
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design to change product specificities and thermostability of cyclodextrin glycosyltransferase from Paenibacillus sp.†
Yu Liabc,
Likun Weic,
Zhangliang Zhuc,
Songtao Lic,
Jian-Wen Wangc,
Qinglong Xinc,
Hongbin Wangabc,
Fuping Lu*abcd and
Hui-Min Qin *abc
*abc
aKey Laboratory of Industrial Fermentation Microbiology, Ministry of Education, No. 29, 13th Avenue, Tianjin Economic and Developmental Area (TEDA), Tianjin, 300457, China. E-mail: lfp@tust.edu.cn; huiminqin@tust.edu.cn; Fax: +86-22-60602298; Tel: +86-22-60601958
bTianjin Key Laboratory of Industrial Microbiology, China
cCollege of Biotechnology, Tianjin University of Science and Technology, China
dNational Engineering Laboratory for Industrial Enzymes, Tianjin 300457, People's Republic of China
First published on 28th February 2017
Abstract
Functional modification of cyclodextrin glycosyltransferase (CGTases) for better product specificity and thermostability is of great importance for industrial applications. In the present study, we aimed to improve enzymatic product specificity and thermostability by rational design using β-CGTase from Paenibacillus sp. (pCGTase) as a model enzyme. pCGTase showed optimal activity in sodium phosphate buffer (pH 7.0) at 60 °C and retained >80% residual activity at pH 6–8 and 40 °C for 30 min. The biochemical data demonstrated that Y100I/T, S145G/P, Y167H, and A315H/R/S mutants increased starch conversion activity, particularly the β-cyclodextrin-forming activity. The structural analysis elucidated that new hydrophobic interactions might be formed with substrates. Furthermore, the substitution of tyrosine with isoleucine/histidine strengthens the hydrophobic reaction. The Ala315 was located in the calcium-binding site, and mutations on Ala315, which is surrounded by a patch of polar residues, appear to increase electrostatic interactions with peripheral residues. Therefore, Ala315 mutants were designed to improve thermostability, and A315D mutant exhibited the highest thermostability at 60–70 °C for 30 min. This study provides an effective approach for improving the product specificity and thermostability of CGTases.
Introduction
The cyclodextrins (CDs), a family of cyclic oligosaccharides, typically contain six (α-CD), seven (β-CD), or eight (γ-CD) D-glucose molecules linked by α-1,4-glycosidic bonds.1,2 CDs adopt a hollow annular shape that has a hydrophilic outer surface and a hydrophobic internal cavity that can form inclusion complexes with various hydrophobic guest molecules.3 The unique structure of the CDs has led to their wide use in food, pharmaceuticals, and related industry products.4 CDs are commonly produced from starch through an enzymatic process that includes a cyclization reaction catalyzed by CD glycosyltransferase (CGTase; EC 2.4.1.19), an extracellular bacterial enzyme belonging to the α-amylase family.5CGTase is an important industrial enzyme that is commonly used to convert starch into CDs by intramolecular transglycosylation. CGTases are classified into three groups on the basis of the main CD product: α-, β-, and γ-CGTases. The 3D structure of CGTase6–8 revealed that these enzymes hold four/five domains, with the active site located at the bottom of an (α/β)8 barrel in the A domain. Substrates bind across the surface of the enzyme in a long groove formed by domains A and B. The active site of CGTase comprises at least nine glucose-binding subsites, labeled −7 to +2.6,7 Structural information has revealed the mechanism of hydrolysis of glycosidic bond and cyclization reactions.7 Glycosidic bond cleavage occurs between subsites −1 and +1, whereas Glu257 acts as a proton donor and Asp229 serves as a general base or nucleophile. The glycosyl intermediate is then transferred to the 4-hydroxyl of its own nonreducing end, forming a new α-1,4-glycosidic bond to yield a cyclic product.9 Paenibacillus sp. produces an intracellular CGTase (pCGTase, 75 kDa), which belongs to the β-CGTase family.10 pCGTase recognizes α-1,4-glucose units, and the hydrolysis activity depends on the size of the oligosaccharides. However, it could not hydrolyze highly branched carbohydrates such as glycogen or dextran. This enzyme was different from other CGTases in its ability to hydrolyze maltose and trehalose because it contained the maltose-binding site.10 Gel filtration of pCGTase gave the same molecular weight suggesting this enzyme to be a monomer. The isoelectric point of the enzyme was calculated to be 5.1. Three calcium-binding sites contributed to the thermostability of this enzyme. The crystal structure of pCGTase has been previously determined (PDB ID: 1UKS). pCGTase contains four subdomains (Fig. 1a) and possesses an (α/β)8 barrel fold. The active site is located in domain A. Three residues, Asp229, Glu257, and Asp328, form a catalytic triad. The substrate carbohydrate and product β-CD are located in the active site groove and are bound in an extended conformation above the periphery of the (α/β)8 barrel at the bottom of the substrate-binding cavity (Fig. 1b). Residues Arg47, Tyr89, His98, Tyr100, Trp101, Ser145, Tyr195, Asp196, Lys232, His233, Phe259, His327, and Asp371 form hydrogen bonds or hydrophobic interactions with the substrate and/or product (Fig. 1c). These residues were considered to play an important role in substrate recognition, and some of these resides (Tyr89, Tyr195, Phe259 etc.) were previously selected as target residues for rational design.2,9
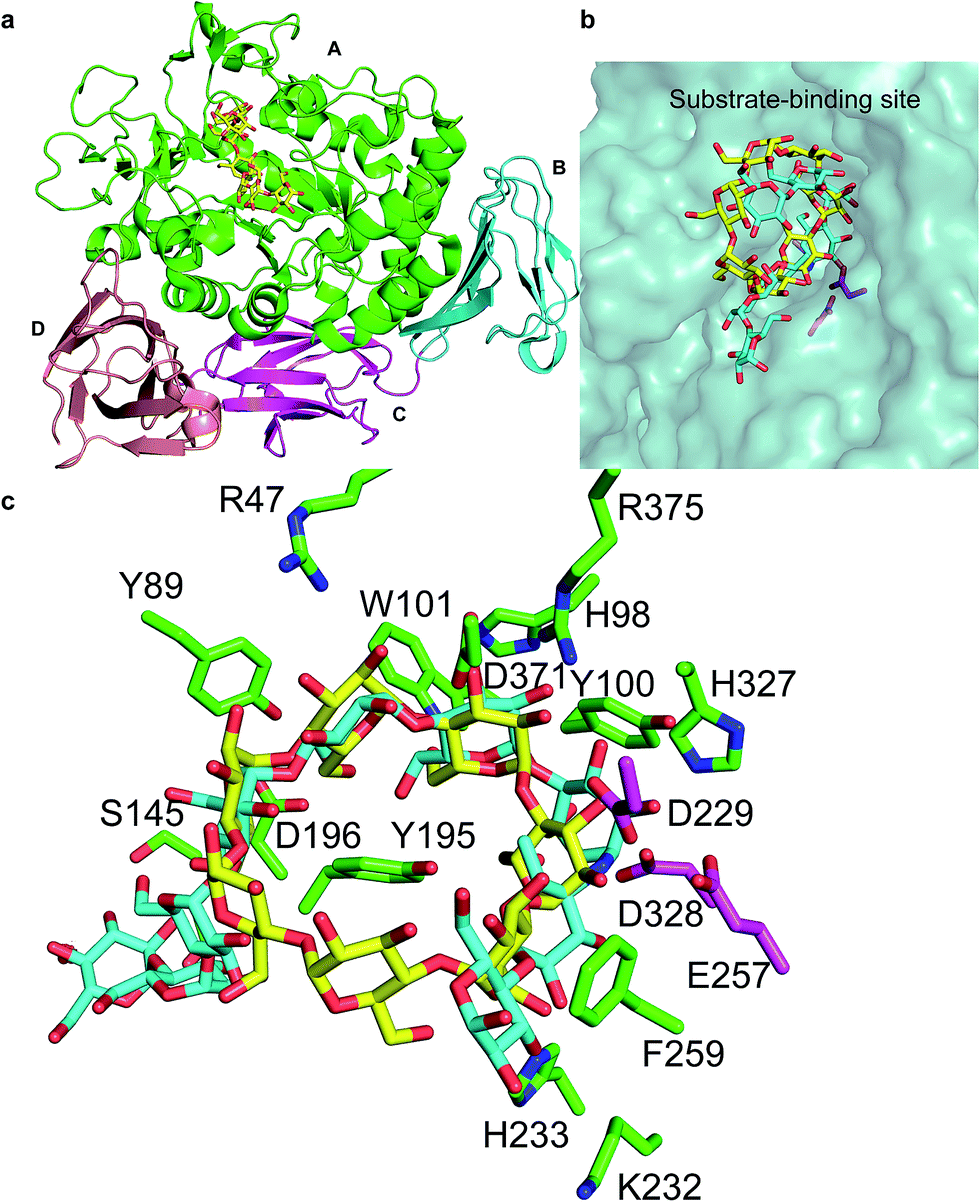 | ||
| Fig. 1 Crystal structure of cyclodextrin glycosyltransferase from Paenibacillus sp. (pCGTase) (PDB ID: 1UKS). (a) pCGTase domains A–D are colored as green, cyan, magenta, and salmon, respectively. (b) Substrate (cyan)/product (yellow) binding at the active site of pCGTase in domain A. (c) Residues in the substrate/product-binding site are shown as green sticks. The catalytic residues are shown as magenta sticks. | ||
One major issue in CD production is that CGTases always produce a mixture of CDs, including α-, β-, and γ-CDs. Selective purification steps are thus required during CD synthesis to obtain high yields of the desired specific product.11 Rational design to produce a preponderance of one particular type of CD would simplify the separation procedure. Many efforts have been made to improve CGTase product specificity through site-directed mutagenesis.12–17 Most of these mutations have targeted amino acid residues located in the active site cleft. For instance, Tyr195 mutants of CGTase from alkalophilic Bacillus sp. 1011 underwent changes in its cyclization characteristics, producing considerably more γ-CD than the wild-type enzyme.18–20 Asp577 variants, which were located in the calcium-binding site of the CGTase from Bacillus circulans STB01, enhanced β-CD production.21 D371R mutation at subsite −3 of T. thermosulfurigenes EM1 CGTase strongly increased γ-CD and lowered α-CD production.22 Mutations on Y89R and D371K at subsite −3 in Paenibacillus macerans enhanced the α-CD yield.15 Mutations on S146P and Y89D at subsite −7 in Bacillus circulans also increased the conversion of starch into α-CD.23 The effects of Ala31 of in the neighborhood of calcium binding site I of CGTase from Bacillus circulans STB01 were investigated, and A31R displayed increase in β-cyclodextrin production with a concomitant decrease in α-cyclodextrin production suggesting the mutant suitable for the industrial production of β-cyclodextrin than the wild-type enzyme.16 Thermostability is also one of the most important parameters that determine the utility and commercial applicability of a target enzyme.5,24 The thermostability of Bacillus sp. G1 CGTase was enhanced by rational mutagenesis at the calcium-binding sites.25 Mutations on A53S and H58I were reported to improve to thermostability Bacillus megaterium α-amylase.26 Li et al. investigated polyethylene glycols of low molecular weights to enhance the thermostability of β-CGTase from B. circulans by 6.5-fold.27
Here we aimed to explore potential industrial biocatalysts for commercial-scale manufacture of CDs. We reported the characterization of pCGTase. We performed site-directed mutagenesis on residues around the substrate-binding site on the basis of the structural information and investigated the effects of the substituted amino acids on CD product specificity and thermostability.
Material and methods
Sources of materials
Escherichia coli JM109 and BL21(DE3) used for DNA manipulations and protein expression were from our laboratory stocks. The CGTase gene was obtained from Paenibacillus sp. (GenBank accession no. JX101460.1). The plasmid extraction kit was obtained from Omega (Norcross, GA, USA). PrimeSTAR max, DpnI, and Taq DNA ligase were purchased from TaKaRa (Dalian, China). Plasmid pET22b(+) (Novagen, Madison, WI, USA) harboring the pCGTase gene was used as a template for site-directed mutagenesis. DNA sequencing was performed by GENEWIZ (Beijing, China). The Ni-NTA agarose resin was purchased from Qiagen (Hilden, Germany). Other reagents were of analytical or biological grade.Protein expression and purification
Recombinant E. coli BL21(DE3) cells were transformed with pET22b(+) harboring the wild-type pCGTase gene and were grown at 37 °C in 5 mL lysogeny broth medium (yeast extract 5 g L−1, peptone 10 g L−1, and NaCl 10 g L−1) containing 100 μg mL−1 of ampicillin. The cultures were then transferred to 25 mL terrific Broth medium [yeast extract 24 g L−1, peptone 12 g L−1, 0.4% (v/v) glycerol, and KH2PO4 2.31 g L−1/K2HPO4 12.54 g L−1] containing 100 μg mL−1 ampicillin and incubated at 37 °C. When the OD600 reached 0.6–0.8, isopropyl β-D-1-thiogalactopyranoside was added to obtain a final concentration of 0.1 mM, and the culture was then further incubated at 16 °C. After cultivation for 24 h, 0.5% (w/v) glycine, 0.5% (v/v) Triton-100, and 0.2 mM CaCl2 were added to increase the permeability of the cell membrane and the stability of the soluble enzyme. The entire induced expression process lasted 64 h.After incubation, the supernatant of pCGTase was harvested by centrifugation at 5000 × g for 15 min at 4 °C. Ammonium sulfate was then added to the crude enzyme solution to obtain 20% saturation. After being stirred for 1 h at 4 °C, the suspension was centrifuged for 20 min at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g. Ammonium sulfate was then continuously added to the supernatant to obtain 65% saturation. After centrifugation, the precipitate including pCGTase protein was redissolved in buffer A (10 mM Na2HPO4, 1.8 mM NaH2PO4, and 140 mM NaCl, pH 7.0). The redissolved pCGTase was trapped on Ni-NTA agarose resin pre-equilibrated with 10 mL buffer A. After washing, the target protein was eluted with 10 mL buffer B (50 mM Na2HPO4 pH 7.0, 200 mM imidazole, 300 mM NaCl). Protein concentrations were determined by the Bradford method after suitable dilutions, using Bio-Rad protein assay reagent kit (Hercules, CA, USA) and bovine serum albumin as the standard for calibration curves. The SDS-PAGE results of wild-type and some mutant pCGTase enzymes are shown in ESI Fig. S1.†
000 × g. Ammonium sulfate was then continuously added to the supernatant to obtain 65% saturation. After centrifugation, the precipitate including pCGTase protein was redissolved in buffer A (10 mM Na2HPO4, 1.8 mM NaH2PO4, and 140 mM NaCl, pH 7.0). The redissolved pCGTase was trapped on Ni-NTA agarose resin pre-equilibrated with 10 mL buffer A. After washing, the target protein was eluted with 10 mL buffer B (50 mM Na2HPO4 pH 7.0, 200 mM imidazole, 300 mM NaCl). Protein concentrations were determined by the Bradford method after suitable dilutions, using Bio-Rad protein assay reagent kit (Hercules, CA, USA) and bovine serum albumin as the standard for calibration curves. The SDS-PAGE results of wild-type and some mutant pCGTase enzymes are shown in ESI Fig. S1.†
Activity assays of pCGTase
The reaction mixture was composed of 0.1 mL 0.3–30 μg of wild-type and mutant enzymes, 0.9 mL of 1% (w/v) soluble starch, and 10 mM phosphate buffer (pH 7.0), and the reaction proceeded at 50 °C for 10 min. The activity of pCGTase was determined on the basis of the production of α-, β-, and γ-CD measured by the methyl orange,28 phenolphthalein,29 and bromocresol green30 methods, respectively. One unit was defined as the amount of enzyme required to form 1 μmol of the CD product per min. Experiments were repeated thrice for each mutant.Characterization of wild-type pCGTase
The β-CD-forming activity was measured to evaluate the activity of pCGTase because it was divided into β-CGTases subfamily and showed high product ratio for β-CD (Table 1). The optimal temperature was determined by measuring the activity of pCGTase at different temperatures ranging from 20 °C to 90 °C. The thermostability was determined by measuring the residual activity after the incubation of the enzyme at different temperatures (20–90 °C) for 30 min. The optimal pH value of the purified enzyme was determined in the following buffers: 0.1 mM citrate buffer (pH 4.0–6.0), 0.1 mM sodium phosphate buffer (pH 6.0–8.0), and 0.1 mM glycine–NaOH buffer (pH 8.0–10.0) at 60 °C for 10 min. The effect of pH on pCGTase stability was determined by incubating the purified enzymes in buffers with different pH values for 1 h at 4 °C.| Mutants | Conversion of starch to CDs (%) | Product ratio (%) | ||
|---|---|---|---|---|
| α-CD | β-CD | γ-CD | ||
| a The value represents the mean of three independent experiments for each mutant (standard deviations, n = 3). | ||||
| Wild type | 41.6 ± 2.8 | 17.5 ± 1.2 | 52.5 ± 3.0 | 30.0 ± 1.8 |
| Y89S | 33.3 ± 2.0 | 19.9 ± 1.5 | 59.4 ± 3.9 | 20.7 ± 2.2 |
| Y100A | 29.4 ± 3.2 | 18.9 ± 3.4 | 36.9 ± 1.2 | 44.2 ± 2.3 |
| Y100D | 37.3 ± 2.8 | 23.4 ± 2.5 | 58.3 ± 2.8 | 18.3 ± 3.6 |
| Y100I | 47.7 ± 1.5 | 14.8 ± 0.8 | 64.8 ± 4.5 | 20.5 ± 1.4 |
| Y100T | 45.7 ± 2.4 | 18.3 ± 3.4 | 53.3 ± 4.2 | 28.4 ± 3.9 |
| S145D | 33.7 ± 2.1 | 32.4 ± 2.5 | 32.6 ± 4.3 | 35.0 ± 3.1 |
| S145G | 47.6 ± 1.8 | 16.1 ± 1.2 | 56.0 ± 2.2 | 27.9 ± 3.6 |
| S145P | 49.5 ± 2.1 | 22.2 ± 4.1 | 45.2 ± 3.1 | 32.6 ± 3.2 |
| Y167H | 49.8 ± 1.4 | 16.5 ± 3.2 | 58.0 ± 3.8 | 25.5 ± 1.0 |
| Y195A | 15.9 ± 2.4 | 43.3 ± 2.3 | 20.1 ± 3.4 | 36.7 ± 2.1 |
| Y195E | 31.4 ± 3.0 | 24.9 ± 0.7 | 20.0 ± 2.7 | 55.2 ± 1.8 |
| Y195V | 23.6 ± 3.7 | 31.9 ± 3.5 | 24.1 ± 3.0 | 44.0 ± 3.7 |
| Y195T | 20.2 ± 3.1 | 19.1 ± 3.2 | 30.4 ± 0.5 | 50.5 ± 1.1 |
| K232D | 37.6 ± 3.7 | 17.8 ± 2.1 | 60.5 ± 2.3 | 21.7 ± 3.9 |
| H233D | 32.8 ± 2.9 | 11.9 ± 2.6 | 59.4 ± 2.1 | 28.7 ± 2.1 |
| A315D | 24.4 ± 3.6 | 11.4 ± 1.3 | 54.1 ± 3.8 | 34.5 ± 4.1 |
| A315H | 50.7 ± 0.9 | 19.9 ± 3.3 | 50.8 ± 1.8 | 29.3 ± 4.4 |
| A315R | 50.6 ± 1.8 | 43.5 ± 1.6 | 26.8 ± 2.6 | 29.7 ± 3.9 |
| A315S | 47.9 ± 2.1 | 19.0 ± 3.0 | 42.4 ± 3.2 | 38.7 ± 2.0 |
HPLC analysis of cyclodextrin products
The substrate (1% soluble starch) was incubated with 2 U pCGTases of CD-forming activity per gram starch at 40 °C for 4 h to determine the product ratio because pCGTase was more stable at 40 °C than at 60 °C. The reaction reached equilibrium conditions, and had little perturbation after 4 h. The residual starch in the reaction mixture was digested into glucose by adding 10 μL glucoamylase for 30 min, and the mixture was then boiled for 10 min and centrifuged at 5000 × g for another 10 min for HPLC analysis. The supernatant was collected and analyzed using HPLC with a Hypersil NH2 column (5 μm particle size; 250 × 4.6 mm). Deionized water (A) and acetonitrile (B) (35/65, v/v) were used as eluents at a flow rate of 1 mL min−1. The products were detected using a refractive index detector (Waters 2414, Milford, MA, USA). The starch conversion was determined as the amount of CD formed in the reaction system. The mutants were also determined at the same reaction conditions.Site-directed mutagenesis
The pCGTase mutants were generated using a one-step PCR method with plasmid pCGTase_pET22b wild-type as the template. The primers used for mutagenesis are summarized in ESI Table S1.† After amplification, the PCR reaction mixtures were treated with DpnI to completely digest the template and then transformed into E. coli JM109 cells. All the mutations were confirmed by DNA sequencing. pCGTase mutants were expressed and purified using the method described above for wild-type enzyme.Results and discussion
Characterization of wild-type pCGTase and cyclodextrin analysis
The purified wild-type pCGTase was active at pH 5.0–9.0, and the maximal activity was observed at pH 6.5–7.5 (Fig. 2a). pCGTase exhibited good stability at pH 6.0–8.0, and more than 80% of the maximal activity was retained in this range (Fig. 2b). The optimal temperature for pCGTase activity was approximately 60 °C. The activity began to decrease when temperature was increased, and there was only 22% residual activity at 90 °C (Fig. 3a). The enzyme activity was also examined after storage at different temperatures in sodium phosphate buffer; 80% of the activity was retained after incubation at 20–40 °C for 30 min (Fig. 3b). The enzyme lost almost all activity when the storage temperature was >70 °C.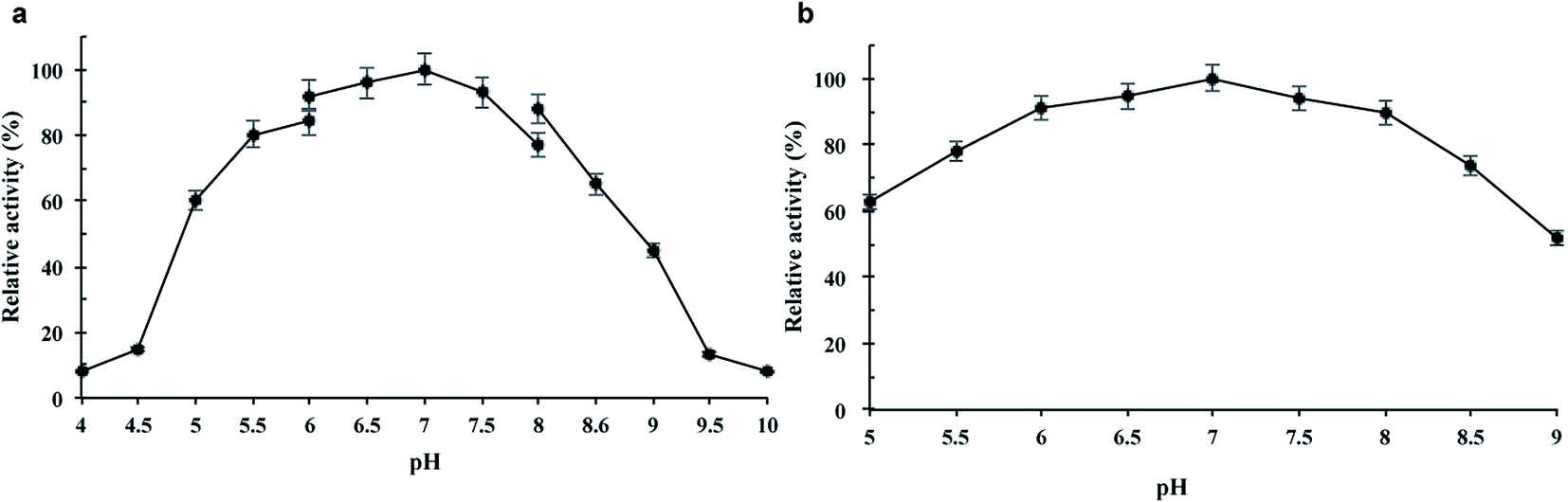 | ||
| Fig. 2 Effect of pH on the activity of pCGTase. (a) pH dependence of pCGTase. Activity was measured in the reaction mixtures adjusted to various pH values with 0.1 mM citrate, sodium phosphate, and glycine–NaOH buffers. (b) Analysis of pH stability of pCGTase. pCGTase was preincubated at different pH values for 1 h at 4 °C. The activities of wild-type pCGTase are represented as 100 in optimal reaction conditions, and the residual activity was determined at various pH conditions. Error bars are standard deviations (n = 3). | ||
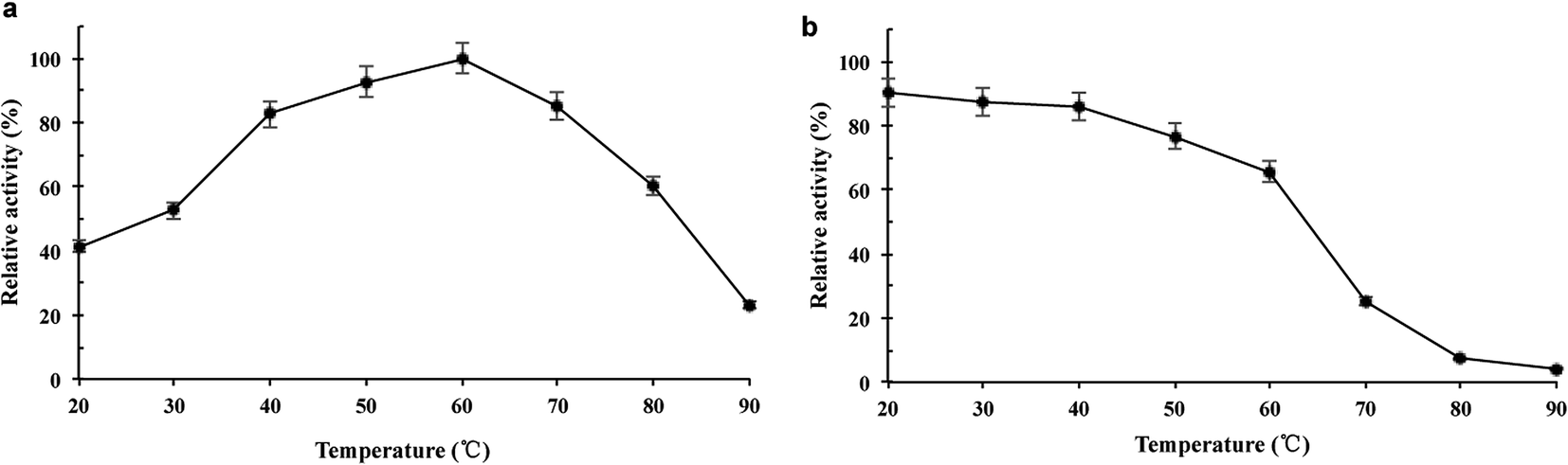 | ||
| Fig. 3 Effect of temperature on the activity of pCGTase. (a) Temperature dependence of PCGTase. Activity was measured at various temperatures in the standard assay conditions. (b) Thermostability analysis of PCGTase. pCGTase was preincubated at various temperatures for 30 min. The activities of wild-type pCGTase are represented as 100 in optimal reaction conditions. Error bars are standard deviations (n = 3). | ||
The CD products were confirmed by HPLC, and the retention times of α-, β-, and γ-CD products were 12.25, 14.75, and 18.5 min, respectively (ESI Fig. S2†). The wild-type pCGTase had high β-CD-forming activity (27.3 U mg−1) compared with α-/γ-CD-forming activity (10.7 and 13.7 U mg−1, respectively). Furthermore, pCGTase (wild type) had a starch conversion ability of approximately 41.6%. The α-, β-, and γ-CD-forming ratios were 17.5%, 52.5%, and 30%, respectively (Table 1), which indicates that pCGTase is a β-CD-specific enzyme. Compared with starch conversion 38.2% (wild type) of cyclodextrin glycosyltransferase from Paenibacillus sp. 602-1 at pH 6.5 and 40 °C for 24 h,19 pCGTase showed better conversion rate in 4 h than the previously reported enzyme.
Protein modification for product specificity
The functional modification of pCGTase has potential benefits to increase product specificity. High thermostability is important for industrial catalysts because enzyme reaction rates and reactant solubility can be significantly improved at higher temperature, which helps to reduce industrial production costs. Compared with random mutagenesis to improve product specificity and enzyme thermostability, rational engineering requires detailed protein structural information.19 Several rational strategies have been applied for the increment of enzyme thermostability, such as improvement of the packing of the hydrophobic core, introduction of disulfide bridges, and engineering of surface salt bridges.31–33Rational design was performed to change the starch conversion, CD-forming activity, and thermostability by analyzing structural information, particularly the residues located in the substrate-binding site. For this purpose, mutants of Tyr89, Tyr100, Ser145, Tyr167, Tyr195, Lys232, His233, and Ala315 were selected as the target residues for designing for rational engineering and aiming at the improvement of product specificity or thermostability. Mutants Y100I/T, S145G/P, Y167H, and A315H/R/S improved starch conversion into the various CDs relative to the wild-type enzyme (Table 1). Notably, Y100I, S145G, and Y167H resulted in a higher yield of β-CD, whereas S145P and A315H/R/S exhibited a higher yield of α-/γ-CD. It was noted that some mutants decreased the starch conversion activity while changing the product CD-forming ratios. In particular, Y89S, Y100D, K232D, H233D, and A315D improved the β-CD-forming ratio. Y100A, S145D, and Y195A/E/V/T reduced starch conversion activities and β-CD-forming specificity and increased α-/γ-CD-forming ratios.
Structural superposition of β-CGTase with α-/γ-CGTase showed that the active site of 3D structures of the CGTases were similar in the CGTase family (Fig. 4a). The substrate-binding cavity is large enough to hold three kinds of products. The residues in substrate-binding sites, such as Tyr89, Tyr100, Tyr167, Tyr195, K232, and H233, are almost identical and highly conserved in this family of enzymes except for Ser145. In pCGTase, the π-stacking interaction of Tyr100 and glucopyranose units plays an important role in substrate recognition and CD-forming ability. Y100I/T showed slightly improved starch conversion activity relative to the wild-type, whereas Y100D/A had decreased the activity. This implied that new CH3/π or hydrophobic interactions might be formed when Tyr100 is mutated to isoleucine or threonine. In particular, the substitution of tyrosine to isoleucine strengthens the hydrophobic reaction. S145G/P and Y167H mutants were considered to form hydrophobic interactions with the substrate. Tyr195 has been previously reported to regulate the cyclization specificity in α-CGTase.18 The α-/γ-CD-forming ratios was increased on mutants of Y195A/E/V/T, although the mutation reduced starch conversion activities. In previous report,19 Y195E retained the same level activity of starch conversion while Y195V decreased the activity. Furthermore, Y195E/V improved the β-/γ-CD-forming ratios. It is possible that cyclodextrin glycosyltransferase from Paenibacillus sp. 602-1 belongs to α-CGTase, and pCGTase is β-CGTase. Tyr195 is located at the bottom of the center-bound CD in the active site, and it forms hydrogen bonds with glucopyranose units (Fig. 1c). It was presumed that the mutants Y195E/T retained the hydrogen bond and increased the space required to form γ-CD. The mutants Y195A/V might form hydrophobic interactions with glucopyranose units of α-CD. It is interesting that A315H/R/S mutants had improved starch conversion activity compared with the wild-type pCGTase, particularly the α-CD-forming activity. This indicates that a charged residue at position 315 is necessary to form alternate pCGTase conformations that are more likely to produce α- or γ-CD rather than β-CD.
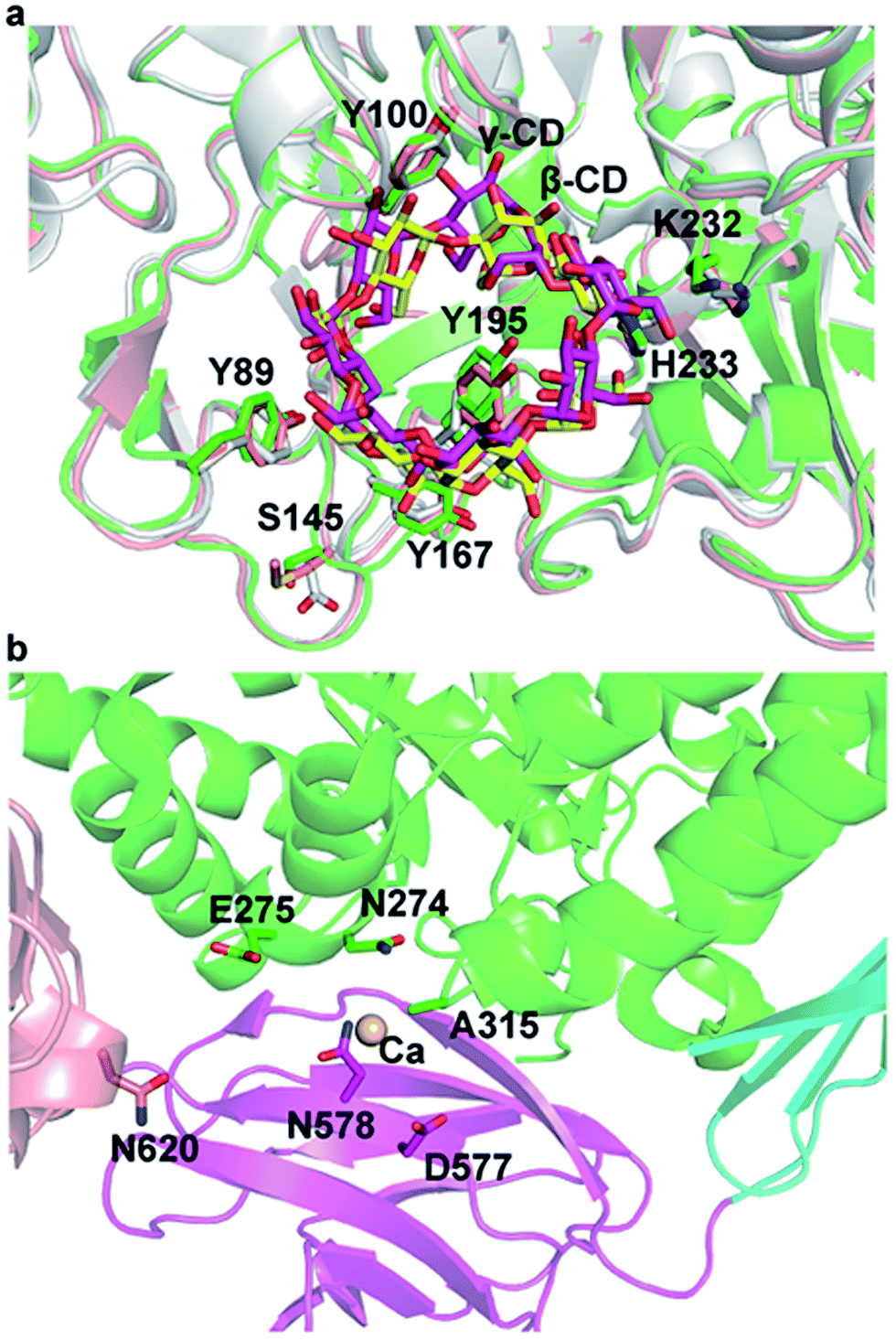 | ||
| Fig. 4 (a) The products [β-cyclodextrin (CD) yellow; γ-CD magenta] binding at the active site of CGTases. (b) Close-up view showing the position of Ala315 in the structure of pCGTase and its interactions with other residues. | ||
Protein modification for thermostability
Mutants of Ala315 exhibited the obviously improved residual activity at 60–70 °C for 30 min compared with the wild-type enzyme (Fig. 5). In particular, A315D retained approximately 90% activity at 60 °C for 30 min (Fig. 5a) and approximately 60% activity at 70 °C for 10 min (Fig. 5b). However, both wild-type enzyme and Ala315 mutants showed drastically reduced activity when incubated at 80 °C for 5 min, with the exception of A315D/H, which retained approximately 30% activity (Fig. 5c). These mutants lost almost all activity (approximately 95%) on incubation at 80 °C for 20 min.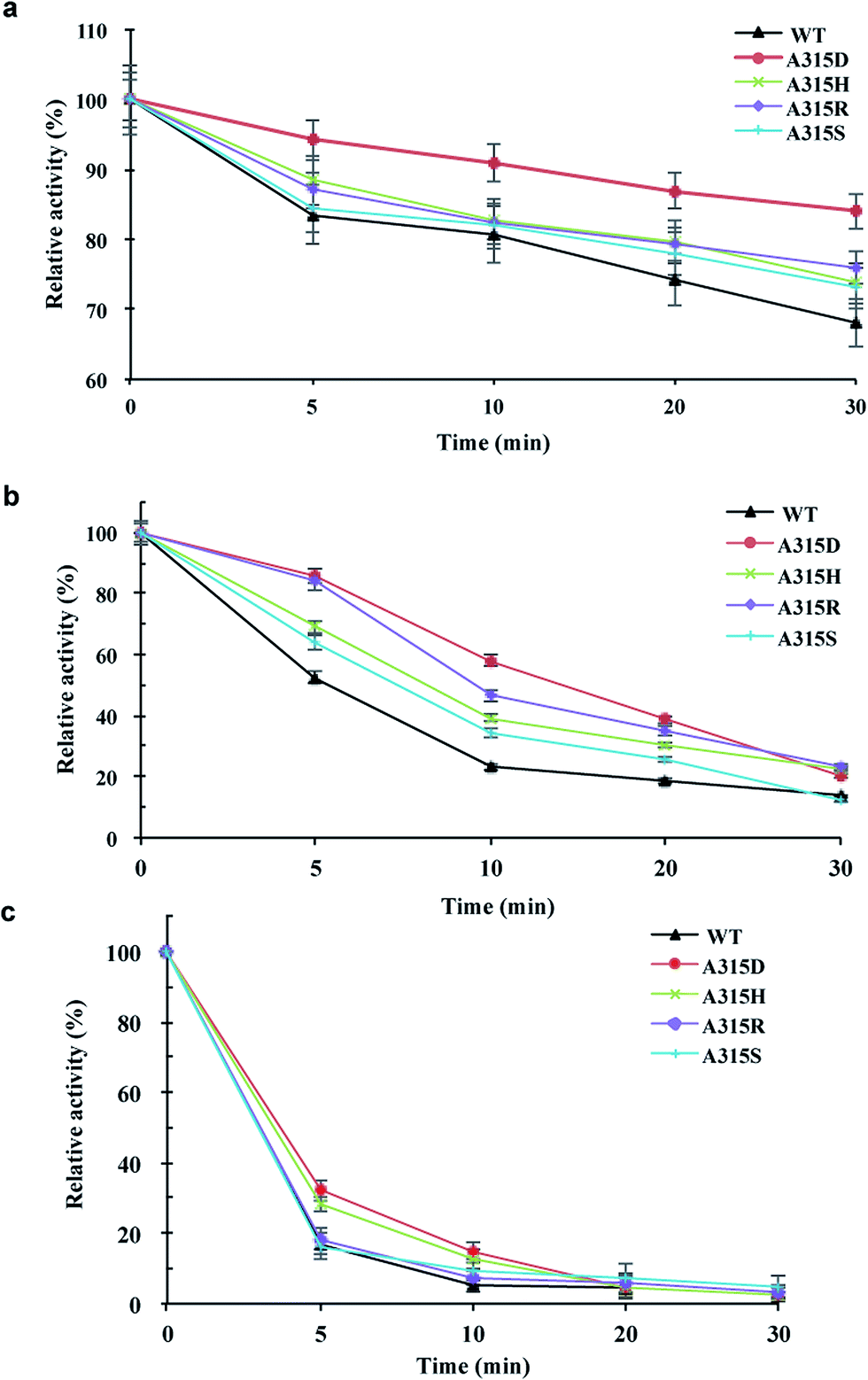 | ||
| Fig. 5 Thermostability analysis of Ala315 mutants of pCGTase. Mutants were preincubated at (a) 60 °C, (b) 70 °C, and (c) 80 °C for 5–30 min. The activities of wild-type pCGTase are represented as 100 in optimal reaction conditions, and the residual activities were then determined in the standard assay conditions. Error bars are standard deviations (n = 3). | ||
Ala315 is located at the substrate entrance of the A/D domain interface and is surrounded by a patch of polar or charged residues including Asn274, Glu275, Asn578, Asp577, and Asn620 (Fig. 1 and 4). The introduction of a charged residue in place of Ala315 appeared to increase electrostatic interactions, and the substrate was considered to move smoothly into the active site. Ala315 was previously reported to be located in the calcium-binding site, and A315D significantly changed the contribution of Ca2+ to β-CGTase thermostability.34 Therefore, Ala315 mutants were designed for increasing the salt bridge/hydrogen bond with peripheral residues to improve enzyme thermostability. A315D/H/R/S mutants demonstrated improved residual activity, particularly at 60 °C and 70 °C, which were consistent with structural predictions. These mutants involving charged or polar residues, i.e., Asp, His, Arg, and Ser, were considered to strengthen the calcium-binding affinity at Ala315 of pCGTase.
Conclusion
The purified pCGTase and mutants were characterized with respect to their CD-forming activities. The optimum pH (7.0) and pH stability (6.0–8.0) of the recombinant pCGTase and the optimum temperature (60 °C) and thermostability (20–40 °C for 30 min) were determined. We successfully applied structure-based rational design to improve the enzymatic activity and thermostability by directly mutating important residues. In particular, Tyr100, Ser145, Tyr167, and Ala315 mutants increased β-CD-forming activity. The Ala315 mutants exhibited higher thermostability at 60–70 °C for 30 min. The hydrophobic interactions and electrostatic interactions attribute to improve the specific activity and thermostability, respectively. The results will be useful for the rational design of a biocatalyst for the industrial production of CDs.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key Research and Development Project (2016YFD0400803), National Natural Science Foundation of China (21306140), the Natural Science Foundation of Tianjin (16JCQNJC09200), and the Overseas High-level Talents Program of Tianjin University of Science and Technology, China. We acknowledge the excellent support of the Tianjin Food Safety & Low Carbon Manufacturing Collaborative Innovation Center 300457, Tianjin, China.References
- B. A. van der Veen, J. C. M. Uitdehaag, B. W. Dijkstra and L. Dijkhuizen, Biochim. Biophys. Acta, 2000, 1543, 336–360 CrossRef CAS.
- R. Han, J. Li, H. Shin, R. R. Chen, G. Du, L. Liu and J. Chen, Biotechnology, 2014, 32, 415–428 CAS.
- D. Li, S. A. Roh, J. H. Shim, B. Mikami, M. Y. Baik, C. S. Park and K. H. Park, J. Agric. Food Chem., 2005, 53, 6516–6524 CrossRef CAS PubMed.
- H. Y. Sun, Y. Bai, M. G. Zhao, A. Y. Hao, G. Y. Xu, J. Shen, J. Y. Li, T. Sun and H. C. Zhang, Carbohydr. Res., 2009, 344, 1999–2004 CrossRef CAS PubMed.
- M. Kobayashi, H. Hondoh, H. Mori, W. Saburi, M. Okuyama and A. Kimura, Biosci., Biotechnol., Biochem., 2011, 75, 1557–1563 CrossRef CAS PubMed.
- B. Strokopytov, R. M. A. Knegtel, D. Penninga, H. J. Rozeboom, K. H. Kalk, L. Dijkhuizen and B. W. Dijkstra, Biochemistry, 1996, 35, 4241–4249 CrossRef CAS PubMed.
- J. C. Uitdehaag, K. H. Kalk, B. A. van Der Veen, L. Dijkhuizen and B. W. Dijkstra, J. Biol. Chem., 1999, 274, 34868–34876 CrossRef CAS PubMed.
- T. Xie, Y. Hou, D. Li, Y. Yue, S. Qian and Y. Chao, J. Biotechnol., 2014, 182–183, 92–96 CrossRef CAS PubMed.
- H. Leemhuis, R. M. Kelly and L. Dijkhuizen, Appl. Microbiol. Biotechnol., 2010, 85, 823–835 CrossRef CAS PubMed.
- J. Kaulpiboon and P. Pongsawasdi, Enzyme Microb. Technol., 2005, 36, 168–175 CrossRef CAS.
- Y. J. Son, C. S. Rha, Y. C. Park, S. Y. Shin, Y. S. Lee and J. H. Seo, J. Microbiol. Biotechnol., 2008, 18, 725–729 CAS.
- H. Costa, A. J. Distéfano, C. Marino-Buslje, A. Hidalgo, J. Berenguer, M. B. de Jiménez Bonino and S. A. Ferrarotti, Appl. Microbiol. Biotechnol., 2012, 94, 123–130 CrossRef CAS PubMed.
- R. M. Kelly, L. Dijkhuizen and H. Leemhuis, Appl. Microbiol. Biotechnol., 2009, 84, 119–133 CrossRef CAS PubMed.
- Z. F. Li, J. Y. Zhang, Q. Sun, M. Wang, Z. B. Gu, G. C. Du, J. Wu and J. Chen, J. Agric. Food Chem., 2009, 57, 8386–8391 CrossRef CAS PubMed.
- Z. Li, J. Zhang, M. Wang, Z. Gu, G. Du, J. Li, J. Wu and J. Chen, Appl. Microbiol. Biotechnol., 2009, 83, 483–490 CrossRef CAS PubMed.
- Z. Li, X. Ban, Z. Gu, C. Li, M. Huang, Y. Hong and L. Cheng, Carbohydr. Res., 2014, 108, 112–117 CrossRef CAS PubMed.
- Y. Nakagawa, M. Takada, K. Ogawa, Y. Hatada and K. Horikoshi, J. Biochem., 2006, 140, 329–336 CrossRef CAS PubMed.
- D. Penninga, B. Strokopytov, H. J. Rozeboom, C. L Lawson, B. W. Dijkstra, J. Bergsma and L. Dijkhuizen, Biochemistry, 1995, 34, 3368–3376 CrossRef CAS PubMed.
- T. Xie, B. Song, Y. Yue, Y. Chao and S. Qian, J. Biotechnol., 2014, 170, 10–16 CrossRef CAS PubMed.
- F. Chen, T. Xie, Y. Yue, S. Qian, Y. Chao and J. Pei, J. Mol. Model., 2015, 21, 92–96 CrossRef PubMed.
- Z. Li, M. Huang, Z. Gu, T. P. Holler, L. Cheng, Y. Hong and C. Li, Int. J. Biol. Macromol., 2016, 83, 111–116 CrossRef CAS PubMed.
- H. Leemhuis, B. W. Dijkstra and L. Dijkhuizen, FEBS Lett., 2002, 514, 189–192 CrossRef CAS PubMed.
- B. A. van der Veen, J. C. Uitdehaag, D. Penninga, G. J. van Alebeek, L. M. Smith, B. W. Dijkstra and L. Dijkhuizen, J. Mol. Biol., 2000, 296, 1027–1038 CrossRef CAS PubMed.
- G. D. Haki and S. K. Rakshit, Bioresour. Technol., 2003, 89, 17–34 CrossRef CAS PubMed.
- P. H. Goh, R. M. Illias and K. M Goh, Int. J. Mol. Sci., 2012, 13, 5307–5323 CrossRef CAS PubMed.
- M. Ghollasi, K. Khajeh, H. Naderi-Manesh and A. Ghasemi, Appl. Biochem. Biotechnol., 2010, 162, 444–459 CrossRef CAS PubMed.
- C. Li, W. Li, T. P. Holler, Z. Gu and Z. Li, Food Chem., 2014, 164, 17–22 CrossRef CAS PubMed.
- A. Lejeune, K. Sakaguchi and T. Imanaka, Anal. Biochem., 1989, 181, 6–11 CrossRef CAS PubMed.
- M. Mäkelä, T. Korpela and S. Laakso, J. Biochem. Biophys. Methods, 1987, 14, 85–92 CrossRef.
- T. Kato and K. Horikoshi, Anal. Chem., 2002, 56, 1738–1740 CrossRef.
- B. van den Burg and V. G. Eijsink, Curr. Opin. Biotechnol., 2002, 13, 333–337 CrossRef CAS PubMed.
- R. Sterner and W. Liebl, Crit. Rev. Biochem. Mol. Biol., 2001, 36, 39–106 CrossRef CAS PubMed.
- C. Vieille and G. J. Zeikus, Microbiol. Mol. Biol. Rev., 2001, 65, 1–43 CrossRef CAS PubMed.
- C. Li, X. Ban, Z. Gu and Z. Li, J. Agric. Food Chem., 2013, 61, 8836–8841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00245a |
| This journal is © The Royal Society of Chemistry 2017 |