
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A graphene oxide-based tool-kit capable of characterizing and classifying exonuclease activities†
Jayeon Songa,
Vo Minh Hoab,
Jungmin Yoob,
Sanghoon Oha,
Hyeryeon Imb,
Daeho Parkb and
Gwangrog Lee *ab
*ab
aDepartment of Biomedical Science and Engineering, Gwangju Institute of Science and Technology, Gwangju, 500-712, Korea. E-mail: glee@gist.ac.kr; Fax: +82-62-715-2484
bSchool of Life Sciences, Gwangju Institute of Science and Technology, Gwangju, 500-712, Korea
First published on 6th March 2017
Abstract
Enzyme kinetics and classification are key topics in modern biochemistry. Here, we have developed a simple, rapid, and quantitative assay via a terminally capped DNA probe, reaction-specific-labeling and the fluorescence-quenching capability of graphene-oxide. Our technique provides not only characterization of kinetic information but also the immediate classification of an exonuclease without needing to design a set of DNA substrates.
Nucleases play vital roles in DNA metabolism, such as DNA replication, recombination, and repair.1 Exonucleases are a class of nucleases that recognize DNA with a very high specificity and digest DNA in a directional manner from either the 3′ or 5′ end. Therefore, they are classified as either 5′ → 3′ or 3′ → 5′ exonucleases. In modern molecular biology and genetic engineering, exonucleases have been extensively used as key tools for genotyping, PCR, and molecular DNA cloning. More importantly, nuclease enzymes are biomarkers of cancer2 and diagnostic targets for a variety of infectious diseases since they participate in the nucleic acid metabolism between the host and pathogen. Thus, bio-sensing platforms for characterization and classification of exonucleases have become needed.3
Various assays have been developed for the detection of nuclease activity, exploiting different tactics, e.g., biochemical,4 colorimetric,5 electrochemical,6 and fluorometric7 methods. The electrophoretic gel mobility shift assay (EMSA)8 and enzyme-linked immunosorbent assay (ELISA)9 are the current standards for studying DNA or RNA nucleases. However, these sophisticated methods involve multiple steps and are rather time-consuming. It is, thus, important to develop rapid, reliable, and sensitive assays with analytical capabilities that can make a large impact on a wide range of applications, from the rapid detection of enzymes to unravelling of enzymatic classifications such as degradation directionality which determine the type of exonucleases.
Graphene oxide (GO) is known to stably interact with single stranded (ss)DNA on its surface via a π–stacking interaction between the ribose rings of the nucleobases and the hexagons of the carbon atoms in GO.10 In contrast, GO does not strongly adhere to double stranded (ds)DNA due to nucleobases that are hidden in the DNA helical structure. GO also functions as an efficient fluorescent quencher when it is in physical contact with organic fluorophore molecules.11 These GO properties have been extensively utilized for the detection of enzymes that convert dsDNA to ssDNA during their functional activities, such as helicases,12 polymerases,13 and nucleases.5,14
DNA is composed of two strands running in an antiparallel fashion. Each strand of DNA is made by connecting the 5′ phosphate (P) and 3′-hydroxyl (OH) group of neighboring nucleotides via phosphodiester polymeric bonds, resulting in both termini of nucleic acids. Most exonucleases recognize either the 5′-P or the 3′-OH group and degrade ssDNA along the nucleic acid phosphodiester backbone in a 3′ → 5′ or 5′ → 3′ direction by directional movement, utilizing chemical energy released from hydrolysis of the backbone. Since the directionality of exonucleases is unique to their enzymatic function, unknown exonucleases can be characterized by this feature. The exonuclease directionality is also important because it accelerates the conversion of dsDNA to ssDNA and enhances the processivity of exonucleases compared to unspecific endonuclease activities.15 Thus, the direct detection of directionality is valuable and frequently required.
Here, we report a simple, rapid, and sensitive technique that not only detects the activity of exonucleases but also immediately reports the enzymatic directionality, which is essential information for exonuclease classification. This reporting is achieved by DNA end-capping via biotin-neutravidin and a bulky fluorophore bound to three of the four ends of the DNA. This terminal protection physically prevents further degradation by exonucleases as shown in Scheme 1. The open uncapped end is, thus, the only site that can initiate exonucleolytic degradation, revealing the enzymatic directionality. Furthermore, this simple method can be used as a quantitative kinetic tool for enzymatic activities and can be expanded to analyse other exo-ribonucleases and screen their inhibitors.
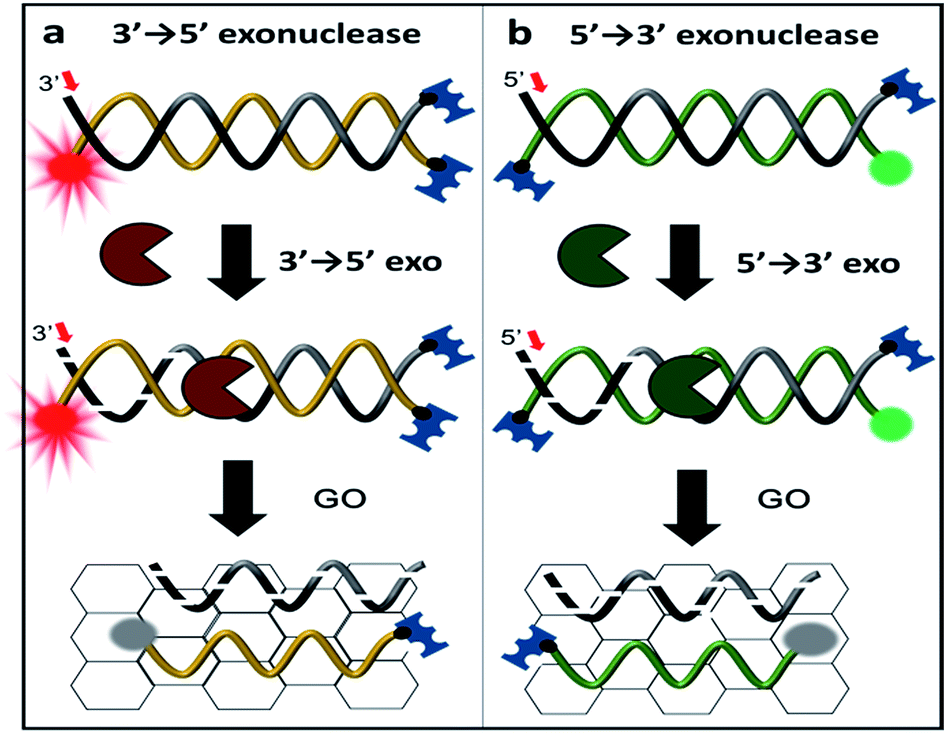 | ||
| Scheme 1 Schematic representation of exonuclease detection, allowing not only for the determination of enzymatic activity but also its classification using graphene oxide (GO)-based fluorometric quenching combined with protection of three DNA termini. Either the 3′ end of a Cy5-labelled DNA probe (denoted 3′-dsDNA) or the 5′ end of a Cy3-labelled DNA probe (denoted as 5′-dsDNA) is recognized and degraded by terminal-specific exonucleases. Upon degradation, the fluorescently labelled complimentary strand is released and adsorbed onto the surface of GO, resulting in fluorescence quenching. | ||
To discriminate the directionality of exonucleases in real time, we developed two types of fluorescent DNA probes (3′-dsDNA and 5′-dsDNA as shown in Scheme 1). Both DNA probes (Table 1 in ESI†) were composed of hydrolysed (gradient black) and non-hydrolysed strands (gold and green in left and right panels of Scheme 1, respectively). One end of the non-hydrolysed strand was blocked by a neutravidin antibody, and the other end was protected by a chemical conjugation with a bulky fluorophore (Cy5 for 3′-dsDNA and Cy3 for 5′-dsDNA as shown in the left and right panels, respectively). One open end of the hydrolysed strand was accessible by exonucleases to initiate enzymatic degradation but the other end was blocked with a neutravidin antibody. Upon the addition of an exonuclease to a buffer containing GO and a DNA probe, the open end of the hydrolysed strand is thus recognized and degraded by the exonuclease, while the fluorescent dye on the non-hydrolysed strand (gold or green strand) is quenched due to the interaction between GO and the non-hydrolysed strand released from the dsDNA after the degradation. The design of both DNA probes was validated by gel assays (Fig. S1-3†), and neutravidin conjugation did not affect the quenching capability (Fig. S4†).
For proof of concept, we tested two different types of frequently used exonucleases. Exonuclease III (ExoIII) and phi29 DNA polymerase (phi29 DNAP) bearing the exo domain (Phi29-Exo) are 3′ → 5′ exonucleases, while lambda exonuclease (λ-Exo) and T7 exonuclease (T7-Exo) are 5′ → 3′ exonucleases. The DNA construct and GO were incubated with one of the exonucleases in the presence of Mg2+ for ∼10 min. During the degradation, the hydrolysed strand is digested from the open end, and the fluorescently labelled un-hydrolysed strand is concomitantly released from the dsDNA construct, resulting in fluorescent quenching due to the adsorption of the strand onto the surface of GO via a hydrophobic interaction. Thus, the graphene-induced quenching of the fluorescence spectra can report the progression of enzymatic activity in real time.
First, we measured the fluorescent spectrum of the Cy5-labelled 3′-DNA probe, and its fluorescence emission peak appeared at 670 nm (Fig. 1a, black curve). When GO was mixed with the DNA probes in the absence of an enzyme, we found that the fluorescence intensity somewhat decreased (Fig. 1a, red curve). This effect was previously reported and is due to a certain amount of interaction between GO and dsDNA, presumably due to thermodynamic spontaneous melting at the end of the DNA duplex.16
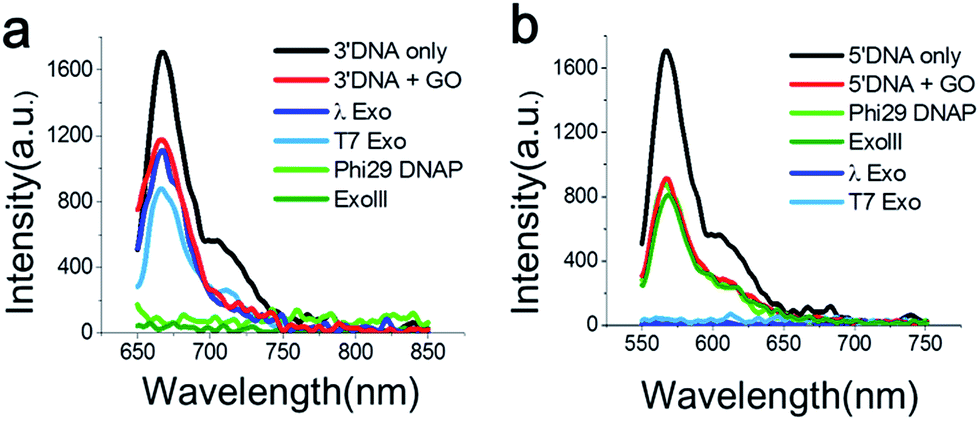 | ||
| Fig. 1 Fluorescence emission spectra measured from Cy5-labelled 3′-dsDNA at λex 670 nm (a) and Cy3-labelled 5′-dsDNA at λex 570 nm (b) after the treatment with various exonucleases in the presence of GO for 30 min. The 3′-dsDNA was degraded only by 3′ → 5′ exonucleases (ExoIII and the exo-domain of Phi29 DNA polymerase), whereas the 5′-dsDNA was degraded only by 5′ → 3′ exonucleases (λ-exo and T7-exo), due to terminal-specific DNA modifications. | ||
To find an optimal GO concentration, the concentration of the DNA probes was maintained at 70 nM, and the GO concentration was increased from 0 to 150 μg ml−1. The fluorescence intensity no longer changed at concentrations greater than 90 μg ml−1 (Fig. S5 and 6†). Thus, 90 μg ml−1 GO and 70 nM DNA probes were used for all experiments, unless otherwise noted. When GO was mixed with the 3′-dsDNA probe without the addition of an enzyme, we noticed that ∼20% quenching occurred, depending on the type of DNA probe (red in Fig. 1a), which was also previously reported.17
The feasibility of our method is signified when the Cy5-labelled 3′-dsDNA probe was added to a reaction buffer containing GO, a 3′ → 5′ exonuclease and a cofactor, Mg2+ (Fig. 1a). The light and dark green intensities obtained from the 3′ → 5′ exonucleases (Phi29-Exo and ExoIII, respectively) were completely quenched, whereas the light and dark blue intensities obtained from the 5′ → 3′ exonucleases (λ-Exo and T7-Exo, respectively) did not decrease significantly (Fig. 1a). This result indicated that the 3′-dsDNA was degraded only by the 3′ → 5′ exonucleases but not by the 5′ → 3′ exonucleases, clearly demonstrating the orthogonality of the DNA probes.
The emission peak of the Cy3-labelled 5′-dsDNA emerged at 570 nm, consistent with the Cy3 fluorescence emission (Fig. 1b). When GO was mixed with the 5′-dsDNA probe, the amount of quenching was ∼48% (red in Fig. 1b). The fluorescence intensities did not change significantly after the treatment of ExoIII or Phi29-Exo with the 5′-dsDNA, but they decreased to the basal level following treatment with λ-Exo or T7-Exo. This result suggested that each DNA probe exhibited selectivity toward its corresponding type of exonucleases.
Since 3′ → 5′ and 5′ → 3′ exonucleases work independently on digesting their respective dsDNA probes, we expected that both DNA probes can be simultaneously used for the multiplex detection of two types of exonucleases (Fig. 2a). To validate our colorimetric approach, we utilized the different colour-labelling scheme as follows: Cy5-labelled 3′-dsDNA and Cy3-labelled 5′-dsDNA as shown in Fig. 1. This two-colour orthogonal detection allows us to determine whether the sample to be tested contains one or more types of exonucleases due to the high abundance of nucleases in cells and/or potential contamination. This combinatorial approach will, thus, shorten the time required for measurement and provide more information with a higher accuracy of exonuclease detection.
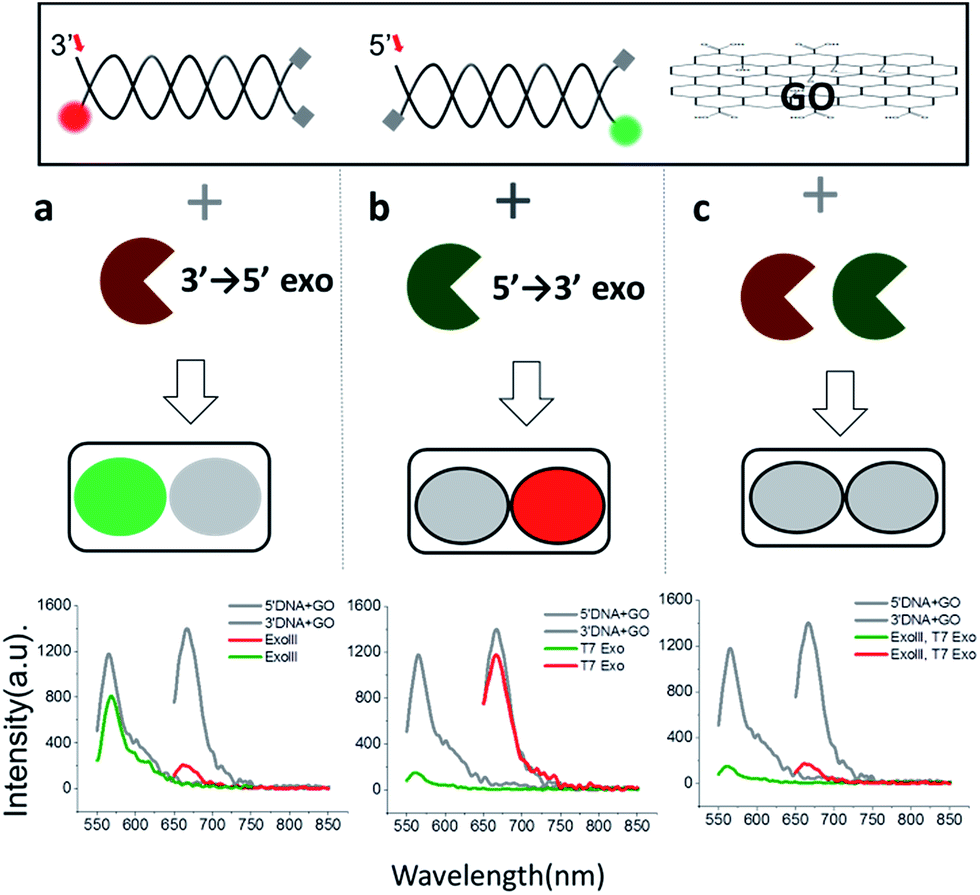 | ||
| Fig. 2 Colorimetric multiplexing detection for two different 3′ → 5′ and 5′ → 3′ exonucleases, acting orthogonally on two dsDNA probes with terminal-specific protection. The green or red fluorescent signal indicates the presence of a 3′ → 5′ (a) or 5′ → 3′ exonuclease (b), respectively, whereas quenching of both signals indicates the presence of both nucleases (c). | ||
When ExoIII, as an example of 3′ → 5′ exonucleases, was added into a buffer at a pH of 7.5 containing both 3′-dsDNA and 5′-dsDNA, the green fluorescence intensity steadily remained steady (with intrinsic quenching), whereas the red fluorescence intensity significantly decreased to a basal level (Fig. 2a). Likewise, when T7 exonuclease, as an example of 5′ → 3′ exonucleases, was added, the green intensity quenched while the red intensity remained steady (Fig. 2b). In contrast, when both ExoIII and the T7 exonuclease were added into the buffer containing 3′-dsDNA and 5′-dsDNA, both the green and the red fluorescent intensities decreased substantially, demonstrating that the simultaneous detection of two different types of 3′ → 5′ and 5′ → 3′ exonucleases is indeed possible (Fig. 2c).
This decrease was not observed without an essential cofactor, Mg2+, and was attributed to the degradation of dsDNA. It is noteworthy that ∼15% of the red fluorescence remained compared to the green fluorescence, suggesting that this multiplex method can be used to directly compare relative turnover rates of two different exonucleases with a single measurement. Based on these data, we propose the following colorimetric code table: a green signal indicates a 3′ → 5′ exonuclease; a red signal indicates a 3′ → 5′ exonuclease; and no fluorescence signal or a basal level denotes the presence of both 3′ → 5′ and 5′ → 3′ exonucleases. Thus, the method offers a convenient classification of exonucleases (Fig. 2a). In summary, this multicolour detection improves not only the accuracy of the sensing capability but also adds a multiplexing capability of enzymatic classification.
Because the fluorimeter-based detection used in Fig. 1 and 2 reads one sample at a time, we used a multimode microplate reader (Flexsation3 in Molecular Devices™) to test the simultaneous detection of a series of different samples in a high-throughput 96-well platform. The fluorescence intensities were measured ∼30 min after a 3′ → 5′ exonuclease was added to a buffer containing GO, Cy5-labelled 3′-dsDNA, and Mg2+. Indeed, a remarkable difference in quenching was observed from the comparison of different exonuclease treatments (Fig. 3a). The decrease in fluorescence intensity obtained from a mixture containing Cy5-labelled 3′-dsDNA, GO, Mg2+ and either ExoIII or Phi29-Exo was significant, which was similar to the intensity of a control experiment measured from a mixture of GO and Cy5-labelled non-hydrolysed ssDNA (i.e., a degradation-mimic product), implying that the complete degradation was effected by the 3′ → 5′ exonucleases. However, the decreases in fluorescence intensities observed after the treatment with the 5′ → 3′ exonucleases λ-Exo and T7-Exo were insignificant (Fig. 3a), illustrating the robustness of the GO-based assay. The average colour codes shown in the white-gray-black scale, reported from the multiplex device clearly showed that the GO-based nuclease assay could be applied to a multiplexing high-throughput screening platform (top in Fig. 3a).
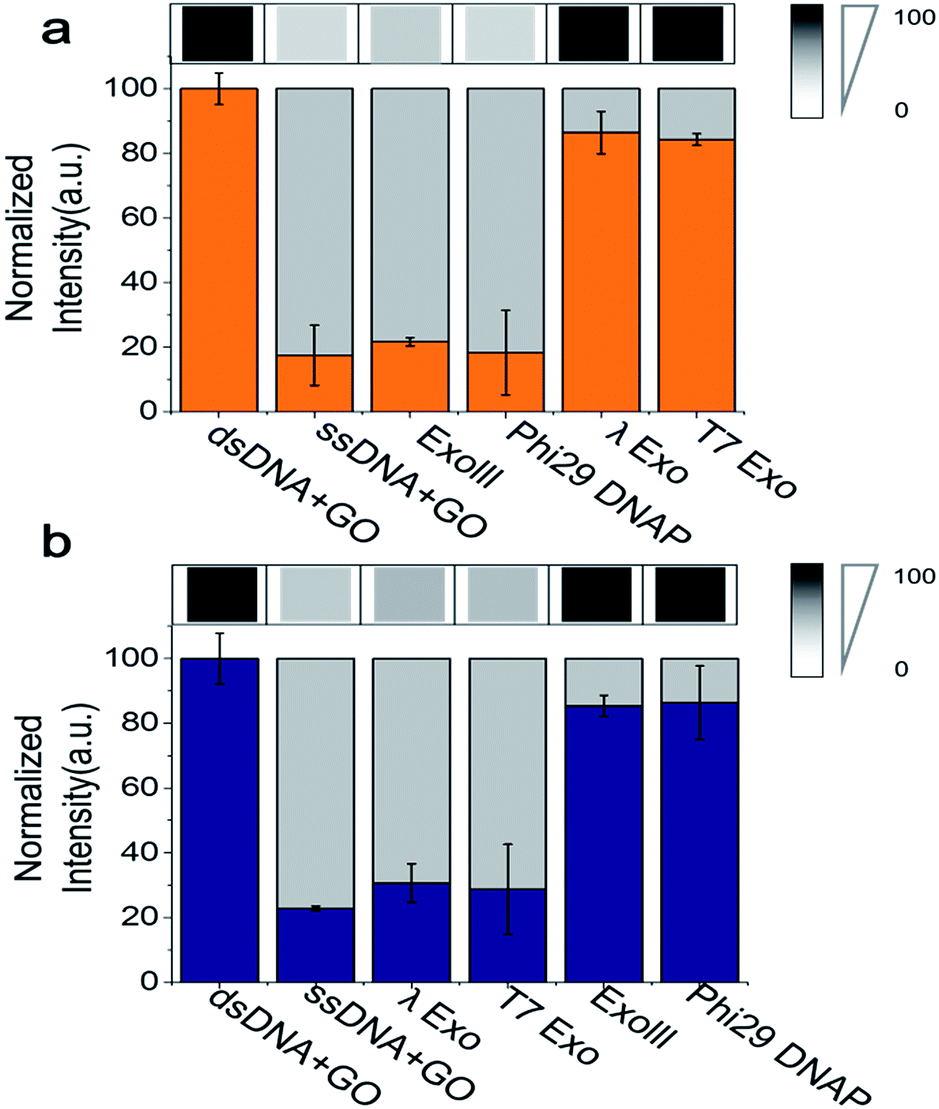 | ||
| Fig. 3 Quantification of GO-based fluorescence quenching after treatment by various exonucleases. (a) Fluorescence intensities of Cy5-labelled 3′-dsDNA were visualized in a gray-scale image (top panel) and were quantified (bottom panel) by a multi-well plate reader (Flexstation™). (b) The same as in (a) except that Cy3-labelled 3′-dsDNA was used instead of the Cy5-labelled 3′-dsDNA. The data demonstrate that the 3′-dsDNA is 3′ → 5′ exonuclease-specific, whereas 5′-dsDNA is 5′ → 3′ exonuclease-specific. | ||
To evaluate the performance of the Cy3-labelled 5′-dsDNA, we repeated the same experiments as shown in Fig. 3a. Following treatment with 5′ → 3′ exonucleases, such as λ-Exo and T7-Exo, the fluorescence intensities dramatically decreased to the basal level, which was similar to the intensity obtained from a mixture of GO and Cy5-labelled ssDNA. In contrast, no significant decrease in fluorescence intensity was observed when the non-corresponding 3′ → 5′ exonucleases ExoIII and phi29-Exo were added to the probe solution containing 5′-dsDNA (Fig. 3b). The large differences in quenching shown in Fig. 3 provide high reliability via the multiplex device in classifying the type of nucleases based on their degradation directionality.
Many GO-based enzyme assays have been demonstrated, but thus far, none have been able to provide quantitative enzymatic kinetic parameters, such as Km (Michaelis–Menten constant) and Vmax (reaction velocity), which are important characteristics in enzymology. The determination of these values requires accurate measurements of time-dependent enzymatic rates. However, GO-based fluorescence-quenching signals typically contain the following three major time periods as shown in Fig. 4: (i) the diffusion time, during which dsDNA is mixed with GO and stays in its immediate vicinity; (ii) the enzyme turnover time, during which the enzymes convert dsDNA to ssDNA; and (iii) the adsorption time, during which ssDNA after degradation binds to the surface of GO.
 | ||
| Fig. 4 A schematic of the exonuclease-induced fluorescence-quenching assay. For accurate kinetic studies, intrinsic fluorescence quenching (i) and binding time onto the GO surface (iii) must be subtracted, and only degradation time (ii) should be considered for enzyme kinetic determination. | ||
The periods represented by (i) and (iii) are the main causes of inaccuracy in determining enzymatic rates by GO-based assays. The following approaches would reduce the non-enzymatic diffusion-delays to improve kinetic measurements: (1) the intrinsic non-enzymatic fluorescence intensity decrease, which results from the interaction between GO and dsDNA, should be reduced; (2) the samples should be rapidly mixed to minimize the diffusion delay for the enzyme to bind to its DNA substrate; and (3) upon conformational conversion of the DNA probes, the diffusion delay for ssDNA to adsorb onto GO should be minimized. To overcome these drawbacks of the GO-based fluorescence platform, we developed a fast-responding GO-quenching assay utilizing a pre-incubation followed by a rapid mixing process of all components, such as the DNA probe, GO, Mg2+ and enzyme. The pre-incubation of dsDNA with GO prior to the addition of an enzyme allowed us to eliminate the intrinsic fluorescence quenching caused by the interaction between dsDNA and GO by separating it, from the actual quenching produced by the enzyme activity via the conversion from dsDNA to ssDNA.
To determine the Michaelis–Menten kinetic curve by the fluorimeter, we first mixed the 3′-dsDNA with GO and incubated the mixture for 1200 s to temporally separate and extract the intrinsic quenching from the actual enzymatic fluorescence quenching (Fig. 5a). ExoIII was then introduced to the reaction cuvette and rapidly mixed to minimize the diffusion time required for the enzyme to bind to the DNA probe for initiation. This experiment was repeated as a function of various concentrations of ExoIII, and the fluorescence emission at ∼650 nm was recorded over time.
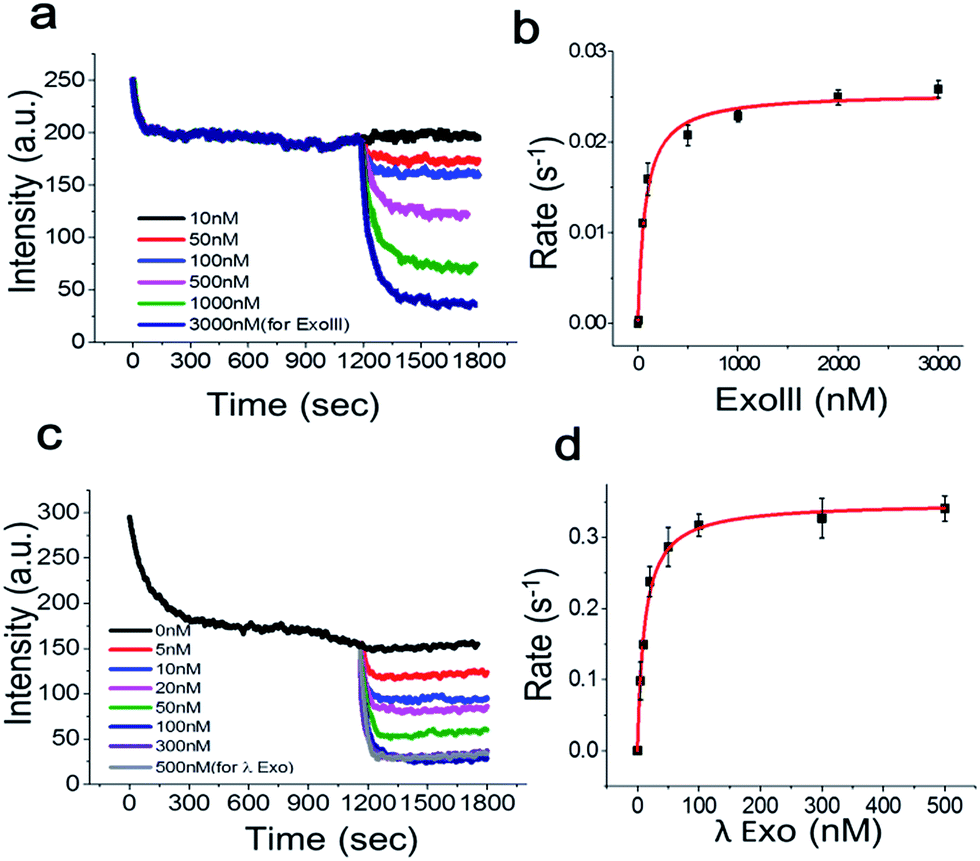 | ||
| Fig. 5 Fluorescence intensity vs. time curves for the real-time kinetic analysis of ExoIII (a) and its Michaelis–Menten fit (b). (c, d) The same as in a and b but for λ-Exo. Only one quenching curve was plotted between 0 and 1200 s for clarity. Error bars indicate the standard error of the mean (SEM) from three independent experiments. | ||
Fig. 5a shows the real-time quenching profiles of various protein concentrations. The 3′-dsDNA probe was premixed with GO in the presence of Mg2+ at room temperature to remove the intrinsic fluorescence quenching of dsDNA by GO, and then, ExoIII was added and quickly initiated by mixing all the components. ExoIII converted dsDNA to ssDNA, and the displaced fluorophore-strand adsorbed onto the GO surface, resulting in a decrease in fluorescence due to GO-induced quenching.
Single exponential fits of the real-time fluorescence quenching curves revealed characteristic decay times, τ, at various protein concentrations (Fig. S7 and 8†). The decay rates were calculated from the inverse of the decay time, τ. The quenching rate vs. protein concentration curve, with the DNA probe concentration fixed at ∼70 nM, displayed a hyperbolic shape, suggesting that the quenching rate was elevated as a function of the protein concentration due to more ssDNA adsorbing onto the GO surface. The equilibrium dissociation constant was determined by plotting the DNA-degradation-induced quenching as a function of the protein concentration (Fig. 5b) and fitting to the data to Michaelis–Menten kinetics with a maximum quenching rate of ∼0.025 s−1, and a Km value of ∼71.3 nM, which was in a good agreement with previous reports that lay in a range between 32 and 140 nM.18
Next, we investigated the degradation activity of λ-Exo as a test case for 5′ → 3′ exonucleases. We observed enzyme fluorescence quenching as a function of the λ-Exo concentration (Fig. 5c). The inverse of the quenching decay time, τ, vs. enzyme concentration also revealed a hyperbolic relationship, and its excellent fit to the Michaelis–Menten curve returned a maximum quenching rate of ∼0.034 s−1 and a Km value of ∼11.6 nM, comparable to the previously measure value of ∼9.13 nM.19
In conclusion, we have developed an exonuclease-specific GO-based fluorometric assay allowing not only the characterization of exonucleases but also immediate classification of the exonuclease type, without further designing a set of DNA substrates as in other systems such as electrophoretic gel mobility shift assay (EMSA).8 Fig. 5 recapitulates the detection limit of our method. Namely, our method is capable of detecting exonucleases at concentrations as low as 10−6 M (e.g., 5 nM in Fig. 5c and d) which is comparable to other popular methods, such as SPR (Surface Plasmon Resonance) and EMSA. Our method is specially designed for detecting various exonucleases regardless of their directionality, but not for detecting endonucleases. Although this work is not the first demonstration for detecting nucleases via GO, it provides additional information such as enzyme directionality and binding affinity of Km. Furthermore, our method can be applied in diverse biological applications, e.g., drug screening, enzymatic assay, and biosensors if combined with the multi-well plate reader as demonstrated in Fig. 3.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NRF-2016R1A2B4014556); by the grant from the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (1320110); and by the GIST Research Institute (GRI) in 2017.Notes and references
- (a) T. M. Marti and O. Fleck, Cell. Mol. Life Sci., 2004, 61, 336 CrossRef CAS PubMed; (b) A. Sancar, in Annual Review of Biochemistry, 1996, vol. 65, p. 43 Search PubMed; (c) I. V. Shevelev and U. Hubscher, Nat. Rev. Mol. Cell Biol., 2002, 3, 364 CrossRef CAS PubMed.
- (a) N. Dai, X. J. Cao, M. X. Li, Y. Qing, L. Liao, X. F. Lu, S. H. Zhang, Z. Li, Y. X. Yang and D. Wang, PLoS One, 2013, 8, e58001 CAS; (b) L. I. Hernandez, V. C. Ozalp and F. J. Hernandez, Chem. Commun., 2016, 52, 12346 RSC; (c) J. H. Shin, S. Choi, Y. R. Lee, M. S. Park, Y. G. Na, K. Irani, S. D. Lee, J. B. Park, J. M. Kim, J. S. Lim and B. H. Jeon, Cancer Res. Treat., 2015, 47, 823 CrossRef CAS PubMed; (d) S. Thakur, B. Sarkar, R. P. Cholia, N. Gautam, M. Dhiman and A. K. Mantha, Exp. Mol. Med., 2014, 46, e106 CrossRef CAS PubMed.
- (a) Z. Guan, J. Liu, W. Bai, Z. Lv, X. Jiang, S. Yang, A. Chen and G. Lv, PLoS One, 2014, 9, e108401 Search PubMed; (b) D. Gerceker, D. Karasartova, E. Elyürek, S. Barkar, M. Kiyan, T. M. Özsan, M. K. Calgin and F. Sahin, J. Gen. Appl. Microbiol., 2009, 55, 291 CrossRef CAS PubMed; (c) A. Economidou-Karaoglou, M. Lans, H. Taper, J. L. Michaux and M. Roberfroid, Blood, 1989, 74, 2730 CAS.
- W. F. Burke Jr and B. S. Slinker, Anal. Biochem., 1980, 108, 320 CrossRef CAS.
- C. H. Lu, J. Li, X. J. Qi, X. R. Song, H. H. Yang, X. Chen and G. N. Chen, J. Mater. Chem., 2011, 21, 10915 RSC.
- S. Sato and S. Takenaka, Sensors, 2014, 14, 12437 CrossRef CAS PubMed.
- (a) Z. Zhou, C. Zhu, J. Ren and S. Dong, Anal. Chim. Acta, 2012, 740, 88 CrossRef CAS PubMed; (b) Y. He, L. H. Xiong, X. J. Xing, H. W. Tang and D. W. Pang, Biosens. Bioelectron., 2013, 42, 467 CrossRef CAS PubMed.
- M. Wakasugi and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6669 CrossRef CAS.
- L. M. Hellman and M. G. Fried, Nat. Protoc., 2007, 2, 1849 CrossRef CAS PubMed.
- (a) N. Varghese, U. Mogera, A. Govindaraj, A. Das, P. K. Maiti, A. K. Sood and C. N. R. Rao, ChemPhysChem, 2009, 10, 206 CrossRef CAS PubMed; (b) S. He, B. Song, D. Li, C. Zhu, W. Qi, Y. Wen, L. Wang, S. Song, H. Fang and C. Fan, Adv. Funct. Mater., 2010, 20, 453 CrossRef CAS.
- (a) R. S. Swathi and K. L. Sebastian, J. Chem. Phys., 2009, 130, 086101 CrossRef CAS PubMed; (b) R. S. Swathi and K. L. Sebastian, J. Chem. Phys., 2008, 129, 054703 CrossRef CAS PubMed.
- (a) H. Jang, S. R. Ryoo, Y. K. Kim, S. Yoon, H. Kim, S. W. Han, B. S. Choi, D. E. Kim and D. H. Min, Angew. Chem., Int. Ed., 2013, 52, 2340 CrossRef CAS PubMed; (b) H. Jang, Y. K. Kim, H. M. Kwon, W. S. Yeo, D. E. Kim and D. H. Min, Angew. Chem., Int. Ed., 2010, 49, 5703 CrossRef CAS PubMed.
- (a) K. Roh, D. M. Kim, E. H. Lee, H. Kim, H. S. Park, J. H. Jang, S. H. Hwang and D. E. Kim, Chem. Commun., 2015, 51, 6960 RSC; (b) J. S. Park, A. Baek, I. S. Park, B. H. Jun and D. E. Kim, Chem. Commun., 2013, 49, 9203 RSC.
- J. Lee and D. H. Min, Analyst, 2012, 137, 2024 RSC.
- M. L. Uhr, Biochem. Educ., 1979, 7, 15 CrossRef CAS.
- D. Jose, K. Datta, N. P. Johnson and P. H. Von Hippel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4231 CrossRef CAS PubMed.
- C. Hong, D. M. Kim, A. Baek, H. Chung, W. Jung and D. E. Kim, Chem. Commun., 2015, 51, 5641 RSC.
- (a) J. Eriksson and Ü. Langel, PLoS One, 2016, 11, e0154099 Search PubMed; (b) M. Takeuchi, R. Lillis, B. Demple and M. Takeshita, J. Biol. Chem., 1994, 269, 21907 CAS; (c) Q. Xu, A. Cao, L. F. Zhang and C. Y. Zhang, Anal. Chem., 2012, 84, 10845 CrossRef CAS PubMed.
- K. Hsieh, Y. Xiao and H. Tom-Soh, Langmuir, 2010, 26, 10392 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00388a |
| This journal is © The Royal Society of Chemistry 2017 |