
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synthesis and mechanistic studies of a highly active nickel phosphide catalyst for naphthalene hydrodearomatization†
Guoxia Yuna,
Qingxin Guan*a and
Wei Li *ab
*ab
aCollege of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin, 300071, China. E-mail: weili@nankai.edu.cn; qingxinguan@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300071, China
First published on 27th January 2017
Abstract
Although HDS and HDN reactions over transition metal phosphides have been widely studied, few publications about aromatic hydrodearomatization (HDA) over transition metal phosphides are found. Using ordered mesoporous Al-MCM-41 (Al-M) as the support, a series of supported nickel phosphide catalysts with different Ni/P molar ratios and loadings have been prepared by a temperature-programmed reduction and characterized. The HDA activity of naphthalene was measured in a fixed bed reactor at 250–330 °C and 3 MPa. The results showed that the catalyst with initial Ni/P molar ratio of 1.25 and 15 wt% loading displayed the highest HDA activity as well as 99.0% of selectivity of decalin at 270 °C, which is even higher than that of 0.5 wt% Pt–Al-M catalyst. As a comparison, the HDA performance of various catalysts was also examined. The results revealed that the presence of framework aluminum favors HDA processes and a synergistic effect between nickel phosphide and the suitable acidity resulted in an improvement of the catalytic activity. Finally, the possible reaction pathway of naphthalene hydrogenation (HYD) on nickel phosphide catalysts was proposed. Taking into consideration the high catalytic activity, Al-M supported nickel phosphide can be considered as a very efficient HDA catalyst to decrease the contents of aromatics in the fuels.
1. Introduction
Population increase, urbanization, and rising living standards have rapidly increased global energy consumption and environmental burdens in the last half century. Energy consumption is expected to continue to increase dramatically in the coming years along with its associated environmental issues. Thus more stringent environmental regulations and fuel specifications on diesel are being tightened in both the United States and Europe, which not only put impelling emphasis on the contents of aromatics in the fuels, but also lay special focus on the density and cetane number.1 Because aromatics in fuels, i.e., diesel and jet fuels, not only lower the quality and produce undesired exhaust emissions but also have potentially hazardous and carcinogenic effects.2 Therefore, HYD of aromatics becomes one of the most important unit operations in modern oil-refinery and conventional hydroprocessing technology is used to reduce aromatics in fuels through the HDA reaction. And HYD can produce cycloalkane from aromatics and thus significantly improve the cetane number of diesel streams and improve the combustion characteristics. The products of aromatics HYD are considered as an ideal component of jet fuel, as they have concurrently appropriate quality and volume caloric value. In this regard, this opinion is consistent with that of Yan et al. They highlight a detailed overview of the development related to energy production and environmental remediation.3Generally, HYD is a highly exothermic reaction and thus thermodynamically favored at lower temperature. A lot of information about naphthalene HYD and formation of decalin have been reported in literature.4 Therefore, searching out excellent HYD catalysts will provide potentiality for decreasing the aromatic content in the fuels and regarding them as high thermal stable jet fuel. Conventional catalysts in aromatic HYD are sulfides based on Mo or W and promoted with Ni or Co supported on alumina.5 The drawbacks of these catalysts include severe operating conditions such as high temperature and high pressure. Furthermore, only moderate levels of saturation of aromatic content under typical hydrotreating conditions can be obtained due to equilibrium limitations. Meanwhile, as proposed by Wang and Zhou et al.,6 many researchs have shown that the noble metal-based catalysts have attracted extensive attention for a wide range of applications in many reactions, as well as the HDA reaction. Noble metal-based catalysts, including Pt,7 Pd,8 Rh,9 or bimetallic systems of these noble metals, show distinctly high catalytic activities towards naphthalene HYD, due to the efficient ability for dissociating hydrogen. However, noble metal has high cost and the presence of sulfur strongly influences their catalytic activity due to the poisoning of active sites. Recently, transition metal phosphides have attracted attention not only because they are very active in the hydrotreating reactions but also because they are much more stable than noble metals and metal carbides and nitrides in the atmosphere of H2S.10 A number of results have shown that Ni2P was the most active catalyst for both hydrodenitrogenation (HDN) and hydrodesulfurization (HDS) reactions over the past several years.11–13 Oyama et al. compared HDS and HDN activity over different phosphides and found that their activity order follows Fe2P < CoP < MoP < WP < Ni2P.
As common supports, silica, alumina, resin, active carbon, clay, and microporous zeolite have been widely used, while diffusion limitation limit their potential applications for catalysing bulky molecules.14 Diffusion and catalytic tests have also proved that mesoporous zeolite, especially MCM-41, can overcome the mass transfer limitation and exhibit excellent catalytic properties for the conversion of bulky molecules.15,16 However, purely siliceous mesoporous materials are electrically neutral frameworks, no acidity was formed on their surface. Recent studies have been proved that the supports with suitable acidity, such as SiO2–Al2O3,17 USY,18 SAPO-11,19 H-ZSM-5,20 mordenites,21 H-beta zeolite,22 etc., have positive effects on the HDA activity. These effects have been attributed to the synergistic effects between acid sites and transition metal phosphides particles. Firstly, it was claimed that electron-deficient metal particles (Mδ+) were formed in a transition metal phosphide/zeolite bifunctional catalyst due to the partial electron transfer from the active particles to acidic sites of the zeolite supports. Secondly, Ni2P behaves somewhat like hydrogenase. A spillover based HYD pathway in which aromatic hydrocarbons were adsorbed on acidic sites and hydrogenated by the hydrogen from active particles would occur in zeolite supported catalyst. Namely, the acid sites of the support provide additional active sites on which aromatic are hydrogenated by the spillover hydrogen.23 Therefore, much effort has been devoted to the enhancement of the surface acidity with the aim of using them as catalyst support or catalyst for acid-catalyzed reactions. Aluminum may be incorporated into the mesoporous structure by adding an aluminum source to the synthesis mixture. The substitution of silicon by aluminum usually unbalances the neutral structure of silica, which may generate acid sites. The nature and strength of such acid sites, as well as the amount of trivalent cation added, may have an impact on the catalytic and adsorption properties of these materials and suit them for applications in several processes.24 Introduction of foreign ions, for instance, aluminum ions, into the silicate framework (that means, a decrease in the Si/Al ratio) has been proved to be an efficient route for modifying the surface acidity of Si-MCM-41 and the Lewis acidity of Si-MCM-41 could be enhanced significantly after modification with aluminum ions. The Brönsted acidity of aluminosilicates generally arises from the presence of accessible hydroxyl groups or in the form of ‘‘bridging’’ Si–(OH)–Al hydroxyl groups (structural hydroxyls) associated with 4-coordination framework aluminium.25 As such a process generally demands a catalyst with high product diffusivity and selectivity for aromatics HYD, molecular sieves with high surface area as well as the pore size and acidity are potential catalyst supports. Furthermore, proper pore structure and acidity favored the dispersion of the active phases.
Although HDS and HDN reactions over transition metal phosphides have been widely studied, few publications about aromatic HDA over transition metal phosphides are found. Here in this work, we prepared the Al-M supported nickel phosphide HYD catalysts with different Ni/P molar ratios and loadings in the precursors. The catalytic performance of the nickel phosphide catalysts and the reaction pathway of naphthalene HYD were studied in detail. Compared to Pt–Al-M, the NixP–Al-M catalyst exhibited much better HYD activity and decalin selectivity.
2. Experimental
2.1 Materials
Nickel nitrate (Ni(NO3)2·6H2O) and ammonium dihydrogen phosphate (NH4H2PO4, 99%) were purchased from Tianjin Guangfu Technology Development Co., Ltd. Chloro platinic acid (H2PtCl6·6H2O, 99%) was obtained from Aladdin Industrial Corporation. The supports, mesostructured hexagonal frame-work, MCM-41 and Al-M were obtained from Tianjin Chem. Sci. Ltd., China and the content of Al in Al-M accounts for about 5 wt%. All of the materials were analytical grade and utilized without further purification.2.2 Catalyst preparation
A series of high-performance catalysts have been synthesized by temperature-programed reduction of nickel phosphate precursors according to the following steps: [1] firstly, the support was dried at 100 °C overnight prior to use. The oxide precursor was prepared by incipient wetness impregnating Al-M with a mixture aqueous solution of Ni(NO3)2·6H2O and NH4H2PO4, followed by drying at 80 °C for 12 h, and calcination at 500 °C for 2 h in air; [2] the oxide precursor was reduced at 650 °C for 2 h under flowing H2 (60 mL min−1) by temperature-programmed reduction (TPR) to obtain the active nickel phosphide catalyst. Finally, the product was cooled to ambient temperature under flowing H2 and was passivated for 1 h under flowing 1% O2/N2. In one series, the loadings on the Al-M was chosen to be 10, 15, 20, 25, and 30 wt% with Ni/P molar ratio of 1.25. In another series, the Ni/P molar ratios of the precursors were 0.5, 1, 1.25, 1.5, 2, and 3 with a 15 wt% loading.In addition, fixing the initial Ni/P atomic molar to 1.25, various supports supported nickel phosphide catalysts with nickel loading of 15 wt% was also prepared following the same procedure. For comparison purposes, Ni–Al-M catalyst with Ni loading of 15 wt% was prepared by a procedure similar to that of the supported nickel phosphides except for the addition of the ammonium dihydrogen phosphate. Catalysts are designated according to the loading (M) of the active phase and the Ni/P ratio (Y) as M wt% NixP–Al-M-(Y), in which x represents the actual Ni/P values after reaction. The Pt loading in catalysts were 0.5 wt%. The supported Pt catalysts were obtained through the wet co-impregnation with an appropriate amount of aqueous H2PtCl6·6H2O. The impregnation lasted for 24 h. After that, the sample was dried in air at 80 °C for 12 h, and finally calcined at 350 °C for 2 h in air. At last the catalyst was cooled to ambient temperature and washed in deionized water for twice followed by drying at 80 °C for 12 h. In this way, Pt supported on Al-M was prepared and denoted as 0.5 wt% Pt–Al-M, which was used as a reference.
2.3 Catalyst characterizations
The catalysts were studied by a series of characterizations as follows:Powder X-ray diffraction patterns (XRD) were recorded by a Bruker D8 focus diffractometer using a Cu-Kα radiation. The measurement was made in the 2θ range of 10–80° at 40 kV and 40 mA. The size and morphology of as-obtained samples were observed by field emission scanning electron microscope (FESEM, JEOL, JSM-7500F) linked with energy-dispersive X-ray spectrometer (EDS). The samples were plated with gold before measurement to avoid the electric charge accumulation. Transmission electron microscopy (TEM) images and the HAADF-STEM mapping analysis were obtained on electron microscope (FEI Tecnai, G2 F20) with a link energy-dispersive X-ray (EDX, EDAX, TEAM) system for elemental mapping operated at an accelerating voltage of 200 kV equipped with a field emission source. The elemental composition of the catalysts was determined by ICP-OES using an SPECTRO ARCOS ICP-OES Thermo Optical Emission Spectrometer. The specific surface area of materials was analysed using nitrogen adsorption–desorption isotherms measured at 77 K on a BELSORP Mini instrument with preprocessing the samples in vacuum at 150 °C for 12 h. The specific surface area data and the pore size distribution analysis were calculated by Brunauer–Emmett–Teller (BET) and nonlocal density functional theory (NLDFT) method from adsorption isotherm, respectively. Temperature-programmed desorption (TPD) experiments were performed with a Micromeritics Chemisorb 2750 instrument. All samples were pretreated in N2 at 300 °C for 2 h with a flow rate of 25 mL min−1. The desorption step was performed from 50 °C to 800 °C with a heating rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) was carried out using a Kratos Axis Ultra DLD spectrometer employing a monochromatic Al Kα X-ray source (hν = 1486.6 eV), hybrid (magnetic-electrostatic) optics, and a multichannel plate and delay line detector with the C 1s peak at 284.6 eV as the internal standard. All XPS spectra were recorded using an aperture slot of 300 × 700 μm, survey spectra were recorded with a pass energy of 160 eV, and high resolution spectra with a pass energy of 40 eV. CO uptake was obtained on Micromeritics Chemisorb 2750 instrument using 100 μL loop ring. CO chemisorption was taken at 30 °C in the flowing He (100 mL min−1). The samples were pre-reduced in flowing H2 at corresponding temperature for 1 h.
2.4 Catalytic activity evaluation
Naphthalene was used to investigate the catalysts HYD performance. HDA reaction was carried out in a continuous flow fixed-bed reactor equipped with a high-pressure pump.The reaction procedure was stated in detail here. The catalysts were pelleted, crushed, and a particle size fraction of 20–40 mesh was used for the test. And catalysts were diluted with quartz sand before loaded into the reaction tube. Before reaction, the catalysts were reduced at the same temperature as preparation for 1 h in flowing H2. After prereduction, the system was adjusted to the desired reaction temperature (250 °C) and pressure (3.0 MPa). The feedstock containing naphthalene in cyclohexane (5 wt%) was continuously pumped into the reactor at the rate of 0.17 mL min−1. The weight hourly space velocity (WHSV) was equal to 1 h−1, and the H2/oil ratio was kept at 500 (v/v).
Liquid products were collected every hour in all assays. The reactant and products were analysed using a gas chromatograph (Agilent 7890A), equipped with a HP-5 capillary column and a flame ionization detector (FID). Products compositions were identified by GC-MS (Agilent 7890A-5977C). The HYD performance of catalysts was determined by the naphthalene conversion and product selectivity.
For TOF calculation, we used the same definition of TOF like other works do, which is moles of reactant converted divided by moles of active site. As follows, the TOF was calculated using eqn (1):
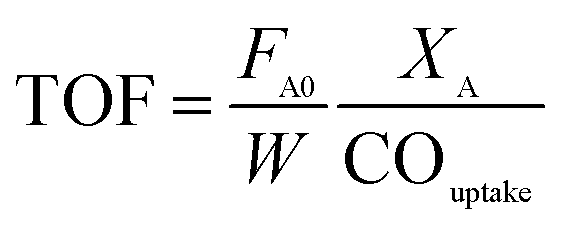 | (1) |
3. Results and discussion
3.1 X-ray diffraction
XRD patterns were used to identify the crystalline phases formed after reduction. In order to study the effect of surface modification on the crystal structure of supported nickel phosphide catalysts, the structures of NixP–Al-M obtained from various initial Ni/P molar ratios at a loading of 15 wt% NixP were determined in Fig. 1(a), as well as a Ni2P and Ni12P5 reference. From the Fig. 1(a), it can be easily found that a broad diffraction line located at 2θ = 23° is due to the amorphous nature of mesoporous structure for all of the catalysts.26 Interestingly, the sample prepared with initial Ni/P ratio of 1/2 shows a pure Ni2P phase and the main diffraction peaks positions clearly appear around 40.7°, 44.6°, 47.4°, 54.2°, and 54.8°, which correspond to the (111), (201), (210), (300), and (211) crystal faces of Ni2P (PDF# 65-1989), respectively. In particular, when initial Ni/P ratio exceeds 1/1 and further increase, the peak of the Ni2P phase declined and the Ni12P5 phase emerged. Diffraction peaks associated with Ni12P5 at 2θ = 38.3°, 41.7°, 46.7°, and 48.9° (PDF# 22-1190) were observed in addition to those of Ni2P. Namely, with increasing Ni/P molar ratio, the excess of P decreases, the conditions for formation of the Ni2P phase become less favorable, so the intensities of the Ni2P diffraction peaks decrease. As previously proposed by Oyama et al., the lower P content led to formation of Ni12P5, which is considered as the intermediate in Ni to Ni2P. And more phosphorus was needed to form Ni2P.27 The formation of Ni12P5 and Ni at the low P content is understandable, as Ni12P5 contain a relatively lower proportion of P than Ni2P. Ni2P is formed by the further reaction between Ni12P5 and phosphorus. This is the result of the partial loss of phosphorous due to the formation of volatile P species, such as PH3, during reducing process, leading to a decrease in the amount of P on the surface. Oyama et al. also found that if the Ni/P ratio of precursor is equal to the stoichiometric ratio of Ni2P, not only Ni2P phase is formed, but also a P-deficient phase Ni12P5 was formed. Therefore, it should be noted that an amount of phosphorus above stoichiometric proportions was necessary in order to obtain the desired stoichiometry. As the Ni/P ratio further increased, with regard to 15 wt% Ni–Al-M, the new diffraction peaks at 44.4°, 51.7°, and 76.3° assigned to (111), (200), and (220) planes of crystalline metal Ni (PDF# 1-120) were clearly observed besides those of supports. These results demonstrate that the initial Ni/P molar ratio in the precursor has profound impact on the structure of NixP catalyst. Namely, different Ni/P ratios can result in different crystal phases of the catalyst and we also found that the catalyst with initial Ni/P ratio of about 1/2 had not an excellent activity. At excessive P content, the active nickel phosphide surface covered by the excess P. This is discussed in below. In a word, only pure Ni2P phase is observed with samples with higher P content, while with the sample with lower P content both Ni2P and Ni12P5 phases were observed. Furthermore, with P content fell to the lowest level, crystalline Ni formed in addition to little Ni12P5.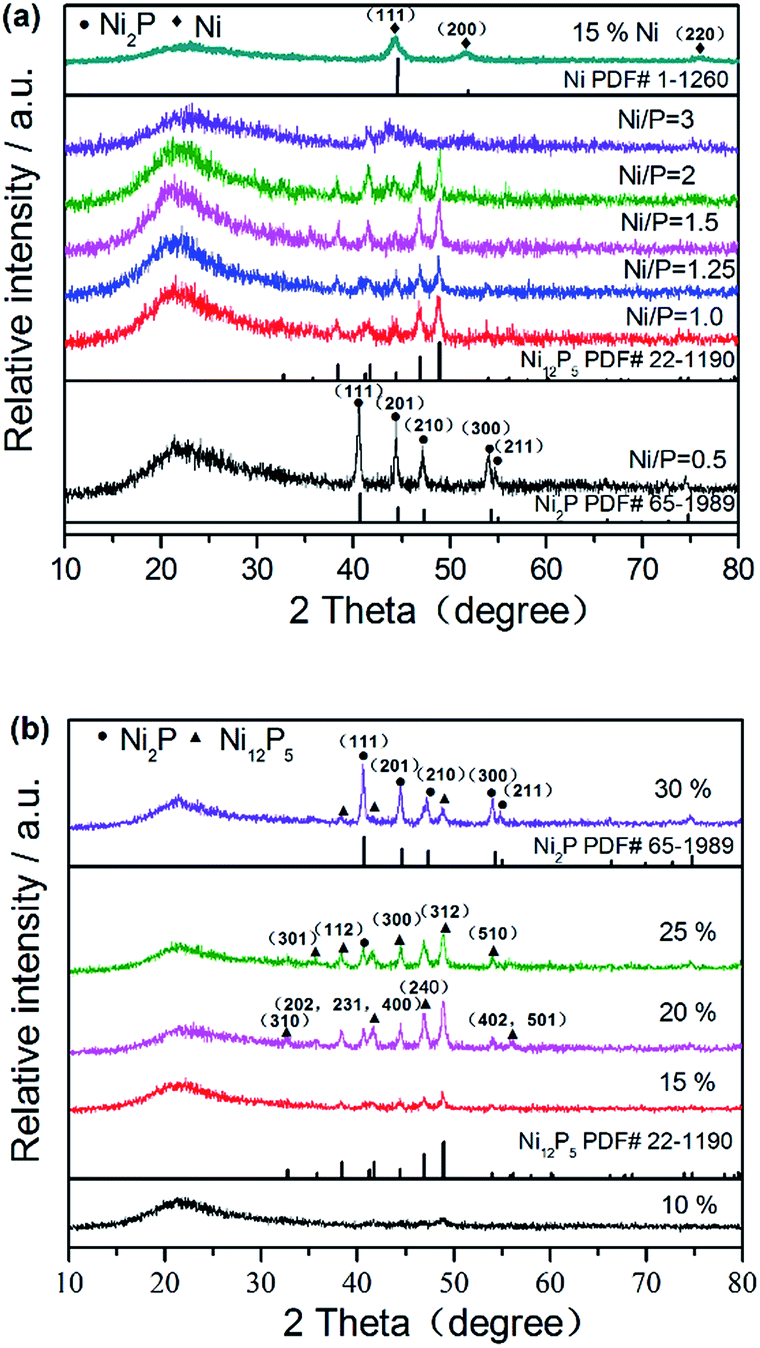 | ||
| Fig. 1 XRD patterns of (a) 15 wt% NixP–Al-M obtained from various initial Ni/P molar ratios and (b) NixP–Al-M-(1.25) with various NixP loadings. | ||
Fig. 1(b) shows the XRD patterns of NixP–Al-M with various NixP loadings from 10 wt% to 30 wt% at an initial Ni/P molar ratio of 1.25. As shown in Fig. 1(b), X-ray line-broadening analysis of the samples indicated a weak trend toward larger crystallite sizes with increasing sample loading. As will be seen, when the NixP loading reached 10 wt%, no prominent NixP diffraction peak was found compared with the other samples due to the low loading and high distribution of NixP on the support. So it was found that a low loading sample was particularly difficult to prepare with the right stoichiometry. This may represent a limitation for the preparation of samples consisting of compounds from two elements like nickel phosphide. As the NixP loading increased from 15 wt% to 20 wt%, a Ni12P5 peak emerged and the intensity of the Ni12P5 diffraction peaks strengthened, finally reached the highest at 20 wt%. With further increase in the NixP loading, the diffraction peak of Ni12P5 obviously decreased. Interestingly, when the NixP loading reached 30 wt%, mainly Ni2P diffraction peaks were observed and the Ni12P5 diffraction peaks nearly disappeared. Moreover, excess loading leads to larger particle sizes of NixP and correspondingly decreases the activity. The HDA activities of the NixP–Al-M catalysts were strongly dependent on the types and dispersion of the nickel phosphide. In this study, the formation of NixP (Ni2P and/or Ni12P5) on the surface of Al-M could be decided by controlling the loading. And it was found that catalysts of intermediate loading had high activity, as shown in below. Besides, low-angle powder X-ray diffraction patterns of the samples are depicted in Fig. S1.† Only the most intense characteristic peak of MCM-41 structure can be observed for 0.5 wt% Pt–Al-M apart from supports. This fact indicates that the structural order of MCM-41 decreases for the supported catalysts, very likely due to the uneven distribution of active phase particles inside the pores. Meanwhile, the wide-angle XRD patterns of Al-M support along with the corresponding Pt-loaded catalyst are displayed in Fig. S2.† For 0.5 wt% Pt–Al-M, diffraction peaks of platinum are hardly visible probably owing to the low content. Crystallite size of various catalysts also have evaluated based on Scherrer equation. The corresponding results are listed in last column of Table S1.† In practical terms, the crystallite sizes of samples are in close agreement with the results of particle size distribution according to TEM images, as described in Fig. 3 below.
3.2 SEM
To further confirm the composition of the catalysts and the effective loading of the NixP in the materials. Fig. 2 shows the SEM and EDS mapping images of different catalysts (red dot is nickel atom and yellow dot is phosphorus). Fig. 2(a), (b) and (d) show that the Ni–Al-M has better metal dispersion than the NixP–Al-M. Moreover, the distribution of NixP particle is relatively uniform, no obvious sintering and aggregation was observed. Compared to Fig. 2(b) and (d) show that Ni and P dispersion decreased significantly with the increase in NixP loading of the same Ni loading. Fig. 2(c) gives the mole ratio of Ni/P = 1.71 measured by EDS. Considering that EDS is a semiquantitative analysis method, and there may be due to a partial loss of the phosphorus during preparation, the mole ratio of Ni/P = 1.71 is a reasonable value. In addition, previous work had shown that phosphorus is removed as PH3 during the reduction procedure and that some P was left on the better dispersed supported Ni2P catalysts, even though higher temperatures were applied for reducing these well dispersed samples.28 This again is consistent with stronger interactions between Ni and P in the smaller crystallites. In addition, Fig. S3† shows the SEM, mapping image (cyan dot refer to platinum atom), and EDS of 0.5 wt% Pt–Al-M catalyst. This study reveals that small Pt particles with a rather homogeneous size distribution are well-dispersed on the Al-M, as shown in Fig. S3.† Besides, the amount of the platinum is 0.46 wt% measured by EDS.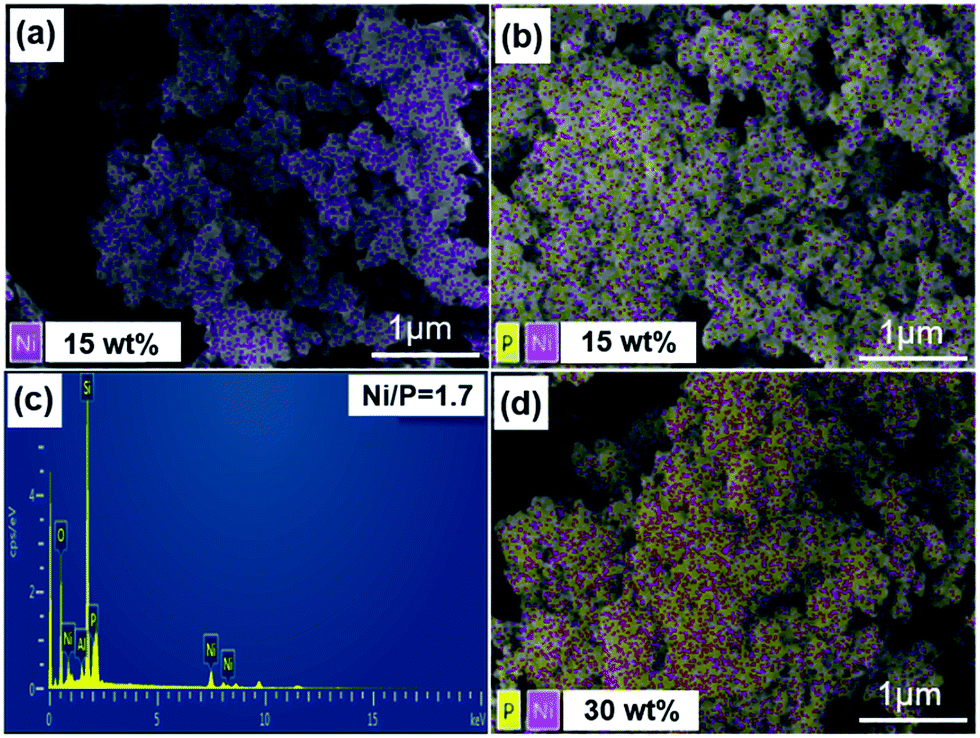 | ||
| Fig. 2 The SEM and EDS mapping images of different catalysts (red dot refer to nickel atom, and yellow dot refer to phosphorus atom), (a) 15 wt% Ni–Al-M, (b) 15 wt% NixP–Al-M-(1.25), (c) 15 wt% NixP–Al-M-(1.25), and (d) 30 wt% NixP–Al-M-(1.25). | ||
3.3 TEM
Fig. 3 shows the representative TEM images of NixP–Al-M catalysts synthesized at 650 °C from Ni(NO3)2·6H2O and NH4H2PO4 precursors with a mole ratio of Ni/P = 1.25, and with different loadings: (a) 10 wt%, (b) 15 wt%, (c) 20 wt%, (d) 30 wt%, and inset is the corresponding particle size distribution histogram. The TEM images exhibited a large number of NixP, fairly uniform globular nanoparticles with a mean particle size of about 5–10 nm, were observed to be supported on Al-M, indicating NixP has good dispersion on the Al-M support. Furthermore, the results clearly show that the distributions of the nanoparticles exhibited poor dispersion and significantly aggregation, along with the increase in the NixP loading from 10 to 30 wt%. Correspondingly, the particle size of nickel phosphides increased, which is similar to the XRD result using the Scherrer formula. A magnified high resolution TEM image of 15 wt% NixP–Al-M approximately yields d-spacing values of 0.221 nm, which was consistent with the {111} crystallographic plane of Ni2P, in good agreement with the calculated values (PDF# 65-1989), as shown in Fig. 3(d). In contrast, the mapping analysis of 15 wt% NixP–Al-M also further confirmed the presence and good distributions of nickel and phosphorus elements in Fig. S4.† Also of note that the highest nanodispersity of the NixP nanoparticles was observed on the 15 wt% NixP–Al-M for all catalysts. It shows a spherical shape with good dispersion of nanocrystalline NixP on the Al-M support. Inset show the consequent size distribution histograms of NixP-based catalysts. The micrographs obtained for each catalyst were analyzed to determine the average particle diameter and size distribution of the NixP nanoparticles. A measurement of at least 200 nanoparticles, assuming virtually spherical and randomly orientated particles was taken for the construction of the size distribution histograms. The NixP nanoparticles are homogeneously distributed over the supports. The sizes of the NixP nanoparticles for all the catalysts are about in the range of 5–80 nm.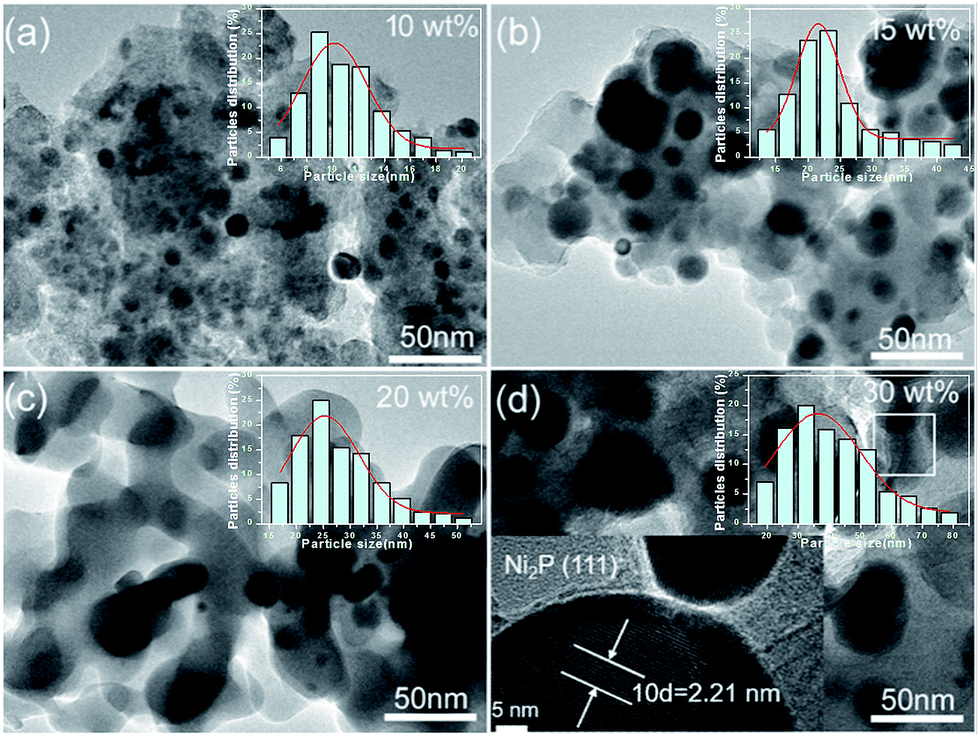 | ||
| Fig. 3 The TEM images of NixP–Al-M catalysts synthesized at 650 °C from Ni(NO3)2·6H2O and NH4H2PO4 precursors with a mole ratio of Ni/P = 1.25, and with different loadings: (a) 10 wt%, (b) 15 wt%, (c) 20 wt%, (d) 30 wt%. Inset is the corresponding particle size distribution histogram. | ||
3.4 ICP-OES
The composition of the catalysts is further measured by ICP-OES and the results were summarized in Table S2.† Obviously, as seen in Table S2,† it is clearly indicated that the experimental values were much larger than theoretical values of Ni/P molar ratio. It might be due to the extra phosphorous except volatilized as PH3 and residual POxn− species during the reduction process, partly situated in the surface of the catalyst, which is further explained in detail below.3.5 N2 sorption–desorption experiment
Fig. S5† shows the N2 adsorption–desorption isotherms of (a) 15 wt% NixP–Al-M with various Ni/P molar ratios, (b) NixP–Al-M-(1.25) with different NixP loadings, (c) MCM-41, Al-M, physically mixed MCM-41 + γ-Al2O3, denoted as (M + Al) supports, and 0.5 wt% Pt–Al-M (d) the corresponding BJH pore size distributions of supports and 0.5 wt% Pt–Al-M catalyst. As can be seen from Fig. S5(a)–(c),† all the samples of N2 isotherms are basically identical to pure supports. The samples present a reversible adsorption–desorption type IV isotherm characteristic with a hysteresis loop according to the IUPAC classification, showing the presence of some mesopores.29 This type of hysteresis corresponds to aggregates or agglomerates of particles which form pores in the form of cleft such as plates or sharp particles such as buckets, with uniform distribution. As for Al-M and (M + Al) supports, it was very similar to that of the pure MCM-41, the adsorption and desorption lines were almost overlapped, nearly leading to no hysteresis loop formation. The coincidence of the adsorption and desorption lines could be ascribed to a similar mechanism for the two processes, which reflects the absence of adsorbate pore blocking. The absence of a hysteresis loop and the sharp curvature along the second stage, suggest the existence of uniform and cylindrical channels throughout the materials. The pore size distributions obtained by BJH analysis of these curves show no marked differences among the samples as we can see from Fig. S5(d).† Pore distribution for all of the samples was mono-modal showing that they belong to the mesoporous materials. The pore diameters for all samples were almost same to original MCM-41, suggesting that NixP particles were basically limited on the external surface of the mesoporous support and only some of the MCM-41 was destroyed after NixP loading. In addition, the corresponding textural characterization of the supports and catalysts are presented in Table S1.† The BET specific surface area and pore volume of the MCM-41 were 893 m2 g−1 and 0.96 cm3 g−1, respectively. However, the incorporation of alumina in the MCM-41 matrix resulted in a slight decrease on the surface area and the pore volume, which can be attributed to the stability of the siliceous material and also a decrease in the pore volume due to the blockage of some from pores of the MCM-41.30 The date also indicated that the deposition of NixP and Pt on the support surface causes a greater drop in the surface area and pore volume. Meanwhile, both the BET surface areas and pore volumes of the samples varied from high to low, with corresponding NixP loadings increasing from 10 to 20 and 30 wt%. One reason is that active component loadings increased the catalyst density so that the proportion of Al-M in the total quality of catalysts correspondingly decreased. The other is that a number of pores of the supports were blocked/filled by NixP during the catalyst preparation. However, no significantly change in the pore diameter is observed from catalysts compared to their corresponding samples, indicating that no additional P is deposited on the walls of the pores.3.6 NH3-TPD
NH3-TPD was performed to elucidate the relationship between the catalysts activity and types of acidic sites on the catalysts. Fig. 4 shows the NH3-TPD profiles of the MCM-41, Al-M supports, and the supported NixP catalysts with different Ni/P molar ratios. In a typical NH3-TPD profile, peaks are generally observed in three temperature regions. The regions of the Tmax (temperature maximum) from 20 to 200 °C, Tmax from 200 to 500 °C, and Tmax higher than 500 °C are referred to as low-temperature, moderate-temperature and high-temperature regions, corresponding to weak acidic sites, intermediate acidic sites, and strong acidic sites, respectively.31 The principal objective of this work is to increase the stability as well as the acidity of the support by adding alumina, which play a vital role in the HDA and product selectivity during hydrotreating. Generally, MCM41 support presented the lowest amount of acid sites. As expected, the addition alumina to the MCM-41 supports, the surface acidity increases with decreasing the Si/Al molar ratio. Desorption peak around low-temperature was assigned to Lewis and Brönsted acid sites of weak acidic strength, while desorption peak about high-temperature was assigned to intermediate Brönsted acid sites (usually bridging Si–(OH)–Al groups).32–34 It is demonstrated that there are the weak and medium strength Brönsted acid sites on Al-M, which correspond to the NH3 desorptions with the peaks at about 120 and 400 °C, respectively, as shown in Fig. 4. However, when Ni or NixP was supported on Al-M, it is composed of two types of weak acid sites, Lewis and Brönsted, which are ascribed to the electron-deficient Ni site and to the P–OH, Si(OH)Al group, respectively. Since the electron-deficiency of these nickel ions is very high and nickel in nickel phosphide exists in positive charges,35 they may play a role of Lewis sites. Also an additional amount of weak acidic sites was found with a shift of the peak to lower temperature shown in Fig. 4. The acid sites on Al-M still maintained, but some of them could be covered by Ni or NixP particles. Besides, as indicated in Fig. 4, Ni–Al-M had more medium strength Brönsted acid sites but less weak strength acid sites than NixP–Al-M. In other words, there is less medium strength Brönsted acid sites on NixP–Al-M, which is due to the coverage of acid sites with P. The amount and strength of acid sites are related not only to the activity of naphthalene HYD but also to the extent of concomitant further cracking and coke disposition. Meanwhile, the total acidity amounts are listed in Table S4.† Moreover, NH3-TPD profiles for supports MCM-41, physically mixed M + Al, Al-M supports, and 0.5 wt% Pt–Al-M catalyst are depicted in Fig. S6.† In general, these profiles for Pt-based catalyst consist of two peaks. One appears at a low temperature range of around 150–200 °C while the other appears at a high temperature range of around 350–450 °C. The low and high temperature peaks correspond to the weak and strong acid sites, respectively.36 In this study, as shown in Fig. S6,† after the addition of Pt, two patterns for NH3 desorption appeared. The first curve was due to the NH3 adsorption on pure mesoporous surface. The second curves were due to the NH3 adsorption on the impregnated Pt species on Al-M.36 Obviously, the NH3 desorption peak temperatures and areas increase, which illustrates that the addition of Pt can effectively enlarge the number and strength of acid sites over 0.5 wt% Pt–Al-M catalyst. In particular, the NH3 amounts desorbed at low temperature reached about 3 times in 0.5 wt% Pt–Al-M compared with pure Al-M.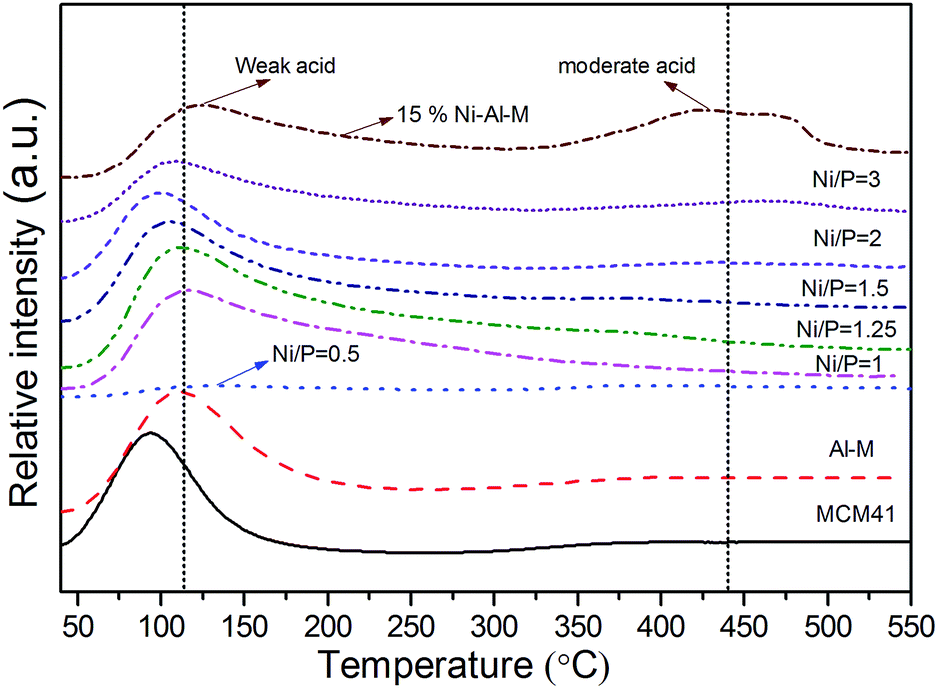 | ||
| Fig. 4 NH3-TPD profiles of the MCM-41, Al-M supports, and the supported NixP catalysts with different Ni/P molar ratios. | ||
3.7 XPS
In order to gain further insight into the surface composition of surface oxidation layer and the valence states of active components, the XPS of catalysts was performed. The XPS Ni 2p and P 2p spectra of 15 wt% NixP–Al-M-(1.25) catalyst are presented in Fig. 5(a) and (b). All the spectra were decomposed by fitting using Origin Pro. 8.5, in which the black lines represent the experimental data, the red lines are the envelopes of decomposed features and the other lines are the Gaussian components peaks, as shown in Fig. 5. According to the result in Fig. 5(a), we can see that the Ni 2p core level spectrum involves two contributions, which correspond to the spin-orbital splitting of the Ni 2p3/2 and Ni 2p1/2 lines (about 17 eV), respectively. Ni 2p3/2 core-level spectrum consists of three components, the first of which centered at 851.6–853.5 eV can be assigned to reduced Niδ+ species in the NixP phase, and the second is at 856.3–857.6 eV, corresponding to Ni2+ ions interacting possibly with phosphate ions as a consequence of a superficial passivation,37 along with the broad satellite peak at approximately 6.0 eV higher than that of the Ni2+ species, and this shake-up peak is ascribed to divalent species. This observation is consistent with that of Korányi et al.38 Other broad peaks on the high binding energy side can be assigned to the Ni 2p1/2 components. The P 2p binding energy spectrum features a low intensity peak at 129.5 eV due to Pδ− of the reduced metal phosphides and a peak at 134.5 eV due to surface metal phosphate species in PO43− arising from the superficial oxidation of the nickel phosphide particles. The result is similar to other report.39 Generally, the higher binding energy indicates a lower electron density. The Ni 2p binding energies in NixP phase are much higher than reported values for Ni metal (852.5–852.9 eV) and lower than those reported for Ni in NiO (854.1 eV),40 which is influenced by the formation of the greater interaction with the support, indicating that the Ni in NixP carries a partial positive charge (δ+), where 0 < δ < 2, whereas the P 2p binding energies are below the value reported for elemental phosphorus (130.2 eV), revealing the small transfer of electron from Ni to P in nickel phosphide and a partial negative charge (δ−) carried on P surface, where 0 < δ < 1.40 Moreover, an active site with a high electron density favors the formation of a π back bond between the aromatic ring and active sites, which promotes the HYD of the aromatic ring. Accordingly, XPS analyses are also used to calculate the surface Ni/P atomic ratios as well as surface compositions of the catalysts before their use. The XPS analysis results for 15 wt% NixP–MCM-41-(1.25) and 15 wt% NixP–Al-M-(1.25) catalysts, given in Table S3.† It can be seen that as prepared catalysts exhibited lower contents values of Ni, P and the surface Ni/P ratios determined using XPS are far below the theoretical Ni/P ratio corresponding to the precursors, which shows that surplus phosphorous tend to distribute on the surface of the catalysts.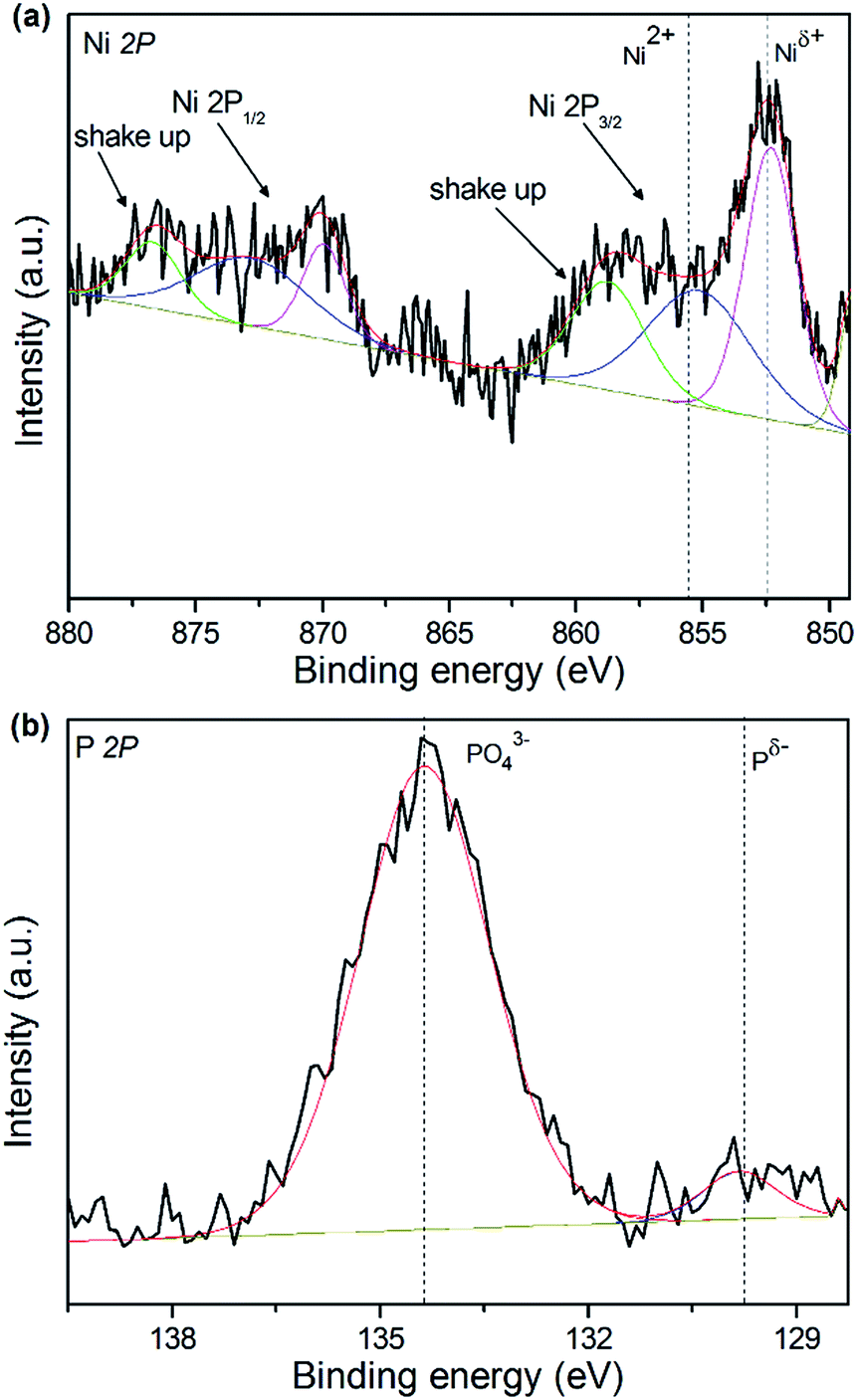 | ||
| Fig. 5 XPS spectra in the Ni 2p and P 2p regions for 15 wt% NixP–Al-M-(1.25) catalyst: (a) Ni 2p core level spectra and (b) P 2p core level spectra. | ||
3.8 Hydrogenation activity
Previous studies have shown that the activity of aromatic HYD depended not only on the amounts of accessible active sites, but also on the surface acid properties.41 Typically, the naphthalene HYD, which was a first-order reaction. There are three major products formed from naphthalene on these catalysts: trans-decalin, cis-decalin and tetralin. Meanwhile, trans- and cis-decalin were integrated as decalin. The NixP loadings and Ni/P ratio had been reported to have an important role in catalytic activity. To investigate the catalytic activity of NixP–Al-M, the HYD of naphthalene was conducted in a fixed bed reactor at the conditions of P = 3 MPa, T = 250–330 °C and WHSV = 1 h−1. Fig. 6(a) shows the influence of NixP loadings on HDA activity and selectivity for naphthalene over NixP–Al-M catalysts at a Ni/P ratio of 1.25 after 6 h on-stream. As depicted in Fig. 6(a), it could be observed that all catalysts showed high naphthalene conversion. With the increasing of the loading, the catalytic activity and selectivity first increases and then decreases. Accordingly, samples with low loadings showed low selectivity to decalin. When the NixP loading was 10 wt%, the selectivity to decalin was only 44.7%, and when the loadings reached 20 wt% and 25 wt%, the selectivity to decalin were 98.0% and 95.9%, respectively. But further increasing of loading led to decline of activity and selectivity. When the NixP loading rises to 30 wt%, the selectivity to decalin rapidly decreased to 36.3%. The HDA properties decrease as a result of the lack of catalytic activity centers, the poor NixP dispersion and the smaller surface area led by excessive load. Combined with the XRD, SEM, and TEM analysis, these results suggest that the loading content and dispersion both have an effect on the HDA properties. Moreover, the one with 15 wt% loading had went through the highest naphthalene conversion and selectivity to decalin, which were 100% and 99.9% at 270 °C. Fig. S7† present HDA activity performed over the temperature range of 250–330 °C. It shows the effect of temperature on the selectivity. Wherein, the decrease in selectivity to decalin with increase in reaction temperature and this appears to be common for all the catalysts, which is probably due to thermodynamic equilibriums limitations and coke formation that can hinder the access to the active sites, as it has been previously found.42 Correspondingly, the conversion toward naphthalene and the selectivity to decalin went through a maximum at about 270 °C. As mentioned earlier, the sample with NixP loading of 15 wt% gave highest activity for decalin formation. Therefore, we studied the effect of initial Ni/P ratio with the NixP loading of 15 wt% on the HYD of naphthalene. The results of HYD of naphthalene over those samples with different Ni/P molar ratios at the level of 15 wt% loading and a reaction temperature of 270 °C were exhibited in Fig. 6(b). As a general trend, it was obvious that the HDA activity altered with the change of the Ni/P molar ratios and the corresponding naphthalene conversion and selectivity towards decalin first increased and then decreased with the increase of the Ni/P molar ratio, as shown in Fig. 6(b). Much the same as above analysis, two main products are detected: decalin and tetralin. No rupture products were detected except tetralin and decalin, indicating the acidity on the NixP–Al-M sample could not catalyze the open-ring reaction of naphthalene. The product selectivity with Ni/P ratio of 1/2 mainly contained tetralin, and the decalin content is lowest, only about 2.0%, and that of other samples are higher. Along with the increase of initial Ni/P ratio, and the selectivity of decalin increases dramatically. The sample with an initial Ni/P ratio of 1.25 has the highest HYD conversion and the selectivity to decalin, about 100% and 99.9%, which is higher than that of the other four samples, with Ni/P ratios of 1.0, 1.5, 2.0, and 3.0. The likely reason is that, on the one hand, with lower P content, some P-deficient Ni12P5 or Ni metal phase are formed, which have been found to be less active in HYD reaction. Nevertheless, on the other hand, at higher P content level, the extra phosphorous partially located at the outer surface of the catalyst,43 further covering the active sites and leading to blocking some apertures of the NixP–Al-M catalysts, as it has been previously mentioned. Moreover, it is possible affected by the relatively large crystallite size of nickel phosphide, which should reduce the number of available centers for HYD activity. Similaring to the optimal Ni/P ratio of 1.25 for the SiO2 supported Ni–P catalyst reported in the published literatures,44 our work also shows the optimal Ni/P ratio is 1.25. Besides, the detailed product distributions obtained with different temperature are also shown in Fig. S8.† It can be seen that product distributions over the six catalysts showed a similar current: the selectivity of decalin increased with reaction temperature until a plateau was noticed at around 270 °C. When temperature was approximately above 300 °C, concentrations of both trans-decalin and cis-decalin declined, and concentration of tetralin slightly increased. This indicates a high temperature does not promote further HYD. Fig. S9† compares the conversions and selectivities obtained from naphthalene HYD over Ni-, NixP-, and Pt-supported catalysts. As shown in Fig. S9,† the result showed that 15 wt% NixP–Al-M has the highest naphthalene conversion and decalin selectivity than 15 wt% Ni–Al-M, 15 wt% NixP–MCM-41, 0.5 wt% Pt–Al-M and physical mixed 15 wt% NixP–(M + Al) samples, which indicates that the support plays important role and this implies again the occurrence of a synergistic effect between the support and NixP. Combined with characterization results mentioned above, it seems that the electronic state of NixP surface, acid site number as well as strength, and particle size provide significant effects on the catalytic performance. The MCM-41 surface is homogeneous and had hardly any acid sites.45 The interaction between the supported NixP and the MCM-41 support is very weak. Some NixP sintering and agglomeration to large particles occurs during calcination and reduction. However, the aluminum promoters affect the acid property and the electron property as well as improving the dispersion of the NixP particles via isolation and anchor effects. The improvement in NixP dispersion, which implies more active sites, is sure to exert an effect on the catalytic activity. Combining the SEM and TEM results with the naphthalene conversions of the catalysts, it is obvious that the higher the NixP dispersion, the better the naphthalene HYD activity. Based on above results, these facts clearly indicated that the surface acidity of supported nickel phosphide catalysts played an important role on the adsorption of naphthalene for the HYD. And a more detailed comparison of the differences in the product distribution over these catalysts with various temperature is displayed in Fig. S10(a)–(e).† As we can see from Fig. S10,† an almost complete conversion of naphthalene into decalin was observed over the reaction temperature range of 250–330 °C. 15 wt% NixP–Al-M catalyst mostly converted naphthalene into decalin at lower temperature, in contrast with the 15 wt% Ni–Al-M catalyst. It also can be obviously seen that the tetralin selectivity steadily increased with the further raise of reaction temperature along with the decrease of naphthalene conversion due to the equilibrium limitation of 15 wt% NixP–MCM-41 and 15 wt% NixP–(M + Al) catalysts. At 270 °C, the 15 wt% NixP–MCM-41 catalyst showed a naphthalene conversion of 99.0% with the tetralin and decalin selectivities of 39.3 and 60.8%, respectively. And at 330 °C, the tetralin and decalins selectivities were almost 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at the naphthalene conversion of 98.7%. The 15 wt% NixP–(M + Al) catalyst showed a moderate level of naphthalene conversion and tetralin selectivity, compared with 15 wt% NixP–MCM-41 and 15 wt% NixP–Al-M. Above results validate the fact that the acidity of the support plays a significant role in the HYD of aromatic molecules and the catalyst prepared by depositing NixP on acidic supports shows higher activity than the catalyst prepared by depositing NixP on non-acidic supports as reported elsewhere. So the higher activity of NixP–Al-M, compared with the 15 wt% NixP–MCM-41 and 15 wt% NixP–(M + Al) catalysts, can be attributed to the stronger surface acidity of the Al-M support which coupled with hydrogen spillover effect favoring the HYD of aromatics. The 15 wt% NixP–Al-M catalysts exhibited high decalin selectivity at high naphthalene conversion level while the reference 0.5 wt% Pt–Al-M catalyst showed a relatively low HYD activity towards naphthalene. This implied the high HYD abilities of the NixP–Al-M catalysts and may take the replace of noble metals in the future. Thus it is easy to understand and conclude that the naphthalene HYD activity is first related to the NixP dispersion. The second factor that affects the catalytic activity is the electron density of the NixP particles. Thirdly, the acid sites and hydroxyls provide additionally active sites and favour spillover hydrogen, which also contributes to HYD activity. Furthermore, long-term stability test for the 15 wt% NixP–Al-M-(1.25) catalyst at 270 °C is displayed in Fig. S11.† As shown in Fig. S11,† during the activity testing, catalysts have not shown deactivation trend on the whole, which indicated that 15 wt% NixP–Al-M-(1.25) catalyst showed good activity and stability. In order to get rid of the influence of different exposed active sites on different catalysts, we chose TOF as a significant the activity of per active site in unit time. It is reported that the numbers of active sites on the NixP surfaces can be titrated by the adsorption of CO since CO species could effectively titrate active centers of NixP via the formation of π back-bonding of higher-electron density NixP species, according to which the turnover frequency (TOF) values for the hydrotreating reactions can be calculated to gain an insight on the number of naphthalene molecules converted to decalin on a catalytic site per second.46 The corresponding CO chemisorption uptakes and turnover frequency (TOF) results based on CO adsorption are presented in the last two columns of Table S4.† Seen from the Table S4,† the TOFs of the all catalysts tested were nearly the same, as anticipated.
:1 at the naphthalene conversion of 98.7%. The 15 wt% NixP–(M + Al) catalyst showed a moderate level of naphthalene conversion and tetralin selectivity, compared with 15 wt% NixP–MCM-41 and 15 wt% NixP–Al-M. Above results validate the fact that the acidity of the support plays a significant role in the HYD of aromatic molecules and the catalyst prepared by depositing NixP on acidic supports shows higher activity than the catalyst prepared by depositing NixP on non-acidic supports as reported elsewhere. So the higher activity of NixP–Al-M, compared with the 15 wt% NixP–MCM-41 and 15 wt% NixP–(M + Al) catalysts, can be attributed to the stronger surface acidity of the Al-M support which coupled with hydrogen spillover effect favoring the HYD of aromatics. The 15 wt% NixP–Al-M catalysts exhibited high decalin selectivity at high naphthalene conversion level while the reference 0.5 wt% Pt–Al-M catalyst showed a relatively low HYD activity towards naphthalene. This implied the high HYD abilities of the NixP–Al-M catalysts and may take the replace of noble metals in the future. Thus it is easy to understand and conclude that the naphthalene HYD activity is first related to the NixP dispersion. The second factor that affects the catalytic activity is the electron density of the NixP particles. Thirdly, the acid sites and hydroxyls provide additionally active sites and favour spillover hydrogen, which also contributes to HYD activity. Furthermore, long-term stability test for the 15 wt% NixP–Al-M-(1.25) catalyst at 270 °C is displayed in Fig. S11.† As shown in Fig. S11,† during the activity testing, catalysts have not shown deactivation trend on the whole, which indicated that 15 wt% NixP–Al-M-(1.25) catalyst showed good activity and stability. In order to get rid of the influence of different exposed active sites on different catalysts, we chose TOF as a significant the activity of per active site in unit time. It is reported that the numbers of active sites on the NixP surfaces can be titrated by the adsorption of CO since CO species could effectively titrate active centers of NixP via the formation of π back-bonding of higher-electron density NixP species, according to which the turnover frequency (TOF) values for the hydrotreating reactions can be calculated to gain an insight on the number of naphthalene molecules converted to decalin on a catalytic site per second.46 The corresponding CO chemisorption uptakes and turnover frequency (TOF) results based on CO adsorption are presented in the last two columns of Table S4.† Seen from the Table S4,† the TOFs of the all catalysts tested were nearly the same, as anticipated.
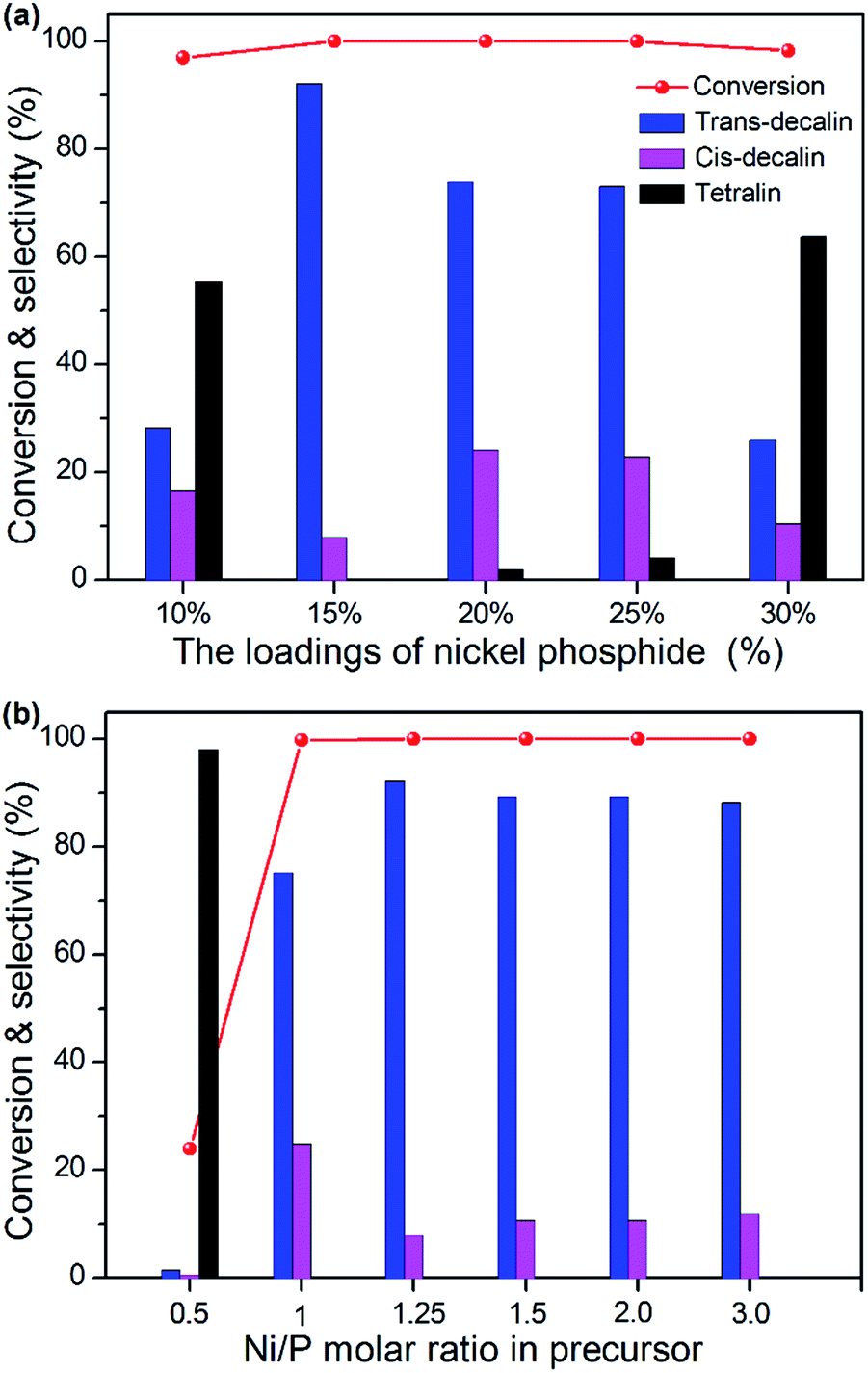 | ||
| Fig. 6 Naphthalene conversion and selectivity to decalin (a) over NixP–Al-M with different NixP loadings and (b) over NixP–Al-M with different Ni/P molar ratio in precursor as 0.5, 1.0, 1.25, 1.5, 2.0, and 3.0 at 270 °C and 3 MPa. | ||
3.9 Possible mechanism
It has been widely reported that the naphthalene HYD occurred in two consecutive steps and a reaction pathway could be figured out as shown in Scheme 1: conversion to tetralin in the first step through sequential cis-addition steps of two dissociatively adsorbed hydrogen atoms, followed by further formation of decalin in the second step according to the following formula:| C10H8 + 2H2 → C10H12 + 3H2 → C10H18 (ref. 44) | (2) |
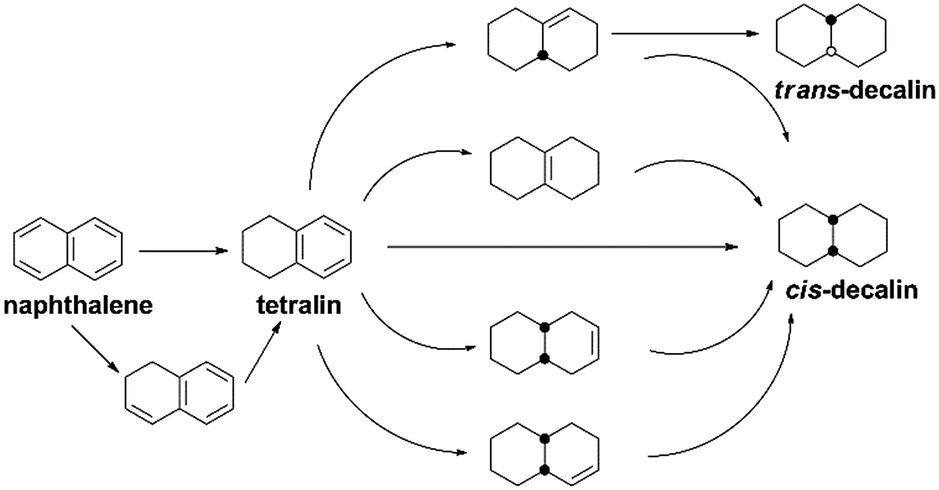 | ||
| Scheme 1 Reaction pathway of naphthalene hydrogenation. | ||
Decalin has two isomers, which are trans- and cis-decalin, and can be used as thermal stable jet fuel in extreme condition. Hence, the selectivity to decalin is used in our study as the main criterion to evaluate the catalytic performance. Pang et al. studied naphthalene HYD and found that the HYD rate of naphthalene to tetralin was an order of magnitude faster than that of tetralin to decalin.47 Some evidence showed that olefins played an important role in the reaction scheme. Naphthalene react to Δ9,10-octalin, which is either directly hydrogenated to cis-decalin at high pressure or isomerized to intermediate Δ1,9-octalin. Δ1,9-octalin is, in turn, hydrogenated to cis- and trans-decalin. Tetralin reacted directly with hydrogen to form cis-decalin, or through the intermediate Δ1,9-octalin. But tetralin could not convert directly into trans-decalin. It needed to convert to Δ1,9-octalin and then to trans-decalin. As aforementioned, Δ1,9-octalin is widely reported to be the most reactive intermediate product in the reaction tetralin → decalin, therefore, the formations of trans- or cis-decalin are closely related to this intermediate. Based on the conformation between the intermediate Δ1,9-octalin and the catalyst surface, the possible formation of trans- or cis-decalin could be inferred considering the syn nature of hydrogen addition, which means that when the H atoms are added to a double bond, both H atoms are always on the same side.48 Dokjampa et al. proposed that the formation mechanisms of trans- or cis-decalin depended on the hydrogen atom in position 10 facing towards or away from the catalyst surface.48 When the hydrogen atom in position 10 of this intermediate is oriented facing towards the catalyst surface, the HYD of the double bond results in the formation of cis-decalin, because hydrogen addition to a double bond is always syn (both H atoms are added on the same side). By contrast, when the hydrogen atom in position 10 is oriented facing away from the catalyst surface, the syn HYD addition results in the formation of trans-decalin. According to our results, the very good catalytic performance is due to the interaction between the support acid sites and the HYD sites on bifunctional catalysts and a possible reaction mechanism for the HYD of naphthalene on nickel phosphide catalyst can be proposed as depicted in Fig. 7. One side, NixP can be ascribed to the effects and behavior of the P sites and the formation of Ni–P bonds. First, in NixP, the concentration of Ni is diluted by the presence of P, the formation of Ni–P bonds induces relatively minor perturbations in the electronic properties of nickel.49 Namely, a weak ligand effect (minor stabilization of the Ni 3d levels and a small Ni → P light charge transfer) that allows a reasonably high activity for the dissociation of naphthalene and molecular hydrogen,49 forming spillover of activated hydrogen from HYD sites to the acidic support. Second, the number of exposed active Ni sites present in the surface decreases due to an ensemble effect of P, which prevents the system from the deactivation.49 Third, the P sites are not simple spectators and provide moderate bonding to the products of the decomposition of naphthalene and the H adatoms necessary for HYD.49 NixP–Al-M is a highly active HDA catalyst by obeying Sabatier's principle: good bonding with the reactants, and moderate bonding with the products. On the other side, a possible explanation for the influence of the acidic sites in the HYD activity of the NixP–Al-M catalyst may be mainly related to the migration of electron charge. Detailed reasons are as follows: firstly, the surface acidity favored the adsorption of naphthalene since naphthalene can be taken as a Lewis base due to the enriched electron density on the aromatic ring with π-bonds.24 Secondly, the NixP supported on the acidic Al-M surface might be electron-deficient due to the electron charge transfer from NixP to acidic sites, leading to changing the surface charge density of nickel and formation of electron deficient metal sites in close vicinity of the support acid sites, increasing the activity. Thus the adsorption of naphthalene on the electron-deficient nickel might be enhanced due to the enhanced electron donor from the naphthalene to nickel atoms. Namely, the electron-deficient surface nickel atoms on the acidic, which favored the donation of electron charges from naphthalene to the electron-deficient d orbitals of surface nickel. Apparently, the surface acidity also favored the adsorption and activation of aromatic rings.50
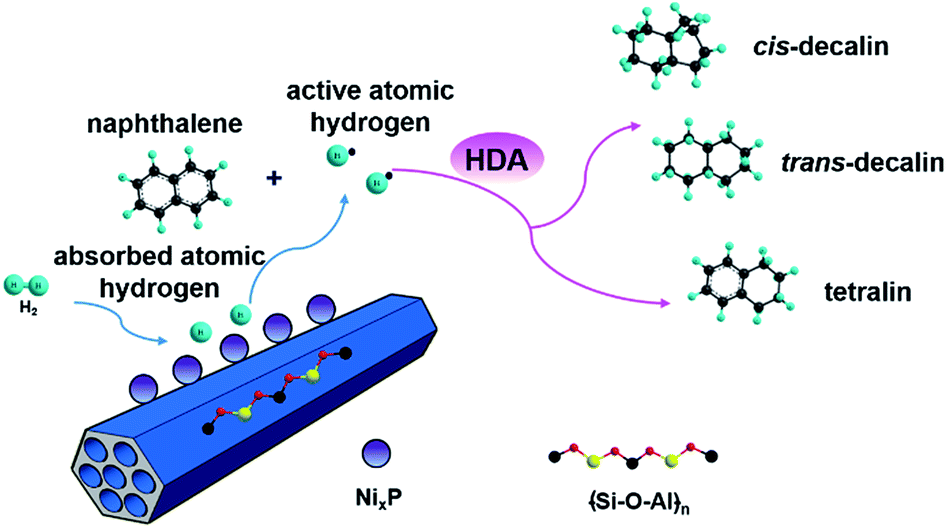 | ||
| Fig. 7 Proposed reaction schematic of naphthalene HDA over highly active NixP–Al-M catalyst. | ||
4. Conclusions
In summary, using Al-M as the support, NixP–Al-M, a series of supported nickel phosphide catalysts with different Ni/P molar ratios and loadings have been prepared by a temperature-programmed reduction method. The naphthalene HDA result shows that both the Ni/P ratio and loading of the catalysts have profound effect on the naphthalene HYD performance. The results showed that the catalyst with initial Ni/P molar ratios of and 15 wt% loading displayed the highest HDA activity as well as 99.0% of selectivity of decalin at 270 °C, which is even higher than that of 0.5 wt% Pt–Al-M catalyst. As a comparison, the HDA performance of various catalysts were also examined. The results revealed that the presence of framework aluminum favors HDA processes and a synergistic effect between nickel phosphide and the suitable acidity resulted in an improvement of the catalytic activity. These results indicated that the catalyst performance is related to its acid property, pore structure, high dispersion, the number of active sites, and the intimate contact between NixP and the support. Finally, the possible reaction pathway of naphthalene HYD on nickel phosphide catalysts was proposed. Considering the high HDS, HDN, and HDA activities of nickel phosphide catalysts, NixP–Al-M can be considered as a very efficient HYD catalyst to produce clean fuels. In our future work, the catalytic activities of nickel phosphide catalysts for HDS and HDA of jet fuel will be studied.Acknowledgements
This work was financially supported by the NSFC (21376123, U1403293, 21603107), MOE (IRT-13R30 and 113016A), NSFT (15JCQNJC05500), and the Research Fund for 111 Project (B12015).References
- A. Zhao, X. F. Zhang, X. Chen, J. C. Guan and C. H. Liang, J. Phys. Chem. C, 2010, 144, 3962–3967 Search PubMed.
- C. S. Song and X. L. Ma, Appl. Catal., B, 2003, 41, 207–238 CrossRef CAS.
- S. De, J. G. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 CAS.
- S. B. Qiu, X. Zhang, Q. Y. Liu, T. J. Wang, Q. Zang and L. L. Ma, Catal. Commun., 2013, 42, 73–78 CrossRef CAS.
- A. C. A. Monteiro-Gezork, A. Effendi and J. M. Winterbottom, Catal. Today, 2007, 128, 63–73 CrossRef CAS.
- Q. Wang, X. C. Cai, Y. Q. Liu, J. Y. Xie, Y. Zhou and J. Wang, Appl. Catal., B, 2016, 189, 242–251 CrossRef CAS.
- W. T. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- B. Pawelec, R. Mariscal, R. M. Navarro, S. van Bokhorst, S. Rojas and J. L. G. Fierro, Appl. Catal., A, 2002, 225, 223–237 CrossRef CAS.
- N. Hiyoshi, A. Yamaguchi, C. V. Rode, O. Sato and M. Shirai, Catal. Commun., 2009, 10, 1681–1684 CrossRef CAS.
- S. T. Oyama, J. Catal., 2003, 216, 343–352 CrossRef CAS.
- S. F. Yang, C. H. Liang and R. Prins, J. Catal., 2006, 237, 118–130 CrossRef CAS.
- R. Wang and K. J. Smith, Appl. Catal., A, 2009, 361, 18–25 CrossRef CAS.
- X. P. Duan, Y. Teng, A. J. Wang, V. M. Kogan, X. Li and Y. Wang, J. Catal., 2009, 261, 232–240 CrossRef CAS.
- A. H. Janssen, A. J. Koster and K. P. de. Jong, J. Phys. Chem. B, 2002, 106, 11905–11909 CrossRef CAS.
- J. C. Groen, W. D. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef CAS PubMed.
- J. Panpranot, K. Pattamakomsan, J. G. Goodwin Jr and P. Praserthdam, Catal. Commun., 2004, 5, 583–590 CrossRef CAS.
- S. Nassreddine, S. Casu, J. L. Zotin, C. Geantet and L. Piccolo, Catal. Sci. Technol., 2011, 1, 408–412 CAS.
- J. I. Park, J. K. Lee, J. Miyawaki, Y. K. Kim, S. H. Yoon and I. Mochida, Fuel, 2011, 90, 182–189 CrossRef CAS.
- M. X. Du, Z. F. Qin, H. Ge, X. K. Li, Z. J. Lu and J. G. Wang, Fuel Process. Technol., 2010, 91, 1655–1661 CrossRef CAS.
- K. B. Sidhpuria, P. A. Parikh, P. Bahadur, B. Tyagi and R. V. Jasra, Catal. Today, 2009, 141, 12–18 CrossRef CAS.
- L. J. Simon, J. G. van Ommen, A. Jentys and J. A. Lercher, Catal. Today, 2002, 73, 105–112 CrossRef CAS.
- T. D. Tang, C. Y. Yin, L. F. Wang, Y. Y. Ji and F. S. Xiao, J. Catal., 2008, 257, 125–133 CrossRef CAS.
- B. Pawelec, R. Mariscal, R. M. Navarro, S. van Bokhorst, S. Rojas and J. L. G. Fierro, Appl. Catal., A, 2002, 225, 223–237 CrossRef CAS.
- R. S. Araujo, D. C. S. Azevedo, C. L. Cavalcante Jr, A. Jimenez-Lopez and E. Rodrıguez-Castellon, Microporous Mesoporous Mater., 2008, 108, 213–222 CrossRef CAS.
- B. J. Liu, Q. L. Meng, H. Wang and Q. M. Meng, Acta Phys.-Chim. Sin., 2010, 26, 3257–3262 CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón and A. Jiménez-López, J. Catal., 2009, 263, 4–15 CrossRef CAS.
- Y. Y. Shu and S. T. Oyama, Carbon, 2005, 43, 1517–1532 CrossRef CAS.
- X. Q. Wang, P. Clark and S. T. Oyama, J. Catal., 2002, 208, 321–331 CrossRef CAS.
- M. S. Li, K. N. Hui, K. S. Hui, S. K. Lee, Y. R. Cho, H. Lee, W. Zhou, S. Cho, C. Y. H. Chao and Y. Y. Li, Appl. Catal., B, 2011, 107, 245–252 CrossRef CAS.
- T. Klimova, M. Calderón and J. Ramıírez, Appl. Catal., A, 2003, 240, 29–40 CrossRef CAS.
- D. Li, P. Bui, H. Y. Zhao, S. T. Oyama, T. Dou and Z. H. Shen, J. Catal., 2012, 290, 1–12 CrossRef CAS.
- D. S. Zhang, R. J. Wang and X. X. Yang, Catal. Commun., 2011, 12, 399–402 CrossRef CAS.
- H. Kosslick, G. Lischke, B. Parlitz, W. Storek and R. Fricke, Appl. Catal., A, 1999, 184, 49–60 CrossRef CAS.
- K. Góra-Marek and J. Datka, Appl. Catal., A, 2006, 302, 104–109 CrossRef.
- M. V. Landau, M. Herskowitz, T. Hoffman, D. Fuks, E. Liverts, D. Vingurt and N. Froumin, Ind. Eng. Chem. Res., 2009, 48, 5239–5249 CrossRef CAS.
- J. H. Jang, S. C. Lee, D. J. Kim, M. Kang and S. J. Choung, Appl. Catal., A, 2005, 286, 36–43 CrossRef CAS.
- T. Chen, B. L. Yang, S. S. Li, K. L. Wang, X. D. Jiang, Y. Zhang and G. W. He, Ind. Eng. Chem. Res., 2011, 50, 11043–11048 CrossRef CAS.
- T. I. Korányi, Z. Vít, D. G. Poduval, R. Ryoo, H. S. Kim and E. J. M. Hensen, J. Catal., 2008, 253, 119–131 CrossRef.
- X. Li, Y. L. Zhang, A. J. Wang, Y. Wang and Y. K. Hu, Catal. Commun., 2010, 11, 1129–1132 CrossRef CAS.
- S. J. Sawhill, K. A. Layman, D. R. Van Wyk, M. H. Engelhard, C. M. Wang and M. E. Bussell, J. Catal., 2005, 231, 300–313 CrossRef CAS.
- S. H. Hu, M. W. Xue, H. Chen and J. Y. Shen, J. Chem. Eng., 2010, 162, 371–379 CrossRef CAS.
- M. Rozwadowski, M. Lezanska, J. Wloch, K. Erdmann, R. Golembiewski and J. Kornatowski, Chem. Mater., 2001, 13, 1609–1616 CrossRef CAS.
- Y. K. Lee, Y. Y. Shu and S. T. Oyama, Appl. Catal., A, 2007, 322, 191–204 CrossRef CAS.
- X. F. Zhang, Q. M. Zhang, A. Q. Zhao, J. Guan, D. M. He, H. Q. Hu and C. H. Liang, Energy Fuels, 2010, 24, 3796–3803 CrossRef CAS.
- M. J. Luo, Q. F. Wang, G. Z. Li, X. W. Zhang, L. Wang and T. Jiang, Catal. Sci. Technol., 2014, 4, 2081–2090 CAS.
- Y. K. Lee and S. T. Oyama, J. Catal., 2006, 239, 376–389 CrossRef CAS.
- M. Pang, C. Y. Liu, W. Xia, M. Muhler and C. H. Liang, Green Chem., 2012, 14, 1272–1276 RSC.
- S. Dokjampa, T. Rirksomboon, S. Osuwan, S. Jongpatiwut and D. E. Resasco, Catal. Today, 2007, 123, 218–223 CrossRef CAS.
- P. Liu, J. A. Rodriguez, T. Asakura, J. Gomes and K. Nakamura, J. Phys. Chem. B, 2005, 109, 4575–4583 CrossRef CAS PubMed.
- V. R. Choudhary and K. Mantri, Langmuir, 2000, 16, 7031–7037 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00250e |
| This journal is © The Royal Society of Chemistry 2017 |