
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CpG and transfer factor assembled on nanoparticles reduce tumor burden in mice glioma model
Yi-Feng Miao†
,
Tao Lv†,
Ran Wang,
Hui Wu,
Shao-Feng Yang,
Jiong Dai and
Xiao-Hua Zhang*
Department of Neurosurgery, Ren Ji Hospital, School of Medicine, Shanghai Jiaotong University, South Campus, No. 2000, Jiangye Road, Shanghai, 201112, China. E-mail: zhangxh_renji@sina.com
First published on 27th September 2016
Abstract
This work describes the use of a transfer factor, a low molecular protein that can transfer cell mediated immunity from donor to recipient, and CpG, a clinically relevant toll-like receptor agonist, for treating glioma. Transfer factor and CpG were assembled onto gold nanoparticles via layer-by-layer assembly. The modified nanoparticles (i.e. particles assembled with transfer factor and CpG) were characterized by size, zeta potential, and loading. An in vivo tumor study revealed that the nanoparticles can inhibit tumor progression more effectively than using either TF or CpG alone or using an equivalent dose of CpG and TF in a soluble mixture. To investigate the anti-tumor mechanism, the modified nanoparticles were interacted with dendritic cells, macrophages. Viability tests showed that the modified nanoparticles did not affect cell viability, and neither did the use of soluble TF or CpG alone. Cell activation assessments showed that the modified nanoparticles can activate DC surface markers (CD80+, CD86+, and CD40+), and promote the production of cytokines (GM-CSF, TNF, and IL-6) from macrophages. In addition, the in vivo study revealed that the modified nanoparticles promoted the production of both inflammatory and effector cytokines in mice serum. Finally, the study also revealed that the production of inflammatory cytokines came primarily from the CpG component, not TF. This study may provide us with a new immune therapeutic strategy for treating glioma.
Introduction
Glioma is one of the most aggressive and lethal tumors that thrives in the human central nervous system – specifically, the brain.1,2 Despite the fact that multimodal treatments (e.g. surgical resection, radiotherapy, and cytotoxic chemotherapy, or a combination of these therapies) have been employed to eradicate this disease, the 5 year survival rate of glioma patients is still extremely low (<5%).3 While glioma immunotherapies are a rapidly expanding frontier,4,5 developing effective immunotherapeutic strategies is hindered by multiple challenges, such as the strong immune suppressive environment from the tumor or lack of glioma specific antigens.6,7 Thus it is necessary to search for new immune therapeutic strategies for this lethal disease.Transfer factors (TFs) are small molecular peptides that are usually dialyzed from immune cells. These peptides possess the capability to transmit cell mediated immunity from sensitized donors to naive recipients.8 Current studies identified that TFs possess a high ratio of tyrosine and glycine that is similar to the N-terminal of some neuropeptides (i.e. encephalin peptide), but more detailed information on TF is still missing.9–12 Although current studies are still unclear as to the molecular mechanism of TFs, multiple clinical trials demonstrated the effectiveness of TFs for treating a wide variety of diseases, such as infectious diseases (i.e. leishmaniasis; toxoplasmosis), immuno-deficiencies (chronic granulomatosis) and even cancers.11,13–15
For example, in one clinical study, TF from lymphocytes of blank donors was employed to treat 356 patients with non-small cell lung cancer (NSCLC); the patients that received TF showed a remarkably improved survival rate, indicating that the administration of TF directly improved lung cancer survival rate.16 In another clinical trial, 5 patients at the advanced stage of breast cancer were treated with TF donated from healthy subjects for 21 to 310 days. While no detectable inflammation or hypersensitivity was detected, regression of the tumor that lasted for 6 months was observed in the recipients, demonstrating the anti-tumor functions of TF.17 CpG is an adjuvant that binds and activates toll like receptor 9 in immune cells. These receptors survey the body for pathogens such as viral RNA and, bacteria that are not common in humans, and they promote the activation of innate and adaptive immunity once they encounter such danger signals. CpG is being employed in multiple clinical trials for the treatment of various cancers.18–20 While these trials are promising, striking discoveries in recent years found that a combination of TLR agonist the tumor antigens can generate synergistic effects in the treatment.21–23 Thus in this study we used CpG and TF together for the treatment of glioma.
In this work, TF was extracted from donor mice implanted with an experimental glioma tumor. CpG and TF were assembled onto gold nanoparticles (NP) via layer-by-layer assembly – a technique that uses electrostatic interactions to assemble oppositely charged material onto substrates.24–26 The assembly of multilayers of materials onto nanoparticles has been widely employed for a variety of applications including radiation therapy, immune therapy and drug delivery,27–30 and showed unique advantages in delivering multiple functional cargos at the nanoscale. We therefore employed this strategy to deliver TF and CpG together for treating glioma.
Materials and methods
Materials
Gold(III) chloride tri-hydrate salt (99.9%) was from VWR. 1× phosphate buffered saline (PBS) was from Sigma. (4-,6-Diamidino-2-phenylindole) (DAPI) was from Invitrogen. The positive isolation beads for isolating dendritic cells were from Miltenyi Biotec. Chitosan (MW = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was from Sigma. Fluorescently labeled antibodies for CD80 (PE), CD86 (PE-Cy7), CD40 (APC) were purchased from Biolegend. RPMI cell culture medium was obtained from VWR.
000) was from Sigma. Fluorescently labeled antibodies for CD80 (PE), CD86 (PE-Cy7), CD40 (APC) were purchased from Biolegend. RPMI cell culture medium was obtained from VWR.
Cells and animals
All experiments that involve mice were approved by the Animal Research Committee Board of Shanghai Jiaotong University, and animal experiments were performed by following the Institutional Animal Care and Use Committee (IACUC) approved by the committee. Mice sacrifice was performed by exposing mice to CO2; the CO2 concentration was increased gradually. Cervical dislocation was employed to ensure a successful sacrifice. C6 glioma cells were obtained from School of Medicine, Shanghai Jiaotong University. The cells were cultured in Dulbecco's modified Eagle's medium plus 10% fetal calf serum. Method for generating C6 glioma cells was established as followed. Briefly, 1 × 106 glioma cells were implanted into mice (10–12 weeks) intraperitoneally. Tumor will be established to sizes ranging from 1 cm2 to 1.5 cm2 in 10 to 15 days. The tumor was then dissected from mice, and minced to pieces with size smaller than 1 mm2. The minced tumor tissues were then processed through a cell strainer (40 μm pore size) by mechanical force to obtain cell suspension. After washing with PBS twice, the cells were collected by centrifuging at 500 rpm for 5 min.Glioma antigen preparation
The freshly dissected tumor tissues were processed into cell suspensions via mechanical forces. After washing with PBS twice, the cells was counted via a cell counter. The cells were frozen to −80 °C and thaw to room temperature. This process was repeated for at least 4 to 5 times to ensure a complete lysis of the cells. The cellular lysate was centrifuged at 1500g for 5 min to remove the large debris from the cells, followed by passing the supernatant to a filter (0.22 μm). The proteins collected was then quantified with a Nanodrop 2000 (Thermoscientific) and stored at −80 °C before use.TF generation
Mice (N = 12) were treated with the glioma antigen (75 μg) plus CpG (25 μg) via intradermal injection on day 0, followed with a boost on day 14. The mice were sacrificed on day 21 with the whole blood collected and treated with EDTA (2 mg mL−1 in blood). The blood was centrifuged at 500g for 25 min to remove the buffy coat. After washing the leukocytes with PBS twice, the cells were frozen to −80 °C and thawed to room temperature. This process was repeated for 10 cycles to ensure a complete lysis of cells. The lysate was collected and dialyzed in a dialysis membrane (MW cut-off = 12000). The TF was dialyzed through the membrane and freezing dried and stored at −80 °C before use. This process also ensures that no pathogen will be collected in the TF since all pathogens have a diameter larger than the dialysis membrane pore.
Nanoparticle synthesis and characterization
The nanoparticles were synthesized according to literature.31 Briefly, 50 mL chitosan was dissolved in acetic acid (1%) at a concentration of 0.5% w/v (pH = 5.1). The chitosan solution was heated to 100 °C and stirred at 650 rpm. Aqueous chloroauric acid (HAuCl4, 1 mM, 75 μL) was added into the chitosan solution. After 30 min, the solution turned into a red wine color. The solution was then moved to 4 °C fridge to stop the reaction. Nanoparticles in solution was stored at 4 °C fridge before use. Surface modification of the nanoparticles was performed through conventional layer-by-layer assembly by following the literatures.24–26 Briefly, the nanoparticles were collected from the solution through centrifuge (15000 rpm, 25 min). The nanoparticles (1.5 mg in 100 μL water, pH = 7.1) were deposited with 1.5 mL TF (200 μg mL−1, pH = 5.1) layer for 10 min. The particles were washed with water (pH = 5.1) to remove the free TFs. Following the deposition of TF, the nanoparticles was then assembled with chitosan layer (1.5 mL, 0.1% in water, pH = 5.3) to generate a positively charged surface on the particles. After washing the particles with water to remove free chitosan, CpG (1.5 mL, 200 μg mL−1, pH = 5.7) was then coated onto the particles as the third layer. These process of deposition was repeated until a desired number of layer was coated onto the particles. Each deposition took 10 min, followed by a washing step with water. The unmodified nanoparticles were imaged using transmission electron microscope (TEM, JEOL JEM-3100F). Zetasizer Nano ZS (Malvern) was employed to measure the size and zeta potential of the particles. The measurement was performed in water (pH = 7.1) and was repeated 3 times. Loading of TF and CpG was performed by an indirect method. Briefly, a certain amount of CpG and TF (i.e. 200 μg for each) was added to the particles. After the assembly, the supernatant was collected by centrifugation (15000 rpm, 25 min). The amount of CpG and TF in the supernatant was character by UV-Vis spectroscopy. The absorbance at 262 nm and 278 nm was used to characterize CpG and TF respectively. The amount of CpG and TF loaded onto the particles were calculated by subtracting the amount of CpG and TF in the supernatant with the original materials added.
In vitro test
For viability test, splenic dendritic cells were collected by sacrificing the mice and collecting the spleen. The spleen was minced to size smaller than 1 mm2, followed by treating with dissociation medium (Miltenyi Biotec), and collected by positive magnetic collection. DCs were then treated with different samples (i.e. TF (25 μg mL−1), CpG (5 μg mL−1), NP (50 μg mL−1) or NP modified with TF and CpG (50 μg mL−1)) for 24 hours, followed by staining with DAPI, assessed with follow cytometry to test their viability. To test the impact of samples to macrophage viability, a same procedure was followed. Activation of DCs were studied by staining the cells with fluorescently labeled antibodies (Invivogen), and assessed via flow cytometry. Production of cytokines from macrophages was performed by using ELISA kits by following the manufacturer's instructions. To test the level of cytokines in serum, C57/BL6 mice (female, 4–8 week old) was immunized with different samples (i.e. TF, CpG, NP or NP modified with TF and CpG) on day 0, and peripheral blood was collected from mice on day 3. The blood was centrifuged at 18000g for 15 min to collect the serum. The serum was stored for ELISA test by following the manufacturer's instructions.
Tumor study
C57/BL6 mice (female, 4–8 week old) were immunized with different samples (i.e. TF, CpG, NP, TF + CpG, or NP modified with TF and CpG) on day 0, followed with a boost on day 15. The mice were implanted with 3 × 105 glioma tumor cells on the flank of mice on day 16. Tumor size was measured every day, and determined by using W × L. Mice were sacrificed when the tumor size reached 1.5 cm2.Results
The first task was to synthesize positively charged gold nanoparticles to act as carriers through the layer-by-layer method reported in literature.31 The particles had a spherical shape, as imaged under transmission electron microscope, although other shapes were also observed (Fig. 1A). The particles were then modified with TF and CpG through layer-by-layer deposition of TF, chitosan, and CpG – the positively charged chitosan served as a bridge that connected the negatively charged TF and CpG onto the nanoparticles. The sizes of the surface modified nanoparticles were studied via dynamic light scattering, in which the particle sizes increased after the deposition of each layer of material. For particles with 0 to 2 trilayers, the average sizes were 18, 91, and 189 nm, respectively (Fig. 1B). Sizes of the particles from TEM is smaller than the number measured by dynamic light scattering probably because the dynamic light scattering tends to collect information from larger particles or aggregates. Zeta potentials of the nanoparticles were also measured. The surface charges of the particles reversed after the deposition of each layer of material, indicating successful deposition. For particles with 0 to 7 single layers of materials, the surface charges were +27.2, −17, +23.7, −15.3, +21.9, −11.9, +20.2, and −12.5, respectively. Briefly, the originally synthesized nanoparticles was positively charged since it was reduced by positively charged chitosan. Once deposited with negatively charged TF, there was a reverse in the surface charge. Another positively charged chitosan was deposited onto the particles to generate a positively charged surface, so that the negatively charged material, CpG, can be deposited onto the particles. Such a deposition was repeated until a desired number of material was assembled (Fig. 1C). The loading levels of the cargo materials (i.e. TF and CpG) were studied by an indirect measurement as well. The particles with 1 and 2 bilayers of (NP/(TF/Chi/CpG)n, n = 1 or 2) had 78.6 μg TF, 126 CpG (for 1 trilayer), and 181.2 μg TF and 251.8 μg CpG (for 2 trilayers), respectively (Fig. 1D).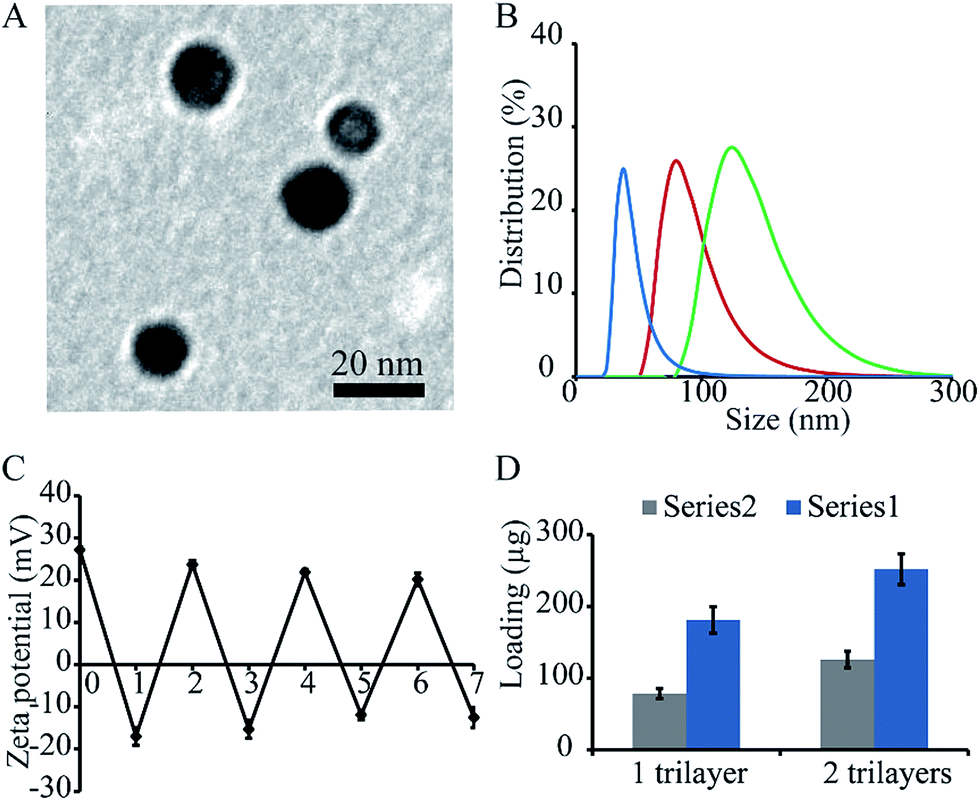 | ||
| Fig. 1 Characterization of nanoparticles modified with transfer factor and CpG. (A) Gold nanoparticles imaged by transmission electron microscopy (TEM). (B) Size of nanoparticles modified with different number layers of materials, i.e. NP/(TF/Chi/CpG)n, n = 1 to 3. Sizes were measured with dynamic light scattering. Sizes of nanoparticles increased with the number of layers deposited. (C) Zeta potential of nanoparticles after the deposition of each layer of materials. There was a reverse of surface charge after depositing each layer of material, indicating the successful deposition of each layer. (D) Loading of TF and CpG on nanoparticles. The nanoparticles had a composition of NP/(TF/Chi/CpG)n, n = 1 to 2. | ||
The modified nanoparticles (i.e. NP/(TF/Chi/CpG)) as well as other samples (TF or CpG) were employed for an in vivo anti-tumor study. The mice with no treatment were used as a negative control (CTRL). The treated mice were injected with these aforementioned samples on day 0 and boosted on day 14. They were then implanted with glioma tumor on day 15 and observed daily. NP/(TF/Chi/CpG) had more dramatic anti-tumor effects compared to the other samples (Fig. 2A). The use of CpG or TF alone also generated an anti-tumor effect – although this tumor growth rate was faster than that in mice treated with modified nanoparticles (Fig. 2A). These anti-tumor effects were measured through tumor weight. On day 18, the mice were sacrificed and measured with tumor weight. The mice treated with NP/(TF/Chi/CpG) had the lowest tumor weight among the four groups, indicating NP/(TF/Chi/CpG) is the most potent in inhibiting tumor progression in all groups (Fig. 2B). Similar to the tumor volume study, TF or CpG alone also reduced the tumor weight in mice compared to CTRL (Fig. 2B). In both measurement, we noticed that a mixture of CpG and TF (i.e. TF + CpG) can also inhibit tumor progression as compared to CTRL or mice treated only either CpG or TF. However, NP/(TF/Chi/CpG) has a stronger potency in tumor inhibition compared to the mixture (Fig. 2A and B).
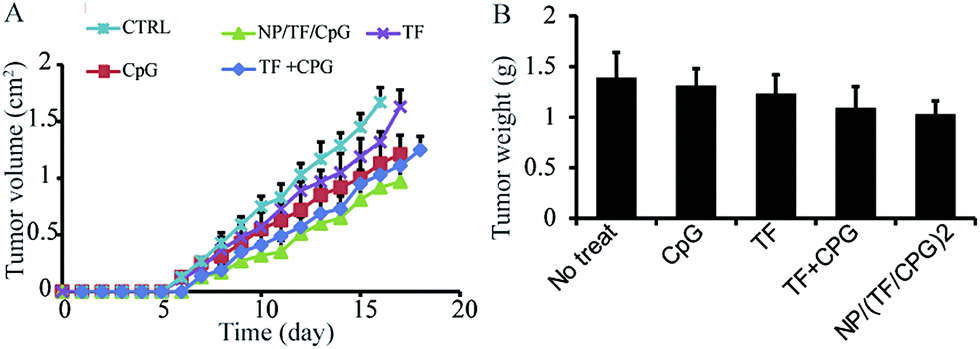 | ||
| Fig. 2 Anti-tumor study using NP/TF/Chi/CpG and other control samples. (A) Daily measurement of tumor in mice treated with different samples. (B) Tumor weight on day 17 after tumor cell implantation. The mice were treated with NP/TF/Chi/CpG or other control samples (soluble TF or CpG, or a mixture of soluble TF and CpG). Those mice with no treatment were used as negative control. | ||
The surface modified nanoparticles (i.e. NP/TF/Chi/CpG) were then utilized to interact with immune cells (i.e. DCs, macrophages, and lymphocytes). The modified nanoparticles were designed for intradermal (i.d.) injection, where dendritic cells and macrophages would be two of the major cells to take up the nanoparticles. Thus, these two cell types were first interacted with NP/TF/Chi/CpG to test the uptake. Fluorescence labeled chitosan (FITC-Chi) as well as TF and CpG were employed to compose the nanoparticles (i.e. NP/(TF/FITC-Chi/CpG)) so as to facilitate flow cytometry analysis. The resulting flow cytometry assay showed that the uptake of nanoparticles by DCs and macrophages was dose dependent (Fig. 3A). When a high dose of nanoparticles was employed (i.e. 1× to 4× dilution), a high percentage (>70%) of cells took the fluorescence labeled nanoparticles; in contrast, when the dose of nanoparticles was diluted 8 to 16 times (i.e. 8× to 16×), a lower percent (∼60%) of DCs and macrophages took the nanoparticles (Fig. 3A). Under flow cytometry, the cells treated with nanoparticles had a stronger fluorescence signal than cells with no treatment (CTRL) (Fig. 3B), again confirming the uptake of nanoparticles by DCs and macrophages (Fig. 3B). A viability assay was used to investigate the impact of nanoparticles on the viability of DC and macrophages (Fig. 3C). Compared to cells with no treatment, the surface modified nanoparticles did not affect the viability of dendritic cells, macrophages (Fig. 3C). Other samples (e.g. CpG, NP, TF, or CpG + TF) were employed to test their impact on cell viability as well. Assessments showed that none of these components affected cell viability (Fig. 3C).
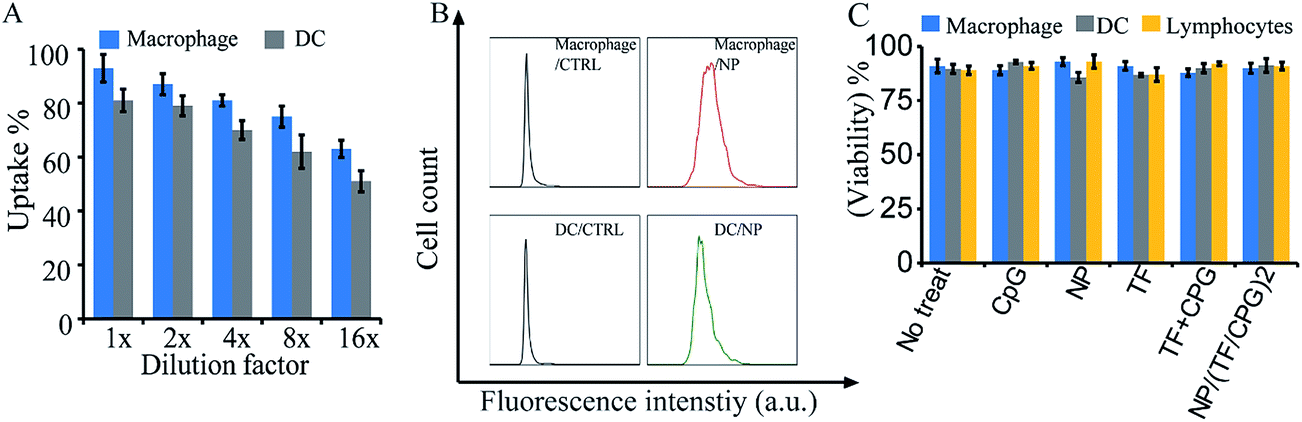 | ||
| Fig. 3 In vitro interactions of NP/TF/Chi/CpG with immune cells for viability test. (A) Dose dependent uptake of NP/TF/Chi/CpG by macrophages and DCs. Chitosan (Chi) was conjugated with fluorescence dye to facilitate flow cytometry detection. The uptake of nanoparticles by the cells were dose dependent. (B) Representative flow cytometry assessment showing the fluorescence shift of macrophages and DCs after nanoparticles uptake. For cells with no nanoparticles treatment, almost no fluorescence signal was detected under flow cytometry; after feeding the cells with nanoparticles, strong fluorescence was detected in the cells, indicating the nanoparticles were uptake by the cells. (C) Viability of macrophages, DCs and lymphocytes after interacting with NP/TF/Chi/CpG. The nanoparticles did not affect viability of the cells. | ||
The surface modified NPs (i.e. NP/TF/Chi/CpG) were used to test their impact on primary DCs – the key antigen presenting cells that bridge adaptive and innate immunity. Briefly, primary DCs isolated from mice spleens were used to interact with NP/TF/Chi/CpG and assessed through a flow cytometry assay to test the activation of major markers (i.e. CD86+, CD80+, CD40+, and CD11c+). Other samples (i.e. no treatment, NPs with no modification, TF, CpG, and TF + CpG in soluble form) were used as controls. Flow cytometry assessments showed that the NP/TF/Chi/CpG promoted CD 86+ activation on DC surfaces (Fig. 4A). Similarly, other important DC activation markers (i.e. CD80+ and CD40+) were also activated by NP/TF/Chi/CpG (Fig. 4B and C). TF in soluble form also slightly activated these surface markers, but the unmodified NP did not have this function compared to CTRL (Fig. 4A–C). Furthermore, compared to TF and CpG in a soluble mixture (TF + CpG), NP/TF/Chi/CpG induced a more potent activation of these markers (Fig. 4A–C). While CD11c+ markers were all at a relatively high level in all DCs, NP/TF/Chi/CpG increased the activation of this marker on DCs compared to TF + CpG in soluble form and CTRL (Fig. 4D). CpG, a toll like receptor that naturally activated all the markers, served as a control in this study, and activated these markers at high efficiency (Fig. 4A–D). These data showed that the co-delivery of TF and CpG can promote the activation of dendritic cells in vivo.
 | ||
| Fig. 4 Activation of DCs by NP/TF/Chi/CpG and other control samples. Expression of (A) CD86+ (B) CD80+, (C) CD40+ (D) CD11c+ on DCs after treating with different samples. Control samples includes, nanoparticles with no modification (i.e. NP), TF or CpG in soluble form, mixture of TF and CpG (i.e. TF + CpG). Those cells with no treatment were employed as control. | ||
NP/TF/Chi/CpG was employed to test their impact on macrophage activation. Briefly, these particles – as well as other controls (i.e. CpG, NP, TF or CpG + TF) – were co-cultured with macrophage cell line J774.A1. ELSA was employed to test the efficacy of these samples on the secretion of three major cytokines: tumor necrosis factor-α (TNF-α), IL-6, and G-CSF, which are three of the major cytokines that are involved in the macrophage associated anti-cancer process. NP/TF/Chi/CpG enhanced TNF-α secretion more than TF + CpG in soluble form, showing the synergistic effect by delivering the two immune cargos in particulate form (Fig. 5A). The use of TF or CpG alone also promoted TNF-α production compared to CTRL group, indicating that both cargos can stimulate the activation of macrophages. Compared to the CpG group, the capacity of TF in stimulating TNF-α production is relatively low (Fig. 5B). Similar trends were also observed in the secretion of another two cytokines (G-CSF and IL-6). The use of CpG alone promoted G-CSF and IL-6 secretion, since CpG was a TLR9 agonist and stimulates macrophages naturally. TF alone also slightly stimulated the production of both cytokines, despite the fact that their production levels were relatively low compared to the CpG group (Fig. 5C and D). The use of CpG + TF in soluble form showed a similar level of G-CSF and IL-6 production compared to the use of CpG alone, indicating that no synergistic effect was generated during the combinational use of G-CSF and IL-6.
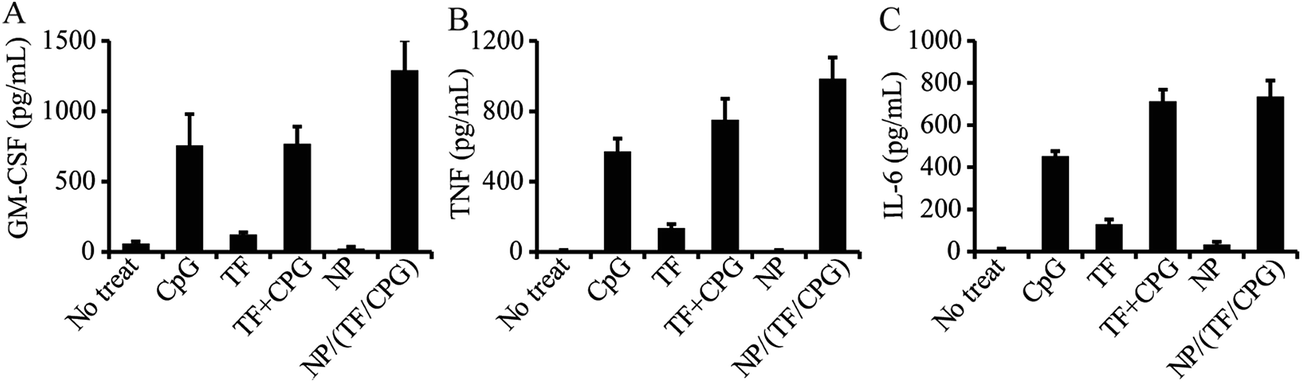 | ||
| Fig. 5 Secretion of cytokines from macrophages treated with NP/TF/Chi/CpG and other control samples. Expression of (A) GM-CSF, (B) TNF and (C) IL-6 by macrophages treated with NP/TF/Chi/CpG. Control samples include TF or CpG in soluble form, nanoparticles with no modification (i.e. NP), TF and CpG in soluble mixture form (i.e. TF + CpG). Cells with no treatment was employed as negative control. Macrophages was treated with the samples for 24 hours. Supernatant was collected for ELISA assessment. | ||
The impact of NP/TF/Chi/CpG on cytokine production in serum was also assessed. To perform this study, mice were immunized with NP/TF/Chi/CpG as well as with other controls (i.e. no treatment, TF, CpG, TF + CpG, and NP) on day 0. Peripheral blood was drained from mice on day 3 and centrifuged at high speed (20000 rpm, 5 min) to collect serum for ELISA. IL-1β level was relatively high in mice treated with NP/TF/Chi/CpG, CpG, or TF + CpG – all of these groups involved the use of CpG. This suggests that CpG plays a significant role in the production of IL-1β. In contrast, IL-1β level in mice treated with TF or NP was relatively low, meaning that neither of these components regulates IL-1β expression (Fig. 6A). Similar trends were observed in the production of another inflammatory cytokine, IL-8. All the groups that involved the use of CpG (i.e. CpG, CpG + TF, and NP/TF/Chi/CpG) showed an enhanced production of IL-8, but the use of TF or NP alone did not show this enhancement (Fig. 6B). As for the effector cytokines (i.e. TNF and IFN-γ), NP/TF/Chi/CpG promoted both cytokines' production in mice; a striking discovery was that TF alone increased TNF and IFN-γ expression compared to CTRL (Fig. 6C and D). In particular, compared to TF + CpG in soluble form, NP/TF/Chi/CpG promoted a higher level of IFN-γ, although this trend was not obvious in the production of TNF (Fig. 6C). It is worth mentioning that the use of NP alone did not promote any of the above-mentioned cytokines (Fig. 6A–D), indicating that the nanoparticles are immune inert.
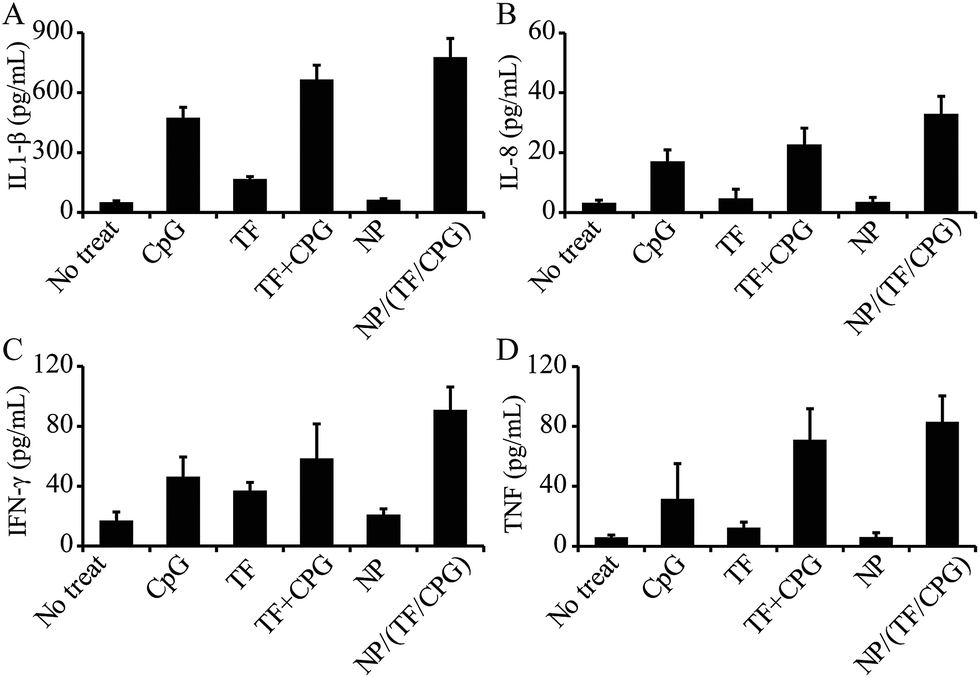 | ||
| Fig. 6 Expression of cytokines in serum of mice treated with NP/TF/Chi/CpG and other control samples. Expression of (A) IL1-β, (B) IL-8, (C) IFN-γ and (D) TNF in mice serum. The mice were treated with NP/TF/Chi/CpG. Control samples include TF or CpG in soluble form, nanoparticles with no modification (i.e. NP), TF and CPG in soluble mixture form (i.e. TF + CpG). Cells with no treatment was employed as negative control. Mice were treated with different samples on day 0 and peripheral blood was collected from mice on day 3. Serum was collected from blood through centrifugation and employed for ELISA assessment. | ||
Discussion
Immune suppression is one of the major issues associated with immune therapy of cancers, including glioma. Cancer progression is usually associated with impaired immune function; specifically, suppressed effector T cell function but enhanced regulatory T cell function.6 The search for effective immune cargos to combat these suppressions has therefore become one of the key goals in the therapeutic treatment of cancers. Broadly, toll-like receptor agonists – a group of “danger” signals (i.e. pathogens) that exists widely in viruses and bacteria but not in humans – have been employed in different ways to treat cancers.32–34 For example, CpG has been conjugated to nanoparticles to activate macrophages for the immune therapy of cancer; the use of CpG enhanced the secretion of cytokines associated with the anti-cancer process.35 In another study, polyIC – a TLR3 agonist – was assembled with a model peptide onto nanoparticles to expand antigen-specific T cells in vivo.30 Similarly, dramatic examples also include the co-use of TLR adjuvants for clinical trials of cancer treatment.32,33 As for glioma – one of the most challenging cancers in the world – immune therapies have also drawn extensive attention in recent years. For example, the delivery of genes that encode cytokines that can modulate the immune suppressed tumor environment have showed promising results by promoting DC activation and effector T cell proliferation.36 In a clinical trial, CpG was used in a phase 1 trial to treat patients with recurrent glioblastoma; preliminary evidence of this study found that two in six patients had a median survival period of 7.2 months.37Transfer factor is a low molecular peptide that can be obtained from humans and animals that have developed immunity against certain type of diseases.38 Studies over transfer factors have drawn considerable attention in the past century.9–11,13,38,39 In particular, multiple clinical trials were carried out for various diseases, ranging from infectious disease to cancers, with both failure and success reported in these trials.9–11,39–41 For example, in one trial, 6 in 9 humans had a reduced hypersensitive immune response to M. Jerpase antigens after being treated with transfer factor.13 In another study, two antigens (i.e. tuberculin, and keyhole limpet hemocyanin) were employed to test for immune specificity and the potency immune response transited from donors to recipients by transfer factor; this study demonstrated in a human trial that the recipient of transfer factor could generate the very same specific immune response as in the donors.38 Cytosine–phosphate–guanine (CpG) is a potent toll like receptor agonist that activates both innate and adaptive immunity. CpG can promote the synthesis of inflammatory cytokines as well as other co-stimulatory molecules; it also promotes effector CD8+ T cell production. CpG shows great potential in combating cancer and has already been employed in several clinical trials.18,42 We therefore combined these two immune cargos in our study to treat glioma. In our study, TF and CpG were assembled onto positively charge nanoparticles via layer-by-layer assembly; this technique uses electrostatic interactions to integrate oppositely charged materials into thin films. Through this technique, we noticed that there was a non-linear increase in particle size after the deposition of each layer of materials (Fig. 1A). This is probably because a certain degree of aggregation was induced during the preparation. We prepared the particles with well-defined properties. The particles showed a reversal of surface charge after the deposition of each layer of material – a result that is expected in the process of layer-by-layer assembly to prove the successful deposition of each layer of material (Fig. 1B). The particle modified with TF and CpG (i.e. NP/TF/Chi/CpG), as well as other control samples, was then employed to treat glioma tumor in mice. NP/TF/CpG had more potent anti-tumor effects compared to using each component alone or a mixture of the two cargos (Fig. 2A). Similar result was confirmed by tumor weight measurement (Fig. 2B).
Flow cytometry showed that the particles were taken up by two major immune cells, macrophages and DCs, indicating that the aggregation did not affect the application of the particles (Fig. 3A and B). Viability tests also confirmed the safety of NP/TF/CpG – neither TF nor CpG impaired cell viability compared to cells with no treatments (Fig. 3C). In this study, TF and CpG were assembled together for treating glioma; this is because previous studies found that the co-delivery of different immune cargos (i.e. different toll like receptor agonists or antigen plus adjuvant) could generate potent synergistic effects much greater than from using each component alone.30 This was consistent with what was demonstrated in our study: the nanoparticles (i.e. NP/TF/Chi/CpG) induced a higher expression of DC surface markers compared to TF + CpG in soluble form (Fig. 4A–C – DC activation). TF alone slightly activated DC surface markers (i.e. CD80+ and CD86+), although the activation level was lower than CpG (Fig. 4A–C). This data indicated that TF was involved in adaptive immunity through interactions with DCs – an important antigen presenting cell that bridges antibody and cellular immunity. On the other hand, TF slightly promoted cytokine production from macrophages – an immune cell that plays an important role in both antibody and cellular immunity (Fig. 5A–C). These data confirmed that TF was involved in adaptive immunity and potentially joined with antibody immunity through macrophages. However, since this study did not investigate the role of B cell functions, the role of TF in antibody immunity has not yet be concluded.
To investigate the in vivo functionality of NP/TF/Chi/CpG, we treated mice with the different samples on day 0 and collected the peripheral blood on day 3 to test the secretion of inflammatory and effector cytokines via ELISA (Fig. 6). NP/TF/Chi/CpG induced a higher level of IL-1β production than the TF + CpG in soluble form (Fig. 6A and B). Similarly, NP/TF/Chi/CpG promoted effector cytokines (i.e. IFN-γ and TNF-α) production to a higher level compared to TF + CpG in soluble form (Fig. 6A–D). This is probably either because the CpG in particulate form has an improved adjuvant effect, or because of the synergistic effect of delivering CpG and TF together. We also noticed that TF alone yielded a very low level of IL-1β and IL-8, indicating TF was not involved in promoting the production of inflammatory cytokines. Instead, TF promoted the production of TNF (Fig. 6C), but not IFN-γ (Fig. 6D). Putting everything together, these data suggest that the TFs involved were involved in effector but inflammatory immune response. While the in vivo tumor study illustrated a synergistic effect of NP/TF/Chi/CpG against glioma, these DC activation and in vivo and in vitro assays partially explained the mechanism underlying this synergistic performance. For a better understanding of using TF and CpG for cancer treatment, further study requires the investigation of the role of the B cell – a major immune cell for antibody immunity in these processes. In addition, studies of the activation pathway of immune cells as well as infiltration of immune cells into tumor tissue will also help us understand the mechanism in this study.
Conclusion
This study investigated the use of TF and CpG for the treatment of glioma. These two immune cargos were loaded onto nanoparticles to ensure the co-delivery of both. An in vivo study showed that particles carrying TF and CpG (i.e. NP/TF/Chi/CpG) had a more potent anti-tumor effect than TF or CpG alone or TF + CpG in soluble mixture. An in vitro activation study showed that NP/TF/Chi/CpG activated DC markers and promoted cytokine productions from macrophages. In vivo serum cytokine secretion analysis showed that NP/TF/Chi/CpG promoted the production of both inflammatory and effector cytokines; among these cytokine productions, our study indicated that TF contributed to the production of effector cytokines but not inflammatory cytokines. These in vitro assays partially explained the immune mechanism involved in the anti-tumor immunity of NP/TF/Chi/CpG. Further studies such as infiltration of immune cells to the tumor as well as B cell immunity will be performed to better understand the mechanism of using TF and CpG together.Conflict of interest
The authors declare no conflict of interest.Ethical standard
All studied that involves animals were performed under the regulation rules of ethical standards of the Institutional and/or National Research Committee.Acknowledgements
This project was supported by the National Nature Science Foundation (No. 81000498).References
- A. Omuro and L. M. DeAngelis, JAMA, J. Am. Med. Assoc., 2013, 310, 1842–1850 CrossRef CAS PubMed.
- T. F. Cloughesy, W. K. Cavenee and P. S. Mischel, Annu. Rev. Pathol.: Mech. Dis., 2014, 9, 1–25 CrossRef CAS PubMed.
- D. R. Johnson, H. E. Leeper and J. H. Uhm, Cancer, 2013, 119, 3489–3495 CrossRef CAS PubMed.
- L. Yang, G. Guo, X. Y. Niu and J. Liu, BioMed Res. Int., 2015, 2015, 717530 Search PubMed.
- N. Ung and I. Yang, J. Neuro-Oncol., 2015, 123, 473–481 CrossRef CAS PubMed.
- T. D. Azad, S. M. Razavi, B. Jin, K. Lee and G. Li, J. Neuro-Oncol., 2015, 123, 347–358 CrossRef CAS PubMed.
- L. W. Xu, K. K. Chow, M. Lim and G. Li, J. Immunol. Res., 2014, 2014, 796856 Search PubMed.
- H. S. Lawrence, J. Clin. Invest., 1955, 34, 219–230 CrossRef CAS PubMed.
- M. S. Ascher, A. A. Gottlieb and C. H. Kirkpatrick, Transfer factor: basic properties and clinical applications: proceedings of the Second International Workshop on Basic Properties and Clinical Applications of Transfer Factor, held at the United States Army Medical Research Institute of Infectious Diseases, October 5–8, 1975, Academic Press, New York, 1976 Search PubMed.
- A. Khan, C. H. Kirkpatrick and N. O. Hill, Immune regulators in transfer factor: [proceedings], Academic Press, New York, 1979 Search PubMed.
- C. H. Kirkpatrick, D. R. Burger and H. S. Lawrence, Immunobiology of transfer factor, Academic Press, New York, 1983 Search PubMed.
- S. J. Rozzo and C. H. Kirkpatrick, Mol. Immunol., 1992, 29, 167–182 CrossRef CAS PubMed.
- W. E. Bullock, M. Brandris and J. P. Fields, N. Engl. J. Med., 1972, 287, 1053–1059 CrossRef CAS PubMed.
- S. Estrada-Parra, A. Nagaya, E. Serrano, O. Rodriguez, V. Santamaria, R. Ondarza, R. Chavez, B. Correa, A. Monges, R. Cabezas, C. Calva and I. Estrada-Garcia, Int. J. Immunopharmacol., 1998, 20, 521–535 CrossRef CAS PubMed.
- G. B. Wilson, J. F. Metcalf and H. H. Fudenberg, Clin. Immunol. Immunopathol., 1982, 23, 478–491 CrossRef CAS PubMed.
- V. Pilotti, M. Mastrorilli, G. Pizza, C. De Vinci, L. Busutti, A. Palareti, G. Gozzetti and A. Cavallari, Biotherapy, 1996, 9, 117–121 CrossRef CAS PubMed.
- H. F. Oettgen, L. J. Old, J. H. Farrow, F. T. Valentine, H. S. Lawrence and L. Thomas, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 2319–2323 CrossRef CAS.
- B. Badie and J. M. Berlin, Immunotherapy, 2013, 5, 1–3 CrossRef CAS PubMed.
- E. Vacchelli, L. Galluzzi, A. Eggermont, W. H. Fridman, J. Galon, C. Sautes-Fridman, E. Tartour, L. Zitvogel and G. Kroemer, OncoImmunology, 2012, 1, 894–907 CrossRef PubMed.
- S. Adams, Immunotherapy, 2009, 1, 949–964 CrossRef CAS PubMed.
- S. P. Kasturi, I. Skountzou, R. A. Albrecht, D. Koutsonanos, T. Hua, H. I. Nakaya, R. Ravindran, S. Stewart, M. Alam, M. Kwissa, F. Villinger, N. Murthy, J. Steel, J. Jacob, R. J. Hogan, A. Garcia-Sastre, R. Compans and B. Pulendran, Nature, 2011, 470, 543–547 CrossRef CAS PubMed.
- D. I. Gabrilovich, S. Ostrand-Rosenberg and V. Bronte, Nat. Rev. Immunol., 2012, 12, 253–268 CrossRef CAS PubMed.
- T. Querec, S. Bennouna, S. Alkan, Y. Laouar, K. Gorden, R. Flavell, S. Akira, R. Ahmed and B. Pulendran, J. Exp. Med., 2006, 203, 413–424 CrossRef PubMed.
- P. Ott, K. Trenkenschuh, J. Gensel, A. Fery and A. Laschewsky, Langmuir, 2010, 26, 18182–18188 CrossRef CAS PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- H. Ai, S. A. Jones and Y. M. Lvov, Cell Biochem. Biophys., 2003, 39, 23–43 CrossRef CAS PubMed.
- P. Zhang, Y. Qiao, C. Wang, L. Ma and M. Su, Nanoscale, 2014, 6, 10095–10099 RSC.
- Y. H. Roh, J. B. Lee, K. E. Shopsowitz, E. C. Dreaden, S. W. Morton, Z. Poon, J. Hong, I. Yamin, D. K. Bonner and P. T. Hammond, ACS Nano, 2014, 8, 9767–9780 CrossRef CAS PubMed.
- A. Elbakry, A. Zaky, R. Liebl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- P. Zhang, Y. C. Chiu, L. H. Tostanoski and C. M. Jewell, ACS Nano, 2015, 9, 6465–6477 CrossRef CAS PubMed.
- H. Huang and X. Yang, Biomacromolecules, 2004, 5, 2340–2346 CrossRef CAS PubMed.
- J. Holldack, Drug Discovery Today, 2014, 19, 379–382 CrossRef CAS PubMed.
- S. Rakoff-Nahoum and R. Medzhitov, Nat. Rev. Cancer, 2009, 9, 57–63 CrossRef CAS PubMed.
- E. Y. So and T. Ouchi, Int. J. Biol. Sci., 2010, 6, 675–681 CrossRef CAS PubMed.
- A. Y. Lin, J. P. Almeida, A. Bear, N. Liu, L. Luo, A. E. Foster and R. A. Drezek, PLoS One, 2013, 8, e63550 CAS.
- S. Behboudi, D. Chao, P. Klenerman and J. Austyn, Immunology, 2000, 99, 361–366 CrossRef CAS PubMed.
- A. Carpentier, F. Laigle-Donadey, S. Zohar, L. Capelle, A. Behin, A. Tibi, N. Martin-Duverneuil, M. Sanson, L. Lacomblez, S. Taillibert, L. Puybasset, R. Van Effenterre, J. Y. Delattre and A. F. Carpentier, Neuro-Oncology, 2006, 8, 60–66 CrossRef CAS PubMed.
- K. S. Zuckerma, J. A. Neidhart, S. P. Balcerza and A. F. Lobuglio, J. Clin. Invest., 1974, 54, 997–1000 CrossRef PubMed.
- A. Uotila, Transfer factor and other immunological activities of human leucocyte dialysate and other dialysates of mammalian tissues, University of Tampere, Tampere, 1979 Search PubMed.
- P. Gröhn, PhD thesis, University of Tampere, Finland, 1976.
- E. Karhumäki, Modulation of infection resistance of mice with dialysates containing transfer factor-like activity derived from leukocytes of man and other mammalia, University of Tampere, Tampere, 1988 Search PubMed.
- B. Jahrsdorfer and G. J. Weiner, Update Cancer Ther., 2008, 3, 27–32 CrossRef PubMed.
Footnote |
| † These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |