
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct C-metallation of N-substituted triazoles promoted by mercury acetate. An alternative route to N-heterocyclic carbene complexes†
Nai-Chieh Lin,
Herbert J. H. Syu*,
Abbas Raja Naziruddin,
Fu-Chen Liu* and
Ivan J. B. Lin
Department of Chemistry National Dong Hwa University, No. 1, Sec. 2, Da Hsueh Rd. Shoufeng, Hualien 97401, Taiwan, Republic of China. E-mail: fcliu@gms.ndhu.edu.tw; d9812001@gms.ndhu.edu.tw
First published on 15th February 2017
Abstract
C-Metallation of neutral mono-substituted triazoles could be induced by Hg(OAc)2 to give a near quantitative yield of C,N-bonded mercury complexes. Initial N-coordination to mercury(II) is confirmed by NMR spectroscopy. Subsequent acetate assisted deprotonation–metallation is a viable route to C-metallated products. Hg(OAc)2 is unique for this reaction. Metal salts of Ag2O, Ag(OAc), Pd(OAc)2 or HgCl2 in combination with bases of NaOH and K2CO3 did not yield triazolate complexes. Further reaction of one of these mercury(II)–C,N-triazolate complexes with a source of methyl cation yielded a monomeric N-heterocyclic carbene complex. Therefore the facile reaction of triazole with Hg(OAc)2 followed by alkylation is a handy alternative way to prepare Hg–NHCs.
Introduction
A plethora of methodologies that facilitate the metallation of N2C carbon of imidazolium cations are known.1 In contrast to the large library of C-metallation of these cations, examples of similar occurrence for mono-N-substituted neutral imidazoles are limited (see Scheme 1 for structural formula of imidazolium cations and imidazoles).2–9 This is because the N2C–H proton of the neutral imidazoles is less acidic than that of the imidazolium cations and hence is more difficult to be deprotonated. A strong base such as BuLi,10–12 KN(SiMe3)2,13,14 KOtBu,15,16 or LiHMDS17 (HMDS = hexamethyldisilazide) is often required to remove the N2C–H proton of mono-N-substituted imidazoles. Alternatively, functionalization of benzimidazoles or imidazoles at N-side arms with tethered phosphine,18–21 pyridine20,21 or olefin22 donor ligands is known to promote the activation of N2C–H bond to give C-metallated products. As a different approach, azoles bearing more reactive N2C–X (X = Cl/Br/I) bonds are also known to undergo oxidative addition into lower valent metal ions to provide azolate complexes, which upon further protonation or methylation could afford NHC complexes.6,7,23,24 In an attempt to use a Rh(I) catalyst to couple N-methyl benzimidazole with iodobenzene, an intermediate complex, [(H-bimy-Me)RhCl(PCy3)2], bearing a protic NHC ligand was isolated.25 In an earlier report, acid has been found to promote the rearrangement of N-metallated Ru(II) into its C-bound tautomer.26 Computational results suggest that there is a thermodynamic preference of C-binding over N-coordination for the second and third row transition metals.27 Recently we have reported the isolation of C,N-bounded, 12-membered mercuramacrocycles from the reactions of N-substituted benzimidazoles with Hg(OAc)2 and NaBr. This reaction presumably proceeds through an initial N-complexation of benzimidazoles to mercury bromide, which in turn favors the deprotonation of N2C–H.28 In the present study, we describe the direct reaction of neutral triazoles with Hg(OAc)2 to prepare mercury–C,N-triazolate complexes. This method could evolve as an important synthetic tool to attain C-metallation of triazoles. A subsequent methylation by [Me3O][BF4] generates mercury(II)–NHC complex, which is possible to form other NHC complexes via the known transmetallation reaction.29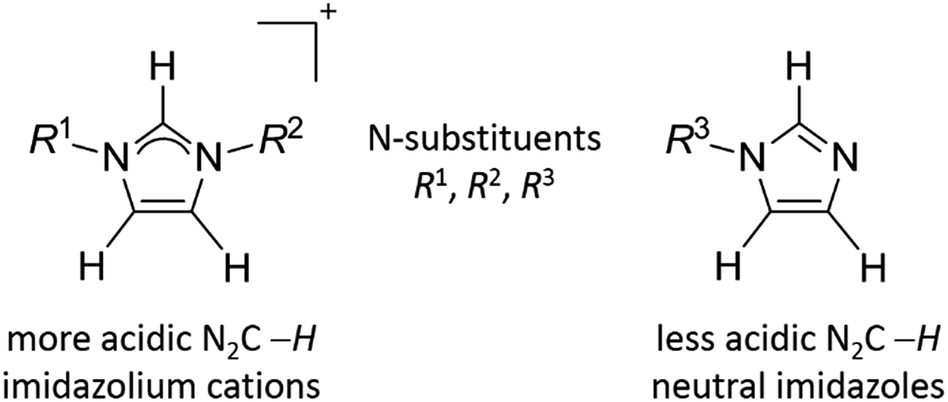 | ||
| Scheme 1 A representation of imidazolium cations and mono-N-substituted neutral imidazoles. | ||
Results and discussion
Ligand precursors of R-TazH, (R = phenyl, pyridyl and methyl) were prepared according to the literature protocols.30,31 Reactions of these ligand precursors with an equi-molar stoichiometry of Hg(OAc)2 in methanol under reflux conditions gave complexes of type [Hg2(R-Taz)2(OAc)2]∞ (1a, R = phenyl; 1b, R = pyridyl; 1c, R = methyl) with yields >95% (Scheme 2). The occurrence of C-metallation at R-TazH was supported by the absence of C5–H proton (see Scheme 2 for triazole atom numberings) resonances and the large downfield shift of the C5 signals at ca. δ 175 ppm in 13C NMR as compared to those of the ligand precursors. Single crystals of 1a were obtained via slow evaporation of its tetrahydrofuran solution. Crystals of 1b and 1c suitable for X-ray diffraction study could not be obtained, in spite of repeated attempts. The structure of 1a is depicted in Fig. 1, along with the important bonding parameters listed in the caption. 1a exhibits a polymeric structure with repeating (Hg-RTaz)2 units. In each unit, one can consider that two Hg(II) centers are bridged by two triazolates with alternating C- and N-bond. Each (Hg-RTaz)2 unit is further linked by four acetates with neighboring units to form a polymer. The C(1)–Hg(1) bond distance of 1a (2.046(6) Å) is comparable to those of reported mercury NHC complexes.32–36 The N(1)–C(1)–N(3) bond angle of 108.5(5)° is larger than those in other related Hg–NHC complexes (ca. 106°).32 The expansion of C–N–C bond angles is attributed to the formation of carbenolate complexes.23 Notably, two trans disposed C(1) and O(1) atoms are in a nearly linear configuration with a C(1)–Hg(1)–O(1) bond angle of 176.8(2)°. This leads to a seesaw type coordination in the Hg center. Unlike 1a, 1b has a pyridyl group, one might wonder whether the pyridyl moiety would participate in the coordination sphere of Hg(II). However, this possibility is very slim as evidenced by the nearly unaltered 1H NMR signals for the pyridyl moiety between 1b and Py-TazH.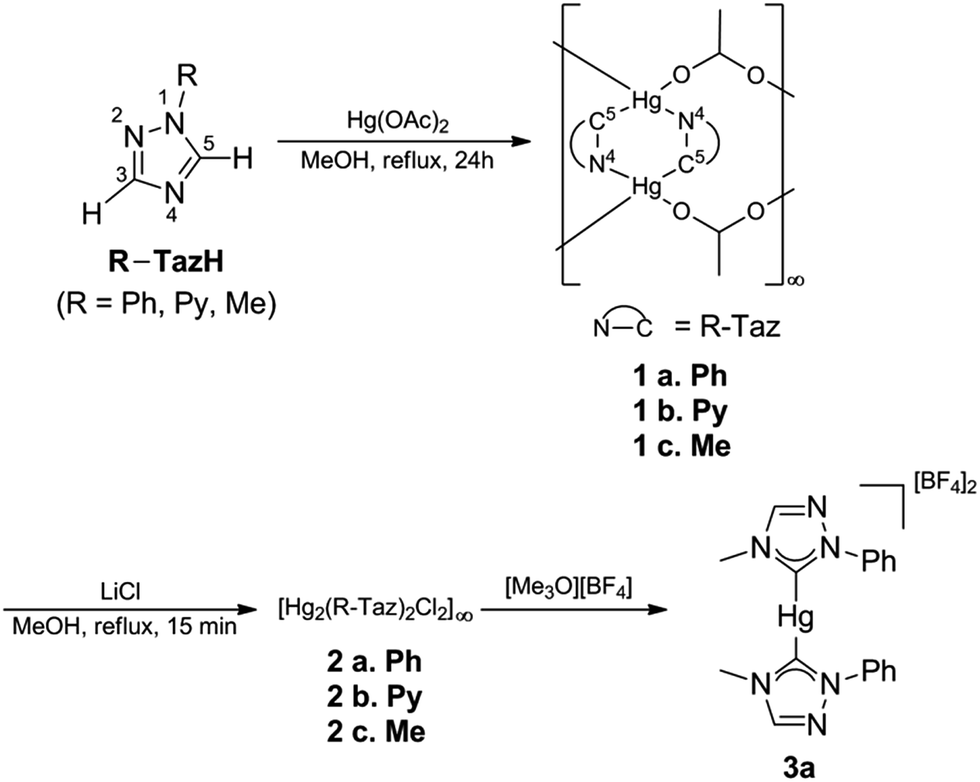 | ||
| Scheme 2 Synthetic scheme depicting the preparation of complexes and atom numberings for triazoles. | ||
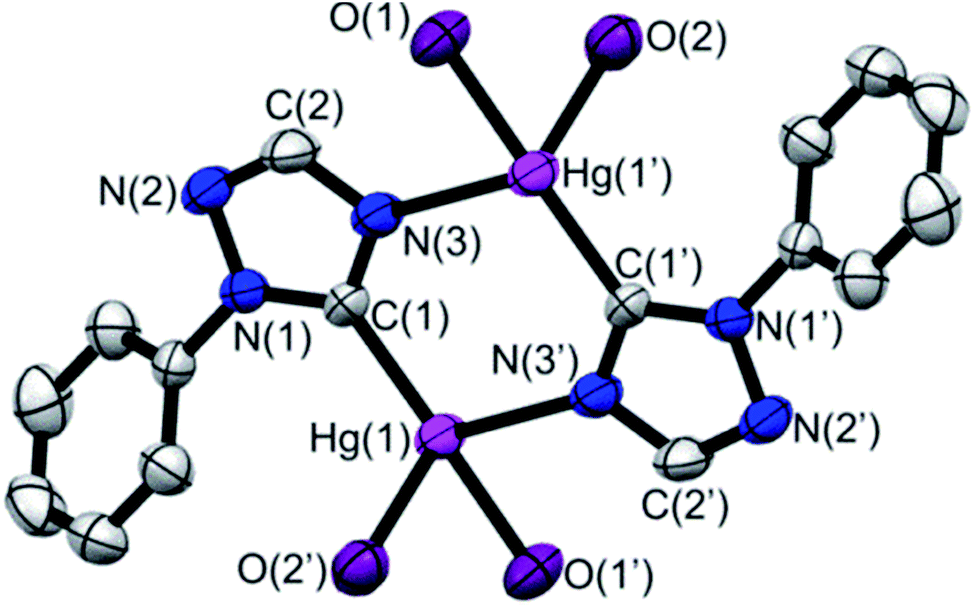 | ||
| Fig. 1 ORTEP of 1a, depicted with 50% polarizability ellipsoids with the carbon atoms in the acetyl moiety and hydrogen atoms omitted for clarity. Selected bond distances [Å] and angles [°]: Hg(1)–C(1) 2.046(6); Hg(1)–O(1) 2.079(4); Hg(1)–N(3′) 2.583(5); N(1)–C(1)–N(3) 108.5(5); C(1)–Hg(1)–O(1) 176.8(2); C(1)–Hg(1)–O(2) 100.3(2); C(1)–Hg(1)–N(3) 100.8(2). | ||
Upon treating 1a, 1b and 1c with LiCl, the bridging acetato ligand was substituted by Cl− ligand, with a nearly quantitative formation of [Hg2(R-Taz)2(Cl)2]∞ (2a, 2b and 2c). Formation of 2a, 2b, and 2c are accompanied by the loss of coordinated acetato singlet resonance in the range of δ 1.92–1.87 ppm, that otherwise would appear in the 1H NMR spectra of 1a, 1b and 1c. The solubility of 2a, 2b or 2c in common organic solvents is poor. In DMSO-d6, increasing the temperature (≈80 °C) could enhance their solubility. However, well resolved 13C signals are achievable only for 2a and 2b. The C5 13C NMR signals of 2a and 2b appear at δ 171.5 and 170.0 ppm, respectively, which are relatively up-field compared to those previously reported Hg(II)–1,2,4-triazol-5-ylidene complexes (δ 183.8 in DMSO-d6)32 due to the relative shielding caused by the anionic nature of the triazolates. We notice that there is a clear shift in the pyridyl 13C signals of 2b relative to those of 1b and Py-TazH. This observation allows us to propose that while the pyridyl wingtip in 1b is dangling, it coordinates to the Hg(II) center in 2b, as also supported by its crystal structure, discussed as follows. Single crystals of 2b suitable for structural determination were obtained by diffusing ethanol vapor into its DMSO solution. We have been unable to obtain crystals of 2a and 2c in spite of repeated attempts. Crystal structure of 2b is depicted in Fig. 2, along with important bonding parameters in the caption. 2b also adopts a polymeric structure composed of repeating single Hg(II) unit. In each four coordinated Hg(II) unit, there is a chelating C^N donor set, a terminal chloride, and an N atom from the neighboring triazolate. These units are thus linked by the bridging triazolates. Most of the bond lengths and angles of 2b are comparable to those of 1a, except a slight bending in the C(1)–Hg(1)–Cl(1) bonds (168.5(7)°), in comparison to the C(1)–Hg(1)–O(1) bond angle of 176.8(2)° in 1a.
 | ||
| Fig. 2 ORTEP of 2b, depicted with 50% polarizability ellipsoids with hydrogen atoms omitted for clarity. Selected bond distances [Å] and angles [°]: Hg(1)–C(1) 2.060(2); Hg(1)–Cl(1) 2.301(6); Hg(1)–N(4) 2.750(2); Hg(1)–N(1′) 2.560(2); N(1)–C(1)–N(3) 108.0(2); C(1)–Hg(1)–N(4) 69.4(8); C(1)–Hg(1)–N(1′) 96.4(8); Cl(1)–Hg(1)–N(1′) 95.0(5); Cl(1)–Hg(1)–N(4) 109.8(5); C(1)–Hg(1)–Cl(1) 168.5(7). | ||
The structure information of 2b cannot be applied to those of 2a and 2c, where no additional donor group at the wingtip is available for the coordination. We believe that the Hg(II) center is also four coordinated in 2a and 2c. To fulfill this condition, a bridging rather than a terminal chloride is likely. Raman spectroscopy was employed to justify this notion. Raman modes involving Hg–Cl–Hg stretching are known to appear bands below 200 cm−1, whereas those of terminal Hg–Cl bonds appear at higher wavenumbers (273–307 cm−1).37 The observation of Raman bands of 2a at 187 cm−1 and 2c at 162 cm−1 is hence consistent to a bridging Hg–Cl–Hg unit, and suggests a four coordinated structure consisting of a C5 and an N atom from two independent triazolates and two bridging chlorides.
Further reaction of 2a with trimethyloxonium–tetrafluoroborate gave 3a featuring neutral NHC ligand. The 13C NMR resonance of the carbenoid carbon atom of 3a occurs at 177.8 ppm which is downfield relative to that with triazolate donors (vide supra). In the crystals structure of 3a, a residue density of 4.37 eÅ−3 could not be fitted. Nonetheless, the main frame of this structure is correct and depicted in Fig. S1.† While 1a featuring an anionic carbon donor adopts a four coordinated Hg atom, 3a bearing a neutral NHC donor adopts a two-coordinated linear geometry at Hg atom, as often observed for other bis-NHC Hg compounds.29,38
To understand how this reaction proceeds, the reaction of Me-TazH and Hg(OAc)2 at room temperature was monitored by 1H NMR. Me-TazH in CD3OD shows three resonances corresponding to ring C5–H and C3–H protons and N–CH3 protons at δ 6.85, 6.40 and 2.38 ppm, respectively, with a relative intensity of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 shown in Fig. 3(a). Addition of an equimolar quantity of Hg(OAc)2 causes a substantial shift of the triazole signals to δ 7.26, 6.65 and 2.48 ppm again with 1:1:3 relative intensity (Fig. 3(b)). N-Coordination apparently occurs and that the larger downfield shift of the signal of C5–H relative to C3–H suggests an N4 rather than N2 coordination. After 1.5 h (Fig. 3(c)), two new signals at δ 6.81 and 2.58 ppm appear, which grow with time (Fig. 3(d–h)), and could be attributed to the formation of the triazolate product. In addition, for those unreacted triazoles the relative intensity of the C5–H to C3–H and CH3 signals gradually decreases along with time. This observation is again consistent with an N4 coordination, which gives a more acidic C5–H proton, such that H–D exchange between the C5–H proton and the deuterated solvent occurs. Then the weak base acetate deprotonates the C5–H proton. However, the role of the acetate ion needs further comment. Reactions of Me-TazH with HgCl2 in the presence of several weak bases were examined. While using NaOAc as a base could indeed produce 2c, other bases such as K2CO3 or NaOH could not yield C-metallation products. Attempts to react R-TazH with other metal sources of Ag2O, Ag(OAc) and Pd(OAc)2 containing basic ligands always produce black metallic powder, but no C-metallation products. Among the few bases and metal ions tried, the combination of Hg(II) and acetate is unique to proceed this facile reaction. A proposed mechanism to account for this reaction is given in Scheme 3. Firstly, N4 coordination of a triazole to Hg(OAc)2 gives a [Hg(OAc)2·R-TazH] adduct. Then two of these adducts form an activation complex, where both (1) weakening of the acidic C5–H bond via hydrogen bonding interaction with neighboring acetate oxygen atom, and (2) forming a partial C5⋯Hg bond with a neighboring Hg atom occur simultaneously. The former interaction is favored by the possible formation of a six-membered HgOCOHC ring. The latter interaction is presumably facilitated by the favorable Hg⋯C-triazolate bond formation. These two factors may contribute to part of the reasons to stabilize the activation complex. Subsequent elimination of HOAc from the coordination sphere via abstraction of the C5–H proton by acetate yields the product. A similar concerted-metallation deprotonation process has been proposed for a Pd(OAc)2 promoted C–C coupling reaction.39
:1:3 shown in Fig. 3(a). Addition of an equimolar quantity of Hg(OAc)2 causes a substantial shift of the triazole signals to δ 7.26, 6.65 and 2.48 ppm again with 1:1:3 relative intensity (Fig. 3(b)). N-Coordination apparently occurs and that the larger downfield shift of the signal of C5–H relative to C3–H suggests an N4 rather than N2 coordination. After 1.5 h (Fig. 3(c)), two new signals at δ 6.81 and 2.58 ppm appear, which grow with time (Fig. 3(d–h)), and could be attributed to the formation of the triazolate product. In addition, for those unreacted triazoles the relative intensity of the C5–H to C3–H and CH3 signals gradually decreases along with time. This observation is again consistent with an N4 coordination, which gives a more acidic C5–H proton, such that H–D exchange between the C5–H proton and the deuterated solvent occurs. Then the weak base acetate deprotonates the C5–H proton. However, the role of the acetate ion needs further comment. Reactions of Me-TazH with HgCl2 in the presence of several weak bases were examined. While using NaOAc as a base could indeed produce 2c, other bases such as K2CO3 or NaOH could not yield C-metallation products. Attempts to react R-TazH with other metal sources of Ag2O, Ag(OAc) and Pd(OAc)2 containing basic ligands always produce black metallic powder, but no C-metallation products. Among the few bases and metal ions tried, the combination of Hg(II) and acetate is unique to proceed this facile reaction. A proposed mechanism to account for this reaction is given in Scheme 3. Firstly, N4 coordination of a triazole to Hg(OAc)2 gives a [Hg(OAc)2·R-TazH] adduct. Then two of these adducts form an activation complex, where both (1) weakening of the acidic C5–H bond via hydrogen bonding interaction with neighboring acetate oxygen atom, and (2) forming a partial C5⋯Hg bond with a neighboring Hg atom occur simultaneously. The former interaction is favored by the possible formation of a six-membered HgOCOHC ring. The latter interaction is presumably facilitated by the favorable Hg⋯C-triazolate bond formation. These two factors may contribute to part of the reasons to stabilize the activation complex. Subsequent elimination of HOAc from the coordination sphere via abstraction of the C5–H proton by acetate yields the product. A similar concerted-metallation deprotonation process has been proposed for a Pd(OAc)2 promoted C–C coupling reaction.39
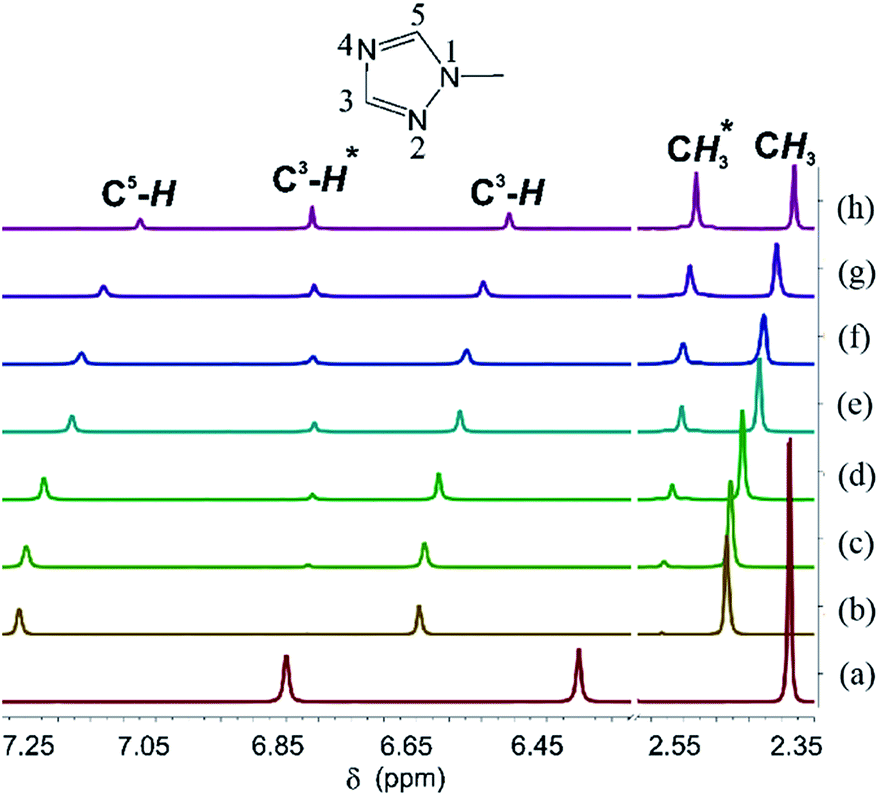 | ||
| Fig. 3 Ambient temperature 1H NMR spectra showing the interaction of Me-TazH (a) with Hg(OAc)2 measured at: (b) t = 0 h, (c) 1.5 h. (d) 6.0 h. (e) 24.0 h. (f) 36.0 h. (g) 48.0 h. (h) 72.0 h. Signals of product (*). | ||
 | ||
| Scheme 3 Proposed mechanism via concerted metallation–deprotonation pathway. | ||
Conclusions
In summary, neutral N-substituted triazoles could react under ambient conditions with Hg(OAc)2 to afford C,N-bound triazole complexes via processes of deprotonation and C-metallation, which otherwise cannot be achieved easily for other metal ions and simple bases. These Hg(II) complexes could be transformed into Hg(II) N-heterocyclic carbene complexes. Furthermore, the transfer of NHC from mercury complexes to other metal centers has been known,29 we thus foresee the role of these Hg-triazolate complexes as an alternative bench top precursor to prepare NR,NR′–NHC complexes, which is a subject for future investigations.Experimental section
General information
Ligand precursors were prepared according to the literature procedures.30,31 Analytical reagent grade chemicals and solvents were purchased from Alfa Aesar, Acros Organics, and Mallinckrodt Chemicals Co. and were used without further purifications. NMR spectra were recorded on Advanced DXP-Bruker spectrometers (300 MHz) and Bruker AvanceII spectrometer (400 MHz). Elemental microanalyses were performed at the Taiwan Instrumentation Center. Single crystal data collection was carried out on a Bruker SMART APEX II diffractometer equipped with a SMART CCD array detector with graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) in ϕ and ω scan modes. All the structures were refined by a full matrix least squares method based on F2 values using the SHELX-97 program.40 Crystallographic data are summarized in ESI.†Synthesis of 1
A mixture of Hg(OAc)2 (0.96 g, 3.0 mmol) and an equimolar amount of the N-functionalized triazole-NHC precursors of Ph-TazH (0.44 g, 3.0 mmol), or Py-TazH (0.44 g, 3.0 mmol), or Me-TazH (0.25 g, 3.0 mmol) was dissolved in methanol (20.0 mL) and heated under reflux for 24 h. Cooling the reaction mixture to room temperature and subsequent filtration afforded the products of 1a, 1b and 1c as colorless solids.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–). Anal. calcd for C5H7HgN3O2: C, 17.57; H, 2.06; N, 12.30. Found: C, 17.27; H, 2.31; N, 12.08%.
O)–O–). Anal. calcd for C5H7HgN3O2: C, 17.57; H, 2.06; N, 12.30. Found: C, 17.27; H, 2.31; N, 12.08%.Synthesis of 2
A solution of 1a (0.61 g, 1.5 mmol), or 1b (0.61 g, 1.5 mmol), or 1c (0.51 g, 1.50 mmol) in ethanol (50.0 mL) was added to a solution of LiCl (0.13 g, 3.0 mmol) in methanol (25.0 mL). The mixture was heated under reflux for 15 minutes and immediately filtered thereafter, to obtain colourless solids, which were further washed with methanol (20 mL × 3), ethanol (20 mL × 3), and subsequently dried in vacuo to obtain the product of 2a, 2b or 2c as colourless solids.Synthesis of [(Ph-Taz-Me)2Hg][BF4]2 (3a)
Reaction was carried out in a dry box. To a suspension of 2a (190.2 mg, 0.50 mmol) in dichloromethane (30 mL), trimethyloxonium tetrafluoroborate (85.0 mg, 0.58 mmol) was added. The resultant cloudy mixture was stirred overnight (12 h) and filtered using a cannula. The filtrate was dried in vacuo, and the resultant solids were washed with about 5 mL of diethyl ether. Solids were again dried in high vacuum to obtain the mercury–NHC complex 3a of [(Ph-Taz-Me)2Hg][BF4]2. Yield 44% (0.76 g) 1H NMR (400 MHz, CD3CN): δ = 8.23 (s, 1H, CH), 7.66 (m, 5H, Ph), 4.05 (s, 3H, CH3). 13C NMR (100 MHz, CD3CN): δ = 177.8 (NCN), 146.8 (taz), 137.7, 131.4, 130.5, 124.4 (Ph), 35.9 (CH3). Anal. calcd for C18H18B2F8HgN6: C, 31.22; H, 2.62; N, 12.13. Found: C, 31.02; H, 2.69; N, 12.03%.Acknowledgements
We thanks the Ministry of Science and Technology, Taiwan (MOST 104-2113-M-259-004-) and (MOST 104-2113-M-259 -010-).Notes and references
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- F. E. Hahn, ChemCatChem, 2013, 5, 419–430 CrossRef CAS.
- S. Burling, M. F. Mahon, R. E. Powell, M. K. Whittlesey and J. M. J. Williams, J. Am. Chem. Soc., 2006, 128, 13702–13703 CrossRef CAS PubMed.
- V. Miranda-Soto, D. B. Grotjahn, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 13200–13201 CrossRef CAS PubMed.
- A. R. Naziruddin, A. Hepp, T. Pape and F. E. Hahn, Organometallics, 2011, 30, 5859–5866 CrossRef CAS.
- T. Kösterke, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2011, 133, 2112–2115 CrossRef PubMed.
- R. Das, C. G. Daniliuc and F. E. Hahn, Angew. Chem., Int. Ed., 2014, 53, 1163–1166 CrossRef CAS PubMed.
- M. C. Jahnke and F. E. Hahn, Coord. Chem. Rev., 2015, 293–294, 95–115 CrossRef CAS.
- M. C. Jahnke and F. E. Hahn, Chem. Lett., 2015, 44, 226–237 CrossRef CAS.
- F. Bonati, A. Burini, B. R. Pietroni and B. Bovio, J. Organomet. Chem., 1989, 375, 147–160 CrossRef CAS.
- H. G. Raubenheimer and S. Cronje, J. Organomet. Chem., 2001, 617–618, 170–181 CrossRef CAS.
- N. Meier, F. E. Hahn, T. Pape, C. Siering and S. R. Waldvogel, Eur. J. Inorg. Chem., 2007, 2007, 1210–1214 CrossRef.
- M. A. Huertos, J. Pérez, L. Riera, J. Díaz and R. López, Angew. Chem., Int. Ed., 2010, 49, 6409–6412 CrossRef CAS PubMed.
- M. A. Huertos, J. Pérez, L. Riera and A. Menéndez-Velázquez, J. Am. Chem. Soc., 2008, 130, 13530–13531 CrossRef CAS PubMed.
- J. Ruiz and B. F. Perandones, J. Am. Chem. Soc., 2007, 129, 9298–9299 CrossRef CAS PubMed.
- J. Ruiz, B. F. Perandones, J. F. Van der Maelen and S. García-Granda, Organometallics, 2010, 29, 4639–4642 CrossRef CAS.
- G. E. Dobereiner, C. A. Chamberlin, N. D. Schley and R. H. Crabtree, Organometallics, 2010, 29, 5728–5731 CrossRef CAS.
- S. E. Flowers and B. M. Cossairt, Organometallics, 2014, 33, 4341–4344 CrossRef CAS.
- M. Brill, J. Díaz, M. A. Huertos, R. López, J. Pérez and L. Riera, Chem.–Eur. J., 2011, 17, 8584–8595 CrossRef CAS PubMed.
- S. Kuwata and T. Ikariya, Chem. Commun., 2014, 50, 14290–14300 RSC.
- K. Araki, S. Kuwata and T. Ikariya, Organometallics, 2008, 27, 2176–2178 CrossRef CAS.
- K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 3202–3203 CrossRef CAS PubMed.
- D. Brackemeyer, A. Hervé, C. Schulte to Brinke, M. C. Jahnke and F. E. Hahn, J. Am. Chem. Soc., 2014, 136, 7841–7844 CrossRef CAS PubMed.
- T. Kösterke, J. Kösters, E.-U. Würthwein, C. Mück-Lichtenfeld, C. Schulte to Brinke, F. Lahoz and F. E. Hahn, Chem.–Eur. J., 2012, 18, 14594–14598 CrossRef PubMed.
- J. C. Lewis, S. H. Wiedemann, R. G. Bergman and J. A. Ellman, Org. Lett., 2004, 6, 35–38 CrossRef CAS PubMed.
- R. J. Sundberg, R. F. Bryan, I. F. Taylor and H. Taube, J. Am. Chem. Soc., 1974, 96, 381–392 CrossRef CAS.
- G. Sini, O. Eisenstein and R. H. Crabtree, Inorg. Chem., 2002, 41, 602–604 CrossRef CAS PubMed.
- M.-H. Yu, H.-H. Yang, A. R. Naziruddin, S. Kanne, B.-H. Wang, F.-C. Liu, I. J. B. Lin and G.-H. Lee, Eur. J. Inorg. Chem., 2016, 4829–4834 CrossRef CAS.
- M. V. Baker, D. H. Brown, R. A. Haque, P. V. Simpson, B. W. Skelton, A. H. White and C. C. Williams, Organometallics, 2009, 28, 3793–3803 CrossRef CAS.
- D. Wang, F. Zhang, D. Kuang, J. Yu and J. Li, Green Chem., 2012, 14, 1268–1271 RSC.
- L. Zhu, P. Guo, G. Li, J. Lan, R. Xie and J. You, J. Org. Chem., 2007, 72, 8535–8538 CrossRef CAS PubMed.
- C. Buron, L. Stelzig, O. Guerret, H. Gornitzka, V. Romanenko and G. Bertrand, J. Organomet. Chem., 2002, 664, 70–76 CrossRef CAS.
- J. C. C. Chen and I. J. B. Lin, J. Chem. Soc., Dalton Trans., 2000, 839–840 RSC.
- S. Budagumpi and S. Endud, Organometallics, 2013, 32, 1537–1562 CrossRef CAS.
- S. Pelz and F. Mohr, Organometallics, 2011, 30, 383–385 CrossRef CAS.
- M. M. D. Roy, M. J. Ferguson and E. Rivard, Z. Anorg. Allg. Chem., 2016, 642, 1232–1235 CrossRef CAS.
- P. Biscarini, L. Fusina and G. D. Nivellini, J. Chem. Soc., Dalton Trans., 1972, 1003–1008 RSC.
- C.-X. Lin, L. Guo, Q.-S. Li, Z.-Z. Zhang, Y.-F. Yuan and F.-B. Xu, J. Organomet. Chem., 2014, 749, 180–187 CrossRef CAS.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1507775 and 1507776. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00163k |
| This journal is © The Royal Society of Chemistry 2017 |