chemistry, Wiley, New York, NY, 1983 Search PubMed.
- E. Juaristi and G. Cuevas, The Anomeric Effect, CRC Press Inc., Boca Raton, FL, 1995 Search PubMed.
- C. J. Cramer, J. Org. Chem., 1992, 57, 7034 CrossRef CAS.
- C. J. Cramer, D. G. Truhlar and A. D. French, Carbohydr. Res., 1997, 298, 1 CrossRef CAS.
- C. L. Perrin, K. B. Armstrong and M. A. Fabian, J. Am. Chem. Soc., 1994, 116, 715 CrossRef CAS.
- A. Lesarri, A. Vega-Toribio, R. D. Suenram, D. J. Brugh, D. Nori-Shargh, J. E. Boggs and J.-U. Grabow, Phys. Chem. Chem. Phys., 2011, 13, 6610 RSC.
- A. Vila and R. A. Mosquera, J. Comput. Chem., 2007, 28, 1516 CrossRef CAS PubMed.
- S. Hosseini and D. Nori-Shargh, Can. J. Chem., 2016, 94, 176 CrossRef CAS.
- E. Juaristi and R. Notario, J. Org. Chem., 2016, 81, 192 CrossRef PubMed.
- J. P. Praly and R. U. Lemieux, Can. J. Chem., 1987, 65, 213 CrossRef CAS.
- D. Nori-Shargh, S. N. Mousavi and H. Kayi, J. Mol. Model., 2014, 20, 2249 CrossRef PubMed.
- M. P. Freitas, Org. Biomol. Chem., 2013, 11, 2885 CAS.
- T. M. Barbosa, R. V. Viesser, R. J. Abraham, R. Rittner and C. F. Tormena, RSC Adv., 2015, 5, 35412 RSC.
- P. Ghanbarpoura and D. Nori-Shargh, RSC Adv., 2016, 6, 46406 RSC.
- G. F. Bauerfeldt, T. M. Cardozo, M. S. Pereira and C. O. da Silva, Org. Biomol. Chem., 2013, 11, 299 CAS.
- N. Hasanzadeh, D. Nori-Shargh, M. Farzipour and B. Ahmadi, Org. Biomol. Chem., 2015, 13, 6965 CAS.
- D. Nori-Shargh, S. N. Mousavi, R. Tale and H. Yahyaei, Struct. Chem., 2016, 27, 1753 CrossRef CAS.
- I. V. Alabugin, J. Org. Chem., 2000, 65, 3910 CrossRef CAS PubMed.
- J. K. Badenhoop and F. Weinhold, J. Chem. Phys., 1997, 107, 5406 CrossRef CAS.
- J. K. Badenhoop and F. Weinhold, J. Chem. Phys., 1997, 107, 5422 CrossRef CAS.
- J. K. Badenhoop and F. Weinhold, Int. J. Quantum Chem., 1999, 72, 269 CrossRef CAS.
- V. F. Weisskopf, Science, 1975, 187, 605 CAS.
- P. A. Christiansen and W. E. Palke, J. Chem. Phys., 1977, 67, 57 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern, K. Raghavachari, V. Rassolov and J. A. Pople, J. Chem. Phys., 1999, 110, 4703 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- D. Cremer, in Encyclopedia of Computational Chemistry, ed. P. V. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A. Kollma, H. F. Schaefer III and P. R., Schreiner, John Wiley, Chichester, 1998 Search PubMed.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS.
- P. J. Hay, J. Chem. Phys., 1977, 66, 4377 CrossRef CAS.
- R. C. Binning Jr and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206 CrossRef.
- L. A. Curtiss, M. P. McGrath, J. P. Blandeau, N. E. Davis, R. C. Binning Jr and L. Radom, J. Chem. Phys., 1995, 103, 6104 CrossRef CAS.
- M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Su, S. J. Nguyen, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, Advances in electronic structure theory: GAMESS a decade later, in Theory and Applications of Computational Chemistry, the first 40 years, C. E. Dykstra, G. Frenking, K. S. Lim and G. E. Scusaria, Amsterdam, Elsevier, 2005 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO Version 5.G., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2004 Search PubMed.
- N. D. Epiotis, R. L. Yates, R. J. Larson, C. R. Kirmayer and F. Bernardi, J. Am. Chem. Soc., 1977, 99, 8379 CrossRef CAS.
- M. J. S. Dewar, The Molecular Orbital Theory of Organic Chemistry, McGraw-Hill, New York, 1969 Search PubMed.
- W. T. Borden, Modern Molecular Orbital Theory for Organic Chemists, Prentice-Hall, Englewood Cliffs, NJ, 1975 Search PubMed.
- M. Wolfsberg and L. Helmholz, J. Chem. Phys., 1952, 20, 837 CrossRef CAS.
- D. D. Radtke and R. F. Fenske, J. Am. Chem. Soc., 1967, 89, 2292 CrossRef CAS.
- F. Weinhold, in Encyclopedia of Computational Chemistry, ed. P. V. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A. Kollman, H. F. Schaefer III and P. R. Schreiner, Wiley, Chichester, U.K., 1998 Search PubMed.
- A. Moyano, M. A. Pericas and E. Valenti, J. Org. Chem., 1989, 54, 573 CrossRef CAS.
- O. N. Faza, C. S. López and I. Fernández, J. Org. Chem., 2013, 78, 5669 CrossRef CAS PubMed.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, J. Org. Chem., 2003, 68, 3884 CrossRef CAS PubMed.
- E. Chamorro, J. Quijano, R. Notario, C. Sánchez, L. A. León and G. Chuchani, Int. J. Quantum Chem., 2003, 91, 618 CrossRef CAS.
- K. J. Glasstone, K. J. Laidler and H. Eyring, The theory of rate processes, McGraw-Hill, New York, 1941 Search PubMed.
- S. W. Benson, The foundations of chemical kinetics, McGraw-Hill, New York, 1969 Search PubMed.
- B. G. Gowenlock, Q. Rev., Chem. Soc., 1960, 14, 133 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00520b |
| This journal is © The Royal Society of Chemistry 2017 |