
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of the binding mode of the PET tracer [18F]ASEM to a chimera structure of the α7 nicotinic acetylcholine receptor†
Guanglin Kuang a,
Yang Zhoua,
Rongfeng Zoua,
Christer Halldinb,
Agneta Nordbergc,
Bengt Långströmd,
Hans Ågrenae and
Yaoquan Tu*a
a,
Yang Zhoua,
Rongfeng Zoua,
Christer Halldinb,
Agneta Nordbergc,
Bengt Långströmd,
Hans Ågrenae and
Yaoquan Tu*a
aDivision of Theoretical Chemistry and Biology, School of Biotechnology, Royal Institute of Technology (KTH), AlbaNova University Center, S-106 91, Stockholm, Sweden. E-mail: yaoquan@kth.se
bKarolinska Institutet, Department of Clinical Neuroscience, Centre for Psychiatric Research, 171 76, Stockholm, Sweden
cDepartment of Neurobiology, Care Sciences and Society, Center of Alzheimer Research, Translational Alzheimer Neurobiology, Karolinska University Hospital, Huddinge, S-141 86, Stockholm, Sweden
dDepartment of Chemistry, Uppsala University, 751 23 Uppsala, Sweden
eInstitute of Nanotechnology, Spectroscopy and Quantum Chemistry, Siberian Federal University, Svobodny pr. 79, 660041 Krasnoyarsk, Russia
First published on 4th April 2017
Abstract
The α7 nicotinic acetylcholine receptor (α7-nAChR) is assumed to be implicated in a variety of neurological disorders, such as schizophrenia and Alzheimer's disease (AD). The progress of these disorders can be studied through imaging α7-nAChR with positron emission tomography (PET). [18F]ASEM is a novel and potent α7-nAChR PET radioligand showing great promise in recent tests. However, the mechanism of the molecular interaction between [18F]ASEM and α7-nAChR is still unclear. In this paper, the binding profile of [18F]ASEM to a chimera structure of α7-nAChR was investigated with molecular docking, molecular dynamics, and metadynamics simulation methods. We found that [18F]ASEM binds at the same site as the crystallized agonist epibatidine but with a different binding mode. The dibenzo[b,d]thiophene ring has a different orientation compared to the pyridine ring of epibatidine and has van der Waals interactions with residues from loop C on one side and π–π stacking interaction with Trp53 on the other side. The conformation of Trp53 was found to have a great impact on the binding of [18F]ASEM. Six binding modes in terms of the side chain dihedral angles χ1 and χ2 of Trp53 were discovered by metadynamics simulation. In the most stable binding mode, Trp53 adopts a different conformation from that in the crystalline structure and has a rather favorable π–π stacking interaction with [18F]ASEM. We believe that these discoveries can be valuable for the development of novel PET radioligands.
Introduction
Nicotinic acetylcholine receptors (nAChRs) are a family of ligand-gated ion channels that are ubiquitously distributed in the central nervous system (CNS), peripheral nervous system and some non-neuronal cells.1 In the human CNS, nAChRs are pentamers composed of various combinations of α and β subunits, with the homopentamer α7-nAChR and heteropentamer α4β2-nAChR being most abundant.1 α7-nAChR is mainly expressed in the hippocampus and cortex regions and plays a critical role in some important physiological functions such as cognition and memory.2 Abundant clinical studies have found that α7-nAChR is implicated in a variety of neurological disorders such as schizophrenia, Alzheimer's disease (AD) and traumatic brain injury.3–6 Post-mortem studies of patients with schizophrenia and AD have demonstrated a reduced density of α7-nAChR in the brain,7,8 which further supports the role of α7-nAChR in these neuropsychiatric diseases. Therefore, α7-nAChR is believed to be an important target to treat these diseases. Many agonists and positive allosteric modulators (PAM) of α7-nAChR are currently under development worldwide in both industry and academia.9–12The mechanism and progress of CNS diseases related to α7-nAChR can be studied with positron emission tomography (PET), a non-invasive method to visualize and quantify receptors in vivo.13,14 PET studies can also facilitate the discovery of drugs targeting α7-nAChR by measuring receptor occupancies and dose–response relationships. In the past decade, several PET radioligands have been developed for the imaging of α7-nAChR, such as [18F]NS14492 (ref. 15), [18F]NS10743 (ref. 16) and [11C]CHIBA-1001 (ref. 17 and 18). However, most of these PET tracers failed in in vivo imaging applications in the human brain due to factors like low affinity, low specificity or low BPND (non-displaceable binding potential).19
[18F]ASEM (3-(1,4-diazabicyclo[3.2.2]nonan-4-yl)-6-[18F]fluorodibenzo[b,d]thiophene 5,5-dioxide, also known as [18F]JHU82132) (Fig. 1), developed by Gao et al.,20 is a PET tracer used for in vivo quantification of α7-nAChR in human CNS.21 It is currently the most promising α7-nAChR PET tracer with excellent affinity, specificity, and BPND values.19–22 [18F]ASEM has a similar framework (a diazobicyclic group connected to an aromatic group) compared with epibatidine (Fig. 1), which is an alkaloid agonist with high affinity to α7-nAChR. However, [18F]ASEM is an antagonist with high affinity and specificity for α7-nAChR. This difference is most probably due to the bulkier aromatic ring of [18F]ASEM, making it adopt a different binding mode with the receptor, which in turn is responsible for the significantly different functions. Up to date, the crystal structure of α7-nAChR with [18F]ASEM is not available. Therefore, it would be helpful to investigate the binding mode of [18F]ASEM with α7-nAChR using molecular modeling methods.
 | ||
| Fig. 1 Structures of epibatidine and [18F]ASEM. | ||
Currently, our understanding of the structure of α7-nAChR is still very limited. Until recently the structural information of α7-nAChR was inferred from the electron microscopic structure of the muscle type nAChR from Torpedo marmorata23 or the acetylcholine binding protein (AChBP) from snails Lymnaea stagnalis,24–26 Aplysia californica,27,28 and Bulinus truncatus.29 These structures provide valuable information about the general configuration and organization of α7-nAChR. However, they have limited usage in the investigation of the function of α7-nAChR, especially ligand binding modes, due to the low sequence identities with α7-nAChR.30 In 2011, a chimera structure of Lys-AChBP and the extracellular domain of the human α7-nAChR with the agonist epibatidine was determined.31 This chimera structure (termed as α7-AChBP) has a sequence identity as high as 64% with the native α7-nAChR and is by far one of the best structure models for α7-nAChR. More importantly, the ligand binding site and surrounding areas of this receptor chimera are lined by α7-nAChR residues,31 making the chimera structure a good model to study the binding profile of α7-nAChR with its ligands. Sequence alignment between α7-nAChR and α7-AChBP shows that binding site residues are almost identical (Fig. S1†). Understanding of the binding mode of [18F]ASEM with α7-nAChR can provide us with insight into the chemical and biological basis of its ideal PET properties and facilitate the development and optimization of future PET radioligands and therapeutic drugs.
In this work, the binding mode of [18F]ASEM with α7-nAChR was first obtained with the induced fit docking (IFD) procedure which allows conformational adjustments of the binding site residues upon ligand binding. The complex structure from IFD was equilibrated with molecular dynamics (MD) simulations. A conserved residue Trp53 was found to be important for the binding of [18F]ASEM and its conformation was then sampled with metadynamics simulations. Below we will discuss the computational techniques implemented as well as the results obtained.
Methods
Induced fit docking
In a standard molecular docking study, the receptor is held rigid, and the ligand can change its position and conformation freely. However, this procedure is problematic when the ligand to be docked is rather different from the crystallized one in shape and size. In reality, the receptor structure will undergo side-chain or backbone movements upon ligand binding to conform to the shape of the ligand, a process known as induced fit.32 The induced fit docking (IFD) workflow of Schrödinger implements this idea through a combination of Glide and Prime jobs, which accounts for the conformational changes of the ligand and receptor, respectively.33 In this work, the structure of the α7-AChBP chimera (PDB code: 3SQ6)31 was used as the protein target. The protein residues within 5 Å of the ligand are optimized in IFD. The α7-AChBP/ASEM complex with the most favorable binding energy was chosen for subsequent analysis. The radionuclide fluorine-18 of ASEM, which is used in PET studies, is not indicated hereafter unless otherwise specified, as radiation does not affect the binding with α7-AChBP.Molecular dynamics simulation
The protonation states of the amino acids of α7-AChBP were determined with the PROPKA module of Schrödinger.34 According to the calculation of PROPKA, all the acidic and basic residues, except histidines, were predicted to adopt the charged states. For histidines, the delta nitrogen of His61 and His112 and the epsilon nitrogen of His148 were protonated, while the two nitrogen atoms of His147 were assigned to be fully protonated. The docked complex was subjected to molecular dynamics (MD) simulations with Gromacs 5.0.4 (ref. 35). The Amber ff99SB-ildn forced field36 was used for the protein and the general Amber force field (GAFF) for ASEM. The partial charges of ASEM were calculated through the restrained electrostatic potential (RESP) fitting procedure using Gaussian (HF/6-31G*)37 and Antechamber.38,39 The complex was solvated in a cubic box with 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 169 TIP3P40 water molecules to have a 12 Å water layer. 140 Na+ and 138 Cl− were then added to neutralize the system and make the ion concentration 0.15 M. The system thus obtained was first minimized with the steepest descent algorithm. Thereafter, it was subjected to temperature equilibration in an isothermal–isochoric (NVT) ensemble at 310 K for 200 ps and density equilibration in an isothermal–isobaric (NPT) ensemble at 310 K and 1 atm for 500 ps. The heavy atoms of the protein and ligand were restrained with a harmonic potential with a force constant of 1000 kJ mol−1 nm−2 in the equilibration steps. The equilibrated system was then submitted to an MD simulation for 100 ns. In the equilibration and production MD simulations, a time step of 2 fs was used and the LINCS41 algorithm was applied to bonds involving hydrogen atoms. A 10 Å cut-off was set for van der Waals and short-range electrostatic interactions and the Particle Mesh Ewald (PME) method42 was used to compute the long-range electrostatic interactions. Three independent MD simulations were implemented.
169 TIP3P40 water molecules to have a 12 Å water layer. 140 Na+ and 138 Cl− were then added to neutralize the system and make the ion concentration 0.15 M. The system thus obtained was first minimized with the steepest descent algorithm. Thereafter, it was subjected to temperature equilibration in an isothermal–isochoric (NVT) ensemble at 310 K for 200 ps and density equilibration in an isothermal–isobaric (NPT) ensemble at 310 K and 1 atm for 500 ps. The heavy atoms of the protein and ligand were restrained with a harmonic potential with a force constant of 1000 kJ mol−1 nm−2 in the equilibration steps. The equilibrated system was then submitted to an MD simulation for 100 ns. In the equilibration and production MD simulations, a time step of 2 fs was used and the LINCS41 algorithm was applied to bonds involving hydrogen atoms. A 10 Å cut-off was set for van der Waals and short-range electrostatic interactions and the Particle Mesh Ewald (PME) method42 was used to compute the long-range electrostatic interactions. Three independent MD simulations were implemented.
Metadynamics simulation
Our MD simulations revealed that Trp53 is an important residue whose conformation has a great impact on the binding of ASEM. In this work, metadynamics simulations were further carried out to sample the conformation of Trp53 using its sidechain dihedral angles χ1 and χ2 as the collective variables (Fig. 2). Well-tempered metadynamics43 implemented in Gromacs5.0.4-plumed2.1.2 (ref. 44) was used for the simulation. The initial Gaussian height was 0.5 kcal mol−1, with a bias factor of 10. The Gaussian widths were 0.05 rad and 0.08 rad for χ1 and χ2, respectively. The time step for metadynamics simulations is 2 fs, and the bias potential was added every 500 steps. The initial Gaussian height was set to be 0.5 kcal mol−1 and the simulations were run for 3 μs for convergence, as shown by the Gaussian HILLS height and the distribution of the collective variables (ESI Fig. S2a and b†). The criteria for judging the convergence of metadynamics simulations have been reported by Sun et al.45,46 | ||
| Fig. 2 Illustration of the side chain dihedral angles χ1 and χ2 of Trp53 which were selected as the collective variables in the metadynamics simulations. | ||
Results and discussion
Binding mode of ASEM with α7-AChBP from induced fit docking
Like the crystallized ligand epibatidine, ASEM has a diazobicyclic head group (Fig. 1) and is protonated under physiological conditions. However, the diazobicyclic head group of ASEM is bulkier than the counterpart of epibatidine (Fig. 1), which should have an impact on its binding with the receptor. Besides, the dibenzo[b,d]thiophene ring of ASEM is also much bigger than the pyridine ring of epibatidine. Actually, these two structural differences make epibatidine and ASEM have different functions, namely epibatidine is an agonist and ASEM an antagonist. This is consistent with the general knowledge that α7-nAChR antagonists such as MLA and α-bungarotoxin tend to be much bulkier than the agonists such as nicotine and acetylcholine.11In a standard docking procedure where the receptor was held rigid, ASEM could not be docked properly, with a very unfavorable docking score (−4.53 kcal mol−1), which is most probably due to the small size of the binding pocket occupied by epibatidine. However, with the induced fit docking (IFD) procedure, ASEM could be docked to the binding site with a much more favorable docking score (−10.8 kcal mol−1). This is straightforward as ASEM is bulkier and would need more space for binding. The residues relaxed most significantly are Trp53, Tyr91, Trp145, Tyr184, Cys186, Cys187, and Tyr191 (Fig. 3).
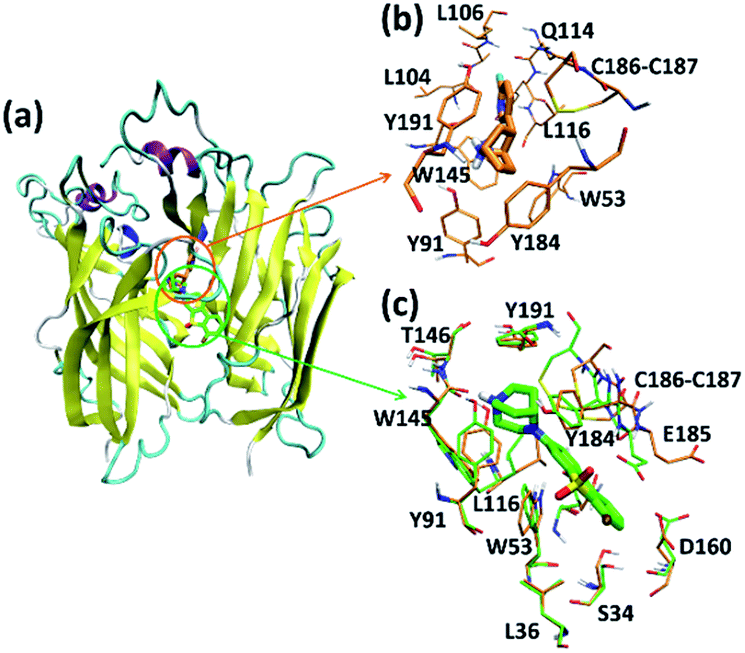 | ||
| Fig. 3 Binding modes of epibatidine and ASEM with α7-AChBP. (a) General view of the binding modes of epibatidine and ASEM. (b) Detailed binding mode of epibatidine. (c) Detailed binding mode of ASEM. Color scheme: oxygen in red, hydrogen in white, nitrogen in blue, fluorine in ochre, chlorine in cyan, sulfur in yellow, carbon (crystal structure) in green and carbon (IFD structure) in green. Non-polar hydrogens are not displayed for clarity. | ||
In the complex structures of epibatidine and ASEM with α7-AChBP, the (di)azobicyclic head groups occupy the same position and have similar interactions with the surrounding residues (Fig. 3). The tip nitrogen (N1, pKa ≈ 9.6) of ASEM is protonated under physiological conditions and has cation-π interactions with the aromatic rings of Tyr91, Trp145, Tyr184, and Try191. These cation-π interactions are believed to be important for the affinity of α7-nAChR ligands.31 The protonated nitrogen also forms a hydrogen bond with the backbone oxygen of Trp145 (Fig. 3). Besides, the diazobicyclic group has extensive van der Waals interactions with the side chains of Tyr91, Trp145, Tyr184, and Try191. Glide docking score decomposition of residues around the binding site shows that van der Waals interactions from Tyr91, Trp145, and Tyr191 have a major contribution to the docking score (Table S2†), which helps to stabilize ASEM in the binding site.
The most significant difference between the binding modes of epibatidine and ASEM was seen in the aromatic tail part (Fig. 3). For epibatidine, the chloro-pyridine ring lies in the cavity formed by Leu106, Gln114, and Leu116 and has van der Waals or hydrophobic interactions with these residues. Besides, the chlorine atom is thought to have halogen-bond interaction with the backbone oxygen atom of Gln114, which also supports the binding of epibatidine.31 However, for ASEM, the dibenzo[b,d]thiophene ring is too big to fit into the place originally occupied by the pyridine ring of epibatidine. As a result, it adopts a different orientation and lies in the cavity on the other side which is formed by Ser34, Leu36, Trp53, Asp160, Gly163, Tyr184, Glu185, Cys186, and Cys187 (Fig. 3). The dibenzo[b,d]thiophene ring is clenched by van der Waals interactions with Glu185, Cys186, and Cys187 from loop C (residues 180–193) on one side and π–π stacking interaction with Trp53 from the complementary subunit on the other side (Fig. 3). Ser34, Leu116, and Asp160 also have some contact with the dibenzo[b,d]thiophene ring. The fluorine and oxygen atoms of ASEM point towards the solvent and do not have much interaction with surrounding residues. With induced fit docking we managed to produce a plausible docking mode of ASEM with α7-AChBP. However, only limited movements of the residues close to the ligand are possible in an induced fit docking procedure, and therefore, it is necessary to analyze the dynamic behavior of the α7-AChBP/ASEM complex in MD simulations where the protein and ligand structures can be fully relaxed.
Three states of Trp53 obtained by molecular dynamics simulations
In order to further relax the α7-AChBP/ASEM complex obtained from the induced fit docking procedure, the complex structure was submitted to 100 ns MD simulations. Three independent MD runs were carried out with different initial velocities in order to account for the chance of randomness.In all three MD simulations, ASEM remained in the binding pocket and did not drift away, which means that the induced fit docking procedure produced a reasonable initial complex structure of ASEM and α7-AChBP. Especially, the diazabicyclic head group bound very stably and was packed tightly by Tyr91, Trp145, Tyr184, and Tyr191. However, the tail part, namely the dibenzo[b,d]thiophene ring, has a different behavior in the MD simulations. In the initial state, this aromatic ring has van der Waals interaction with Glu185, Cys186, and Cys187 on one side and π–π stacking interaction with Trp53 on the other side (Fig. 3). Trp53 lies on the complementary face of the binding site and is the only residue that is able to form π–π stacking interaction with the dibenzo[b,d]thiophene ring of ASEM. Therefore, its conformation should have a great impact on the binding of ASEM. In this work, we found that Trp53 reached three different final states (states 1–3, Fig. 4 and 5) in the MD simulations, which reflects the flexibility of Trp53 in the binding site. The conformation of Trp53 is determined by the interactions with ASEM. In state 1, Trp53 has a similar conformation to that in the initial state, which is roughly parallel to ASEM (Fig. 3b and 4a). However, the indole ring and the dibenzo[b,d]thiophene ring are not strictly parallel but have a crossing angle of about 30 degrees, which is possibly stabilized by the hydrogen bond formed between the polar hydrogen of the indole ring and the backbone carbonyl of Leu116 (Fig. 4a). This packing mode was rather stable in simulation 1 but was changed in simulations 2 and 3 after about 25 ns. As indicated by the χ1 and χ2 plots in Fig. 5b and c, Trp53 was stabilized in two different conformations (states 2 and 3) in simulations 2 and 3. In state 2, Trp53 rotates by about 60 degrees to the left side. As a result, it points away from Leu116 and towards Tyr91 instead. The hydrogen bond interaction between Trp53 and Leu116 is broken, and a new water-mediated hydrogen bond is formed between Trp53 and Tyr91. ASEM has favorable interactions with Trp53 in this state and, therefore, adopts a similar conformation as in state 1 (Fig. 4a and b). In state 3, Trp53 rotates by about 90 degrees towards ASEM and points towards the disulfide bond formed between Cys186 and Cys187 (Fig. 4c). The flipping of the indole ring also causes the dibenzo[b,d]thiophene ring to rotate by about 60 degrees. Consequently, the dibenzo[b,d]thiophene ring has weaker interactions with the disulfide bond than in states 1 and 2 and turns to contact Leu36 with hydrophobic interactions.
 | ||
| Fig. 4 Binding modes in states 1–3 (a–c) observed in three independent MD simulations. ASEM and Trp53 are shown in thick stick mode while other residues in thin stick mode. Hydrogen bonds are represented by black dotted lines. Color scheme: oxygen in red, hydrogen in white, nitrogen in blue, fluorine in ochre, sulphur in yellow, carbon (ASEM) in green, carbon (principal face) in cyan and carbon (complementary face) in pink. Non-polar hydrogens are not shown for clarity. | ||
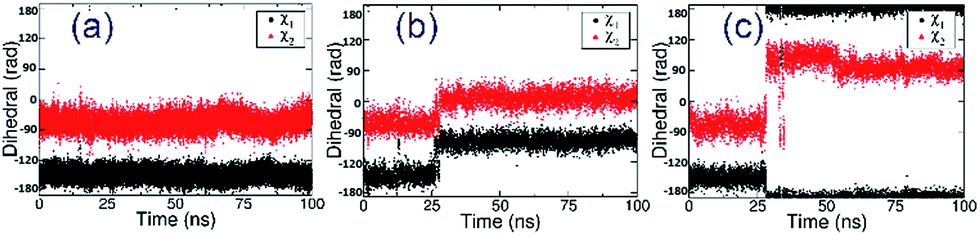 | ||
| Fig. 5 Evolution of the side chain dihedral angles χ1 and χ2 of Trp53 in three independent MD simulations (a–c) starting from the α7-AChBP/ASEM complex structure obtained from induced fit docking. | ||
The transitions from state 1 to state 2 or state 3 observed in the MD simulations suggest that the energy barriers between these states are not high and can be overcome by the thermodynamic fluctuations in unbiased MD simulations. On the other hand, as shown in ESI Table S1,† states 2 and 3 were very stable in unbiased MD simulations and did not transit to other states, which suggests that the system was trapped in these two states and was unable to transit to other minima. To investigate the free energy profile of the conformational changes of Trp53, to explore other stable states and to find the global minimum, metadynamics simulations were implemented using the side chain dihedral angles χ1 and χ2 of Trp53 as the collective variables.
Free energy surface of Trp53 obtained with metadynamics simulations
Metadynamics simulation is an advanced method to enhance conformational sampling in MD simulations and can be used to construct the free energy surface (FES) from a few selected collective variables.47 In this work, metadynamics simulations were used to enhance the sampling of χ1 and χ2 of Trp53 and to construct the related FES by adding history-dependent biased potentials to the system. As shown in Fig. 6, states 1–3 can be clearly identified in the constructed FES. States 2 and 3 are energetically more favorable than state 1 with moderate energy barriers (3–5 kcal mol−1, from state 1), which explains that state 1 can transit to state 2 or state 3 in unbiased MD simulations. However, the energy barriers from state 2 or state 3 back to state 1 are much higher (>6 kcal mol−1), which is the reason why state 2 and state 3 were trapped in unbiased MD simulations. Besides, there are three more states (states 4–6) in the FES, which were not observed in the unbiased MD simulations starting from initial IFD complex structure. Moreover, state 5 is the global minimum, which is separated from states 1–3 with rather high energy barriers (>10 kcal mol−1) (Fig. 6). Therefore, the system did not arrive at the global minimum in the initial MD simulations. In these states, ASEM has similar binding modes, with the diazabicyclic head group tightly fixed by Tyr91, Trp145, Tyr184, and Tyr191 (Fig. 7). The most obvious difference is the interaction pattern of the dibenzo[b,d]thiophene ring of ASEM with the indole ring of Trp53. The stability of each state was examined by three independent 100 ns MD simulations (ESI Table S1†).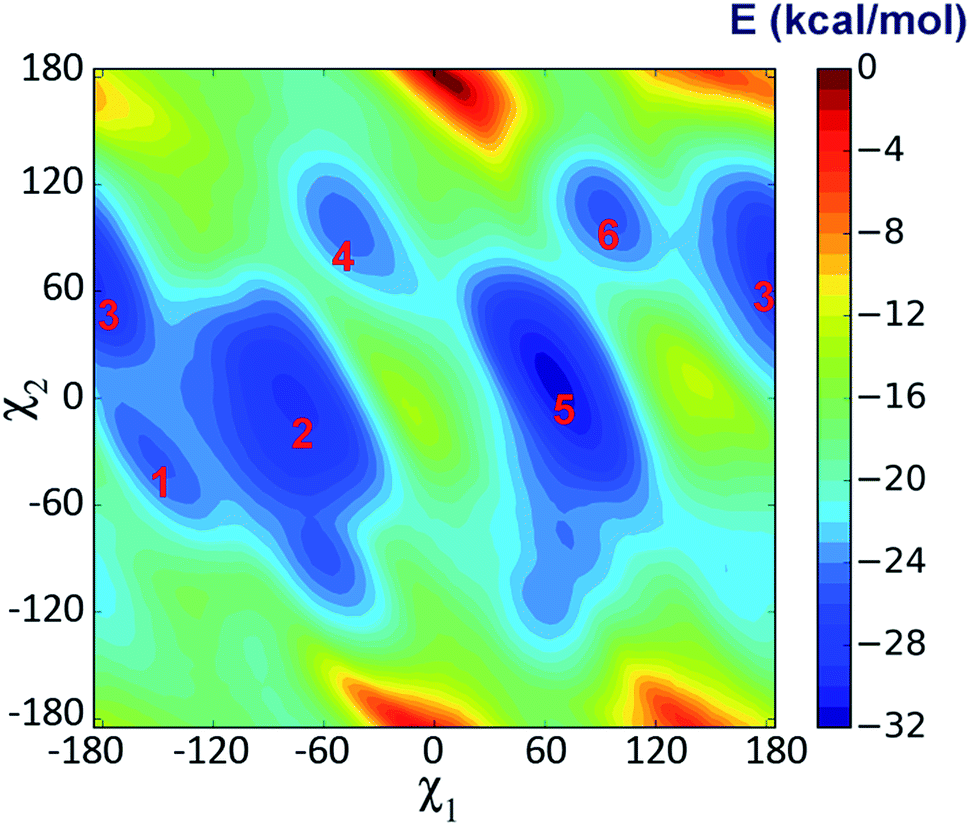 | ||
| Fig. 6 Free energy surface (FES) with respect to the side chain dihedral angles χ1 and χ2 of Trp53 obtained from metadynamics simulation. | ||
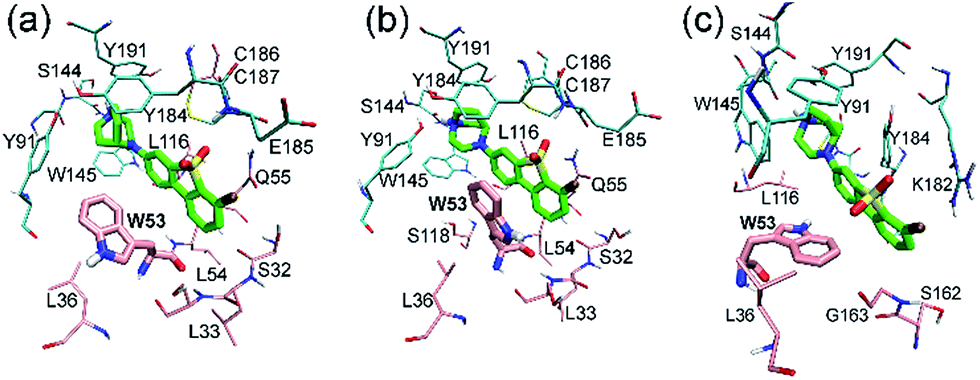 | ||
| Fig. 7 Binding modes of states 4–6 (a–c) discovered by metadynamics simulation. ASEM and Trp53 are shown in thick stick mode while other residues in thin stick mode. Color scheme: oxygen in red, hydrogen in white, nitrogen in blue, fluorine in ochre, sulfur in yellow, carbon (ASEM) in green, carbon (principal face) in cyan and carbon (complementary face) in pink. Non-polar hydrogens are not shown for clarity. | ||
In state 4, the indole ring of Trp53 adopts an edge-to-face π–π stacking mode with the dibenzo[b,d]thiophene ring of ASEM. In the FES shown in Fig. 6, state 4 has a higher energy than states 1, 2, 3, and 5, where parallel face-to-face π–π stacking was seen. This result reflects that edge-to-face π–π stacking is energetically less favorable than face-to-face π–π stacking in solution, possibly due to fewer contact areas. State 4 is an unstable state and can transit to state 5 at the early stage of unbiased MD simulations (ESI Table S1†). In state 5, Trp53 is rotated by about 90 degrees upwards and has face-to-face π–π stacking interactions with ASEM. Besides, it is obvious from Fig. 4 and 7 that the π–π stacking interaction in state 5 is more favorable than that in all other states as the indole ring and the dibenzo[b,d]thiophene ring are in perfect parallel orientation. The results of docking score and MM/GBSA decomposition, which are common methods to estimate the contributions of individual residues,48,49 also show that Trp53 has very favorable interactions with ASEM in state 5 (Tables S2 and S4†). As shown in Fig. 6, state 5 has the lowest energy and, therefore, corresponds to the global minimum, which means that ASEM most probably adopts this binding mode with α7-AChBP in reality. The Glide docking score (an estimation of binding free energy using empirical methods)50 of ASEM is −13.46 kcal mol−1 in state 5, which is consistent with the nanomolar binding affinity of ASEM in the experiment.20 This docking score is much more favorable than the rigid docking score (−4.53 kcal mol−1) and the induced fit docking score (−10.8 kcal mol−1) mentioned above, meaning that the complex structure in state 5 is much more reasonable than the counterparts obtained by the docking procedures. Therefore, state 5 can serve as a suitable model for structure-based design and optimization of the compounds having similar properties as ASEM.
Similar to the case in state 4, Trp53 adopts an edge-to-face π–π stacking mode with the dibenzo[b,d]thiophene ring of ASEM also in state 6. However, the indole ring is now rotated by about 180 degrees towards ASEM (Fig. 7c). The flipping of the indole ring causes the dibenzo[b,d]thiophene ring to move away from loop C, which is similar to the movement seen in state 3 (Fig. 4c). State 6 is an energetically less favorable state and is close to state 3 in the FES with a small energy barrier (∼3 kcal mol−1) (Fig. 6). As shown in ESI Table S1,† state 6 can transit to state 3 in the unbiased MD simulations.
We have carried out an additional simulation for the apo-system. The free energy surface demonstrates that there are also several intermediate states for Trp53 in the apo-system (ESI Fig. S3c†). However, the energy barriers separating these states are much lower than those for the holo-system (Fig. 6). Therefore, the metadynamics simulation for the apo-system converges much faster (ESI Fig. S2a and b†). The higher energy barriers for the holo-system are caused by the steric effect of ASEM and π–π stacking interaction with the dibenzo[b,d]thiophene ring of ASEM. Due to the limitation of unbiased MD simulations, the system may stay in a local minimum or transit to another close minimum in a limited simulation time. Nonetheless, with the combination of metadynamics and unbiased MD simulations, we have acquired a better understanding of the binding mode of ASEM with α7-AChBP, which indeed will be crucial for future structure-based drug design.
Conclusions
Owing to the importance of developing potent PET radioligands which can be used to study the roles of the α7 nicotinic acetylcholine receptor (α7-nAChR), to facilitate drug discovery and to monitor the progress of diseases related to α7-nAChR, molecular modelling methods have been used in this work to investigate the binding profile of [18F]ASEM (a promising PET radioligand) and α7-AChBP (a structural homologue of the extracellular domain of α7-nAChR). We found that [18F]ASEM binds at the orthosteric site of α7-AChBP. Several structural details of this binding are found to be important. The diazabicyclo[3.2.2]nonane ring has cation-π and extensive van der Waals interactions with Tyr91, Trp145, Tyr184, and Try191, which fixes [18F]ASEM tightly in the binding site. The dibenzo[b,d]thiophene ring turns to the other side of the pyridine ring of epibatidine (the crystallized agonist) and has van der Waals interactions with residues from loop C on one side and π–π stacking interaction with Trp53 of the complementary subunit on the other side. The conformation of Trp53 has a great impact on the binding mode of [18F]ASEM. Six states in terms of the sidechain dihedral angles χ1 and χ2 of Trp53 were predicted by metadynamics simulation. State 5 is the global minimum, where Trp53 is rotated to have favorable π–π stacking interactions with [18F]ASEM. These findings at the atomistic level of the binding mechanism of [18F]ASEM with α7-AChBP can have wide ramifications for the development of novel PET radioligands for α7-nAChR.Acknowledgements
The authors acknowledge support from the Swedish Foundation for Strategic Research (SSF) through the project “New imaging biomarkers in early diagnosis and treatment of Alzheimer's disease (RB13-0192)” and the support from the Stockholm Country Council through the project “Biomolecular profiling for early diagnosis of Alzheimer's disease (K1764-2013)”. Computer time for this work was awarded by a grant from the Swedish Infrastructure Committee (SNIC) for the project “Modeling of protein–ligand binding” (m.2015-1-396). The figures containing molecular structures were rendered with PyMol 1.3 (ref. 51) and VMD 1.9.2 (ref. 52). The work was partly sponsored by the China Scholarship Council (CSC).References
- P. B. Sargent, Annu. Rev. Neurosci., 1993, 16, 403–443 CrossRef CAS PubMed.
- C. Gotti and F. Clementi, Prog. Neurobiol., 2004, 74, 363–396 CrossRef CAS PubMed.
- K. Sugaya, E. Giacobini and V. A. Chiappinelli, J. Neurosci. Res., 1990, 27, 349–359 CrossRef CAS PubMed.
- W. R. Kem, Behav. Brain Res., 2000, 113, 169–181 CrossRef CAS PubMed.
- G. K. Lloyd and M. Williams, J. Pharmacol. Exp. Ther., 2000, 292, 461–467 CAS.
- D. D’hoedt and D. Bertrand, Expert Opin. Ther. Targets, 2009, 13, 395–411 CrossRef PubMed.
- A. Wevers, L. Burghaus, N. Moser, B. Witter, O. K. Steinlein, U. Schutz, B. Achnitz, U. Krempel, S. Nowacki and K. Pilz, et al., Behav. Brain Res., 2000, 113, 207–215 CrossRef CAS PubMed.
- A. Marutle, X. Zhang, J. Court, M. Piggott, M. Johnson, R. Perry, E. Perry and A. Nordberg, J. Chem. Neuroanat., 2001, 22, 115–126 CrossRef CAS PubMed.
- J. A. Lieberman, G. Dunbar, A. C. Segreti, R. R. Girgis, F. Seoane, J. Beaver, N. H. Duan and D. A. Hosford, Neuropsychopharmacolgy, 2013, 38, 968–975 CrossRef CAS PubMed.
- S. N. Haydar, C. Ghiron, L. Bettinetti, H. Bothmann, T. A. Comery, J. Dunlop, S. La Rosa, I. Micco, M. Pollastrini and J. Quinn, et al., Bioorg. Med. Chem., 2009, 17, 5247–5258 CrossRef CAS PubMed.
- A. A. Mazurov, J. D. Speake and D. Yohannes, J. Med. Chem., 2011, 54, 7943–7961 CrossRef CAS PubMed.
- H. Fan, R. Gu and D. Wei, Adv. Exp. Med. Biol., 2015, 827, 353–365 CrossRef CAS PubMed.
- J. Toyohara, J. Wu and K. Hashimoto, Curr. Top. Med. Chem., 2010, 10, 1544–1557 CrossRef CAS PubMed.
- M. Piel, I. Vernaleken and F. Rosch, J. Med. Chem., 2014, 57, 9232–9258 CrossRef CAS PubMed.
- A. Ettrup, J. D. Mikkelsen, S. Lehel, J. Madsen, E. O. Nielsen, M. Palner, D. B. Timmermann, D. Peters and G. M. Knudsen, J. Nucl. Med., 2011, 52, 1449–1456 CrossRef CAS PubMed.
- W. Deuther-Conrad, S. Fischer, A. Hiller, G. Becker, P. Cumming, G. Xiong, U. Funke, O. Sabri, D. Peters and P. Brust, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 1541–1549 CrossRef CAS PubMed.
- K. Hashimoto, S. Nishiyama, H. Ohba, M. Matsuo, T. Kobashi, M. Takahagi, M. Iyo, T. Kitashoji and H. Tsukada, PLoS One, 2008, 3, e3231 Search PubMed.
- J. Toyohara, M. Sakata, J. Wu, M. Ishikawa, K. Oda, K. Ishii, M. Iyo, K. Hashimoto and K. Ishiwata, Ann. Nucl. Med., 2009, 23, 301–309 CrossRef CAS PubMed.
- A. G. Horti, Biochem. Pharmacol., 2015, 97, 566–575 CrossRef CAS PubMed.
- Y. Gao, K. J. Kellar, R. P. Yasuda, T. Tran, Y. Xiao, R. F. Dannals and A. G. Horti, J. Med. Chem., 2013, 56, 7574–7589 CrossRef CAS PubMed.
- D. F. Wong, H. Kuwabara, M. Pomper, D. P. Holt, J. R. Brasic, N. George, B. Frolov, W. Willis, Y. J. Gao and H. Valentine, et al., Mol. Imaging Biol., 2014, 16, 730–738 CrossRef PubMed.
- A. G. Horti, Y. Gao, H. Kuwabara, Y. Wang, S. Abazyan, R. P. Yasuda, T. Tran, Y. Xiao, N. Sahibzada and D. P. Holt, et al., J. Nucl. Med., 2014, 55, 672–677 CrossRef CAS PubMed.
- N. Unwin, J. Mol. Biol., 2005, 346, 967–989 CrossRef CAS PubMed.
- K. Brejc, W. J. van Dijk, R. V. Klaassen, M. Schuurmans, J. van Der Oost, A. B. Smit and T. K. Sixma, Nature, 2001, 411, 269–276 CrossRef CAS PubMed.
- P. H. N. Celie, S. E. van Rossum-Fikkert, W. J. van Dijk, K. Brejc, A. B. Smit and T. K. Sixma, Neuron, 2004, 41, 907–914 CrossRef CAS PubMed.
- Y. Bourne, T. T. Talley, S. B. Hansen, P. Taylor and P. Marchot, EMBO J., 2005, 24, 1512–1522 CrossRef CAS PubMed.
- P. H. N. Celie, I. E. Kasheverov, D. Y. Mordvintsev, R. C. Hogg, P. van Nierop, R. van Elk, S. E. van Rossum-Fikkert, M. N. Zhmak, D. Bertrand and V. Tsetlin, et al., Nat. Struct. Mol. Biol., 2005, 12, 582–588 CAS.
- S. B. Hansen, G. Sulzenbacher, T. Huxford, P. Marchot, P. Taylor and Y. Bourne, EMBO J., 2005, 24, 3635–3646 CrossRef CAS PubMed.
- P. H. Celie, R. V. Klaassen, S. E. van Rossum-Fikkert, R. van Elk, P. van Nierop, A. B. Smit and T. K. Sixma, J. Biol. Chem., 2005, 280, 26457–26466 CrossRef CAS PubMed.
- A. Karlin, Nat. Rev. Neurosci., 2002, 3, 102–114 CrossRef CAS PubMed.
- S. X. Li, S. Huang, N. Bren, K. Noridomi, C. D. Dellisanti, S. M. Sine and L. Chen, Nat. Neurosci., 2011, 14, 1253–1259 CrossRef CAS PubMed.
- W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner and R. Farid, J. Med. Chem., 2006, 49, 534–553 CrossRef CAS PubMed.
- Induced Fit Docking Protocol 2014-1, Glide version 6.1, Prime version 3.4, Schrödinger, LLC, New York, NY, 2014 Search PubMed.
- M. H. M. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. Vanderspoel and R. Vandrunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- L. Wickstrom, A. Okur and C. Simmerling, Biophys. J., 2009, 97, 853–856 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. W. Sousa da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 367 CrossRef PubMed.
- J. M. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- D. J. Price and C. L. Brooks, J. Chem. Phys., 2004, 121, 10096–10103 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- H. Y. Sun, Y. Y. Li, S. Tian, J. M. Wang and T. J. Hou, PLoS Comput. Biol., 2014, 10, e1003729 Search PubMed.
- H. Y. Sun, P. C. Chen, D. Li, Y. Y. Li and T. J. Hou, J. Chem. Theory Comput., 2016, 12, 851–860 CrossRef CAS PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 826–843 CrossRef CAS.
- T. J. Hou, N. Li, Y. Y. Li and W. Wang, J. Proteome Res., 2012, 11, 2982–2995 CrossRef CAS PubMed.
- H. Y. Sun, Y. Y. Li, D. Li and T. J. Hou, J. Chem. Inf. Model., 2013, 53, 2376–2389 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, et al., J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- L. L. C. Schrodinger, The PyMOL Molecular Graphics System, Version 1.3r1, 2010 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00496f |
| This journal is © The Royal Society of Chemistry 2017 |