
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simply packaging Ni nanoparticles inside SBA-15 channels by co-impregnation for dry reforming of methane†
Songbai Qiu ,
Qiao Zhang,
Wei Lv,
Tiejun Wang*,
Qi Zhang and
Longlong Ma
,
Qiao Zhang,
Wei Lv,
Tiejun Wang*,
Qi Zhang and
Longlong Ma
Key Laboratory of Renewable Energy, Guangdong Key Laboratory of New and Renewable Energy Research and Development, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, PR China. E-mail: wangtj@ms.giec.ac.cn; Fax: +86 20 87057789
First published on 5th May 2017
Abstract
The highly dispersed Ni nanoparticles inside SBA-15 channels (Ni@SBA-15) were synthesized simply by co-impregnation using ethylene glycol (EG). During co-impregnation, the EG additive in the metal nitrate aqueous solution could successfully deliver and package the nickel species into the channels of mesoporous SBA-15. The characterization results suggest that drying after impregnation plays a critical role in the distribution of the precursor. Compared with conventional impregnation, redistribution and agglomeration of the Ni precursor were efficiently avoided due to the lower surface tension and high viscosity of the concentrated solution during drying, and super-small Ni nanoparticles with higher dispersions and narrow particle size distributions were predominantly confined over the internal surface of mesoporous channels instead of its external surface. The Ni@SBA-15 catalyst using co-impregnation demonstrated excellent sintering-resistance and stability in the dry reforming of methane due to its strong metal-support interaction and the confined location of Ni nanoparticles.
1. Introduction
In recent years, the dry reforming of methane (DRM) has received much attention because it can effectively convert the two dominant greenhouse gases, CO2 and CH4, into synthesis gas and hydrogen.1–3 This process provides a promising technology for producing suitable feedstock for Fischer–Tropsch synthesis or hydrogen power.3–5 As was recently reviewed, different supported metal catalysts, e.g. Pd, Ru, Pt, Ir, Rh, Ni and Co, have been extensively investigated in the DRM reaction;1–7 Ni-based catalysts have been demonstrated as the most attractive candidates, due to their high activity and abundant supply at low cost.1,8 Nevertheless, the major drawback of Ni-based catalysts is their easy tendency toward rapid catalyst deactivation, caused by heavy coke deposition and Ni particle sintering during the harsh DRM reaction.7–9To improve the catalytic properties, current research efforts have focused on the development of more stable catalysts with high resistance to coking and sintering. It is generally acknowledged that the small Ni nanoparticle size and the strong metal-support interactions (SMSI) can effectively suppress coking and sintering.10–12 From this point of view, many strategies have been proposed to enhance the catalytic stability, such as introducing different metal promoters, preparation methods, support materials and pre-treatment conditions.1,9,13 Among them, the confinement of Ni nanoparticles in mesoporous SBA-15 has been proven to be a good approach for dispersing and controlling the Ni particle size, as well as suppressing carbon formation and nanoparticle agglomeration.6,7 Generally, the conventional impregnation is preferred for preparing supported metal catalysts, due to its simplicity and practicability. Unfortunately, the Ni precursor is hard to fully drive and package into the mesoporous channels, and the obtained NiO particles are detected both inside and outside the pores, resulting in poor dispersions due to redistribution and agglomeration during the subsequent drying and calcination.14,15
Formerly, it was reported that polyols such as EG can deliver and immobilize the nickel species into the SBA-15 channels for producing the highly dispersed Ni nanoparticles.16 Now, we have simplified this preparation route and have developed a more simple and practical preparation method of co-impregnation with the nickel nitrate aqueous solution containing the appropriate amount of EG for preparing the Ni@SBA-15 catalyst. Compared with the conventional wetness impregnation, the only difference of co-impregnation is that there is the need to add proper EG into the nickel nitrate aqueous solution, and the other preparations have the same procedures. Herein, we have disclosed that the nickel species could also be effectively introduced and confined to the SBA-15 channels using the co-impregnation pathway. Fig. 1 displays a schematic illustration of the preparation process via co-impregnation for preparing the highly dispersed NiO nanoparticles entrapped in the porous channel. Particularly, the simplified preparation route could avoid not only the drying in vacuum, but also the carbonization under an inert atmosphere. The EG in the nickel nitrate aqueous solution could assist the Ni precursor in easily entering the SBA-15 channels by capillary force, and as the solvent was continually evaporating, the additive EG was adsorbed onto the internal surface of the mesoporous channels, which inhibited the redistribution and agglomeration of the impregnated solution upon drying of the support.16–19 As a result, the highly dispersed Ni nanoparticles were almost anchored in the SBA-15 channels with strong metal-support interactions, and were simply acquired by co-impregnation using ethylene glycol (EG). The confinement effect of mesoporous channels was strongly beneficial for preventing the sintering of Ni nanoparticles and suppressing carbon deposition so that the acquired catalysts presented higher activity and excellent stability in the severe DRM reaction.
 | ||
| Fig. 1 Schematic of the Ni@SBA-15 catalyst using co-impregnation with EG. | ||
2. Experimental
2.1. Catalyst preparation
Pure siliceous SBA-15 and nano-SiO2 (average particle size 20 nm) were supplied by Nanjing XFNANO Materials Tech Co., Ltd. (Nanjing, China) as support materials. The metal loading of nickel was set as 10.0 wt% for all catalysts. The Ni supported on SBA-15 catalysts were prepared using co-impregnation according to the previously established procedure.17 Prior to co-impregnation, the support needed to be calcined at 550 °C for 4 h in air. In a typical preparation, the quantitative EG and nickel nitrate (Ni(NO3)2·6H2O) were added into a defined amount of deionized water (Ni/EG ratio = 1) and stirred for dissolution, then the mixed solution was impregnated in the SBA-15 support. After that, the sample was kept standing for 12 h, followed by drying at 100 °C in air overnight, and was finally calcined at 450 °C for 2 h with a heating rate of 2 °C min. The catalysts obtained via co-impregnation were denoted as Ni@SBA-15. For comparison, the samples, denoted as Ni/SBA-15, were also prepared by conventional incipient wetness impregnation. The Ni supported on the nano-SiO2 sample was prepared by an ammonia evaporation method (the mole ratio of NH3/Ni was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and denoted as Ni/nano-SiO2 AE.
:1) and denoted as Ni/nano-SiO2 AE.
2.2. Catalyst tests
The DRM reaction was carried out in a fixed-bed quartz reactor with an inner diameter of 6 mm. The catalyst powder needed to be pelletized, crushed and then sieved into 60–80 mesh. Normally, 0.1 g of catalyst was diluted using 1.0 g of quartz sand with identical mesh size, and loaded on a quartz wool bed. Prior to each reaction, the Ni-based catalysts were reduced at 700 °C for 1 h under flowing H2 (60 mL min−1), and then purged by Ar. The activities of the catalysts were evaluated in the temperature range of 600–800 °C at an increment of 50 °C with a heating rate of 10 °C min. When the reaction temperature was elevated to the preset level, the reactor was subsequently purged with the reactant gas. Furthermore, the reaction temperature was maintained for 30 min before collecting the experimental data for each test. The DRM reaction was tested under atmospheric pressure (CH4/CO2/N2 = 45/45/10, N2 was the inner reference, gas hourly space velocity (GHSV) of 36000 mL g−1 h). For catalytic stability test experiments, the reaction temperature was kept constant at 700 °C for 30 h; otherwise, conditions were the same. The products were analyzed using an on-line gas chromatograph (Shimadzu GC-2010) equipped with a TDX-01 column and a thermal conductivity detector (TCD).
2.3. Catalyst characterization
The BET (Brunauer–Emmet–Teller) specific surface area, average pore diameter and pore volume of catalysts were determined by N2 isothermal adsorption using a QUADRASORB SI analyzer equipped with the QuadraWin software system. X-ray diffraction (XRD) patterns were recorded by an X'pert Pro Philips diffractometer using Cu Kα radiation (0.1541784 nm). The measurement conditions were in the range of 2θ = 5–80°, step counting time 10 s, and step size 0.0167° at 298 K. TEM images were determined by a JEOL JEM-2010 microscope with an electron beam of 200 kV. Each specimen was dispersed ultrasonically in ethanol for 30 min and then dropped onto a copper grid. The XPS analysis was performed on a ThermoFisher Scientific ESCALAB 250 spectrometer. The spectra were excited by the monochromatized Al Kα source (1486.6 eV). All the electron binding energies were calibrated using the C 1s peak at 284 eV as the reference. Hydrogen temperature-programed reduction (H2-TPR) studies of the different catalysts were carried out on a Micromeritics Autochem apparatus (model 2910) with a thermal conductivity detector (TCD). Before the test, 50 mg of the sample were pretreated in a He atmosphere at 300 °C for 1 h. All H2-TPR runs were performed from 50 to 850 °C under a 5% H2/N2 flow with a heating rate of 10 °C min.3. Results and discussion
3.1. Catalyst characterization
In the case of mesoporous SBA-15, the impregnation solution should be easily sucked up by the mesoporous channels and generally fill the pores quickly, due to the capillary pressure.20 However, the SBA-15 support was not fully hydrophilic, which was attributed to the relatively low hydroxyl density of mesoporous silica, resulting in incomplete wetting.21 Thus, inhomogeneous filling of pores during impregnation was inevitable, and the degree of wetting and infiltration into the mesoporous channels and subsequent thermal treatment appeared particularly important. As shown in Tables 1 and 2, upon gradual removal of the solvent, the surface tension and the viscosity of impregnation solutions led to an obvious change. It should be noted that addition of EG into the nickel nitrate aqueous solution has been shown to decrease the surface tension of impregnation solutions, and there was a gradual decrease upon evaporation of the solvents. Interestingly, EG could work as a surfactant; as the solvent was gradually removed during drying, the decreased surface tension of impregnation solutions favored better wetting of the support, and accelerated diffusion and infusion into the deeper mesoporous channels of the catalyst support. The figure also confirmed that the viscosity increased slowly during the initial stage of drying, and then it exhibited a rapid increase in the viscosity of the impregnation solutions, especially late in drying.| Samples | Water contentc (wt%) | Surface tensiond (10−3 N m−1) |
|---|---|---|
| a The impregnation solution was used for preparing Ni/SBA-15 and Ni@SBA-15, respectively.b Pure EG (AR, 98%).c In the solutions, the mole ratio of EG/Ni(NO3)2·6H2O was 1:1.d The surface tension was measured for different solutions using the pending drop method, with the “contact angle system OCA” (OCA40 Micro) from DataPhysics Instruments GmbH, Germany. |
||
| Solution 1 with NO EGa | 94.3% | 71.07 |
| Solution 2 with EGa | 93.1% | 66.83 |
| Solution 3 with EG | 62.2% | 58.61 |
| Solution 4 with EG | 50.7% | 56.62 |
| Solution 5 with EG | 6.2% | 48.53 |
| Pure EGb | — | 43.16 |
| Samples (25 °C) | Water content (wt%) | Dynamic viscosity (mPa·s)c |
|---|---|---|
| a The impregnation solution with EG was used for preparing the Ni@SBA-15 sample.b Solution 5 contained the same amount of water and Ni(NO3)2·6H2O as in solution 4, except for EG. The test temperature was set at 51 °C so that the solute of nickel nitrate could be dissolved thoroughly.c The dynamic viscosity of different solutions was measured using a NDJ-1 Rotary Viscometer from Shanghai Tianmei Balance Instruments Co. Ltd., China. | ||
| Solution 1 with EGa | 93.1% | 1.5 |
| Solution 2 with EG | 50.7% | 3.3 |
| Solution 3 with EG | 34.1% | 5.5 |
| Solution 4 with EG | 6.2% | 45.0 |
| Solution 5 with NO EGb (51 °C) | 7.4% | 10.0 |
The above results contributed to the physical picture of drying:20 as drying proceeded, the concentrated solvent with the lower surface tension could diffuse deeply at first and be transported toward the internal surface of the mesoporous channels via capillary forces; towards the end of drying, the enhanced viscosity could lead to a strong interaction with the internal surface of the support and inhibit the redistribution of the impregnation solution during drying, thus favoring a uniform distribution of the active component in the mesoporous channels. In contrast, the precursor has a weak interaction with the support without EG, and the concentrated solvent sucked up by the mesoporous pores could be back-diffused by higher surface tension and migrate toward the external surface of the mesoporous channels, resulting in a concentration buildup on the external rim of the support. As a consequence, inhomogeneous distributions of the precursor appeared.
The TEM images of the Ni-based catalysts further produced clear evidence to support the above views, after drying at 100 °C without calcination. Fig. 3 shows that inhomogeneous distributions of the precursor were formed after drying, even if there was no subsequent calcination and reduction, while the use of EG as a co-impregnation solvent resulted in especially homogeneous distributions and no aggregates were found, which was attributed to the lower surface tension and higher viscosity of the concentrated impregnation solution during drying. Furthermore, Fig. 3B(b) TEM images showed that the SBA-15 pores were not completely filled with the precursor, suggesting that the SBA-15 support was not fully hydrophilic, due to the relatively low hydroxyl density of mesoporous silica, resulting in incomplete wetting.
Moreover, ESI SFig. 4† shows that the addition of EG during co-impregnation changed the thermal decomposition behavior of the samples, i.e., the heat released from thermal decomposition of the adsorbed EG on the SBA-15 surface was quickly absorbed by impregnated nickel nitrate to supply energy for decomposition, which led to the increasing of the nickel nitrate decomposition in the low temperature range (at 221 °C). This could efficiently prevent further aggregation of the precursor, contributing to improving the NiO dispersion inside the SBA-15 channels.
The high-angle and low-angle XRD patterns of the Ni-based catalysts before and after reduction are shown in Fig. 4 (reduction conditions: 700 °C, 1 h, 60 mL min−1 H2). The corresponding textural parameters are presented in Table 3. The Ni metal dispersion and crystallite sizes of the different Ni-based catalysts were measured using the hydrogen-temperature program desorption technique (H2-TPD).22
| Samples | Impregnation solvent | Metal loadingd (wt%) | SBETb (m2 g−1) | Pore volumeb (cm3 g−1) | Average pore diameterb (nm) | Dispersione (%) | NiO particle sizea (nm) |
|---|---|---|---|---|---|---|---|
| a NiO particle size was calculated using Scherrer's equation, with the Bragg angle of 2θ = 43.3°.b SBET and pore volumes and diameters of the samples were calculated using standard BET and BJH theory, respectively.c The samples were prepared by co-impregnation with EG.d The metal loadings were determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES).e The dispersion and crystallite sizes of metallic Ni were determined from the amount of H2 desorption in the H2-TPD at 50 °C, in which the H2 adsorption was assumed to be Nisurface/H = 1 and the supported metal was fully reduced. | |||||||
| SBA-15 | — | — | 678 | 1.10 | 6.43 | — | — |
| Ni/SBA-15 | Water | 9.62 | 511 | 0.85 | 5.66 | 7.8 | 15.2 (12.9)e |
| Ni@SBAc-15 | EG + water | 8.70 | 570 | 0.92 | 5.66 | 19.0 | 4.1 (5.3)e |
The loading of Ni on the SBA-15 support obviously decreased the accessible specific surface area and pore volume. Compared to the pore diameter of the original SBA-15 (6.43 nm), that of Ni-based samples got slightly smaller due to the shrinkage of silica walls by the heat treatment or partial blockage of mesoporous channels by NiO nanoparticles, which is in good agreement with the observation of the peak shift in the XRD patterns. As shown in the low-angle XRD patterns, each spectrum exhibited three well-resolved diffraction peaks in the region of 2θ = 0.5–2.5°, which were assigned to the (100), (110), and (200) reflections, revealing that the support material was characteristic of the highly ordered 2D-hexagonal mesoporous structure (p6mm).19 After Ni loading, there was only a slight shift toward higher angles, relative to the low-angle peaks of the SBA-15 support, proving that the catalyst preparation had no significant effect on the mesoporous structure.
The N2 adsorption/desorption isotherms of the samples and the pore size distribution are presented in Fig. 2. These isotherms are typical of a reversible type IV with a hysteretic loop based on the IUPAC classification scheme for mesoporous materials, with a narrow pore size distribution of the cylindrical pores, well in agreement with XRD at low angle and TEM observations. However, new steps were clearly present on the desorption branch of the Ni@SBA-15 sample, which generated delays in the closing of the hysteresis loops at approximately 0.4–0.6. This indicates that a new porous system was present in tubular SBA-15, according to the capillary condensation model, which was previously indicated as possible in ink-bottle mesopores.23 At the same time, two porous systems were obviously noted on the Ni@SBA-15 sample (Fig. 2B). Primary mesopores had an average diameter of 5.66 nm, and the diameter of the new mesopores was 3.38 nm. These two porous systems were the direct evidence of the blockage of primary mesopores with confined NiO nanoparticles, which is in line with the above measurements. Thus, these new mesopores appeared as a result of the partial blocking of the SBA-15 channels with confined NiO nanoparticles.
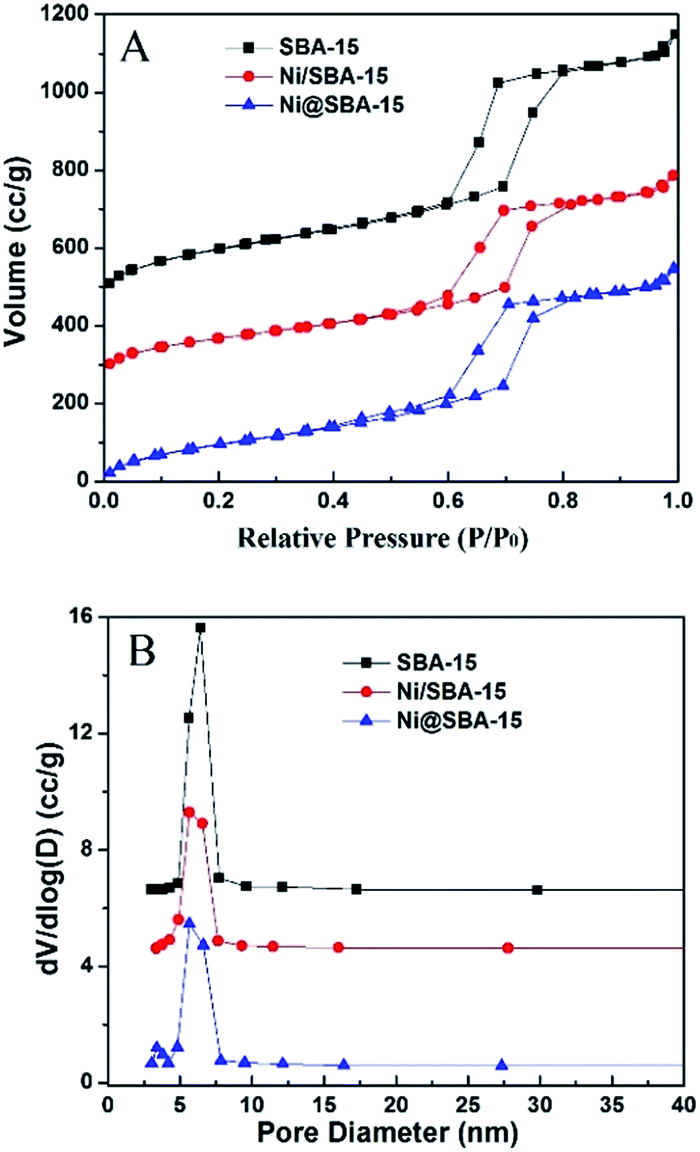 | ||
| Fig. 2 (A) Nitrogen adsorption isotherms and (B) pore size distribution of the Ni-based catalysts. | ||
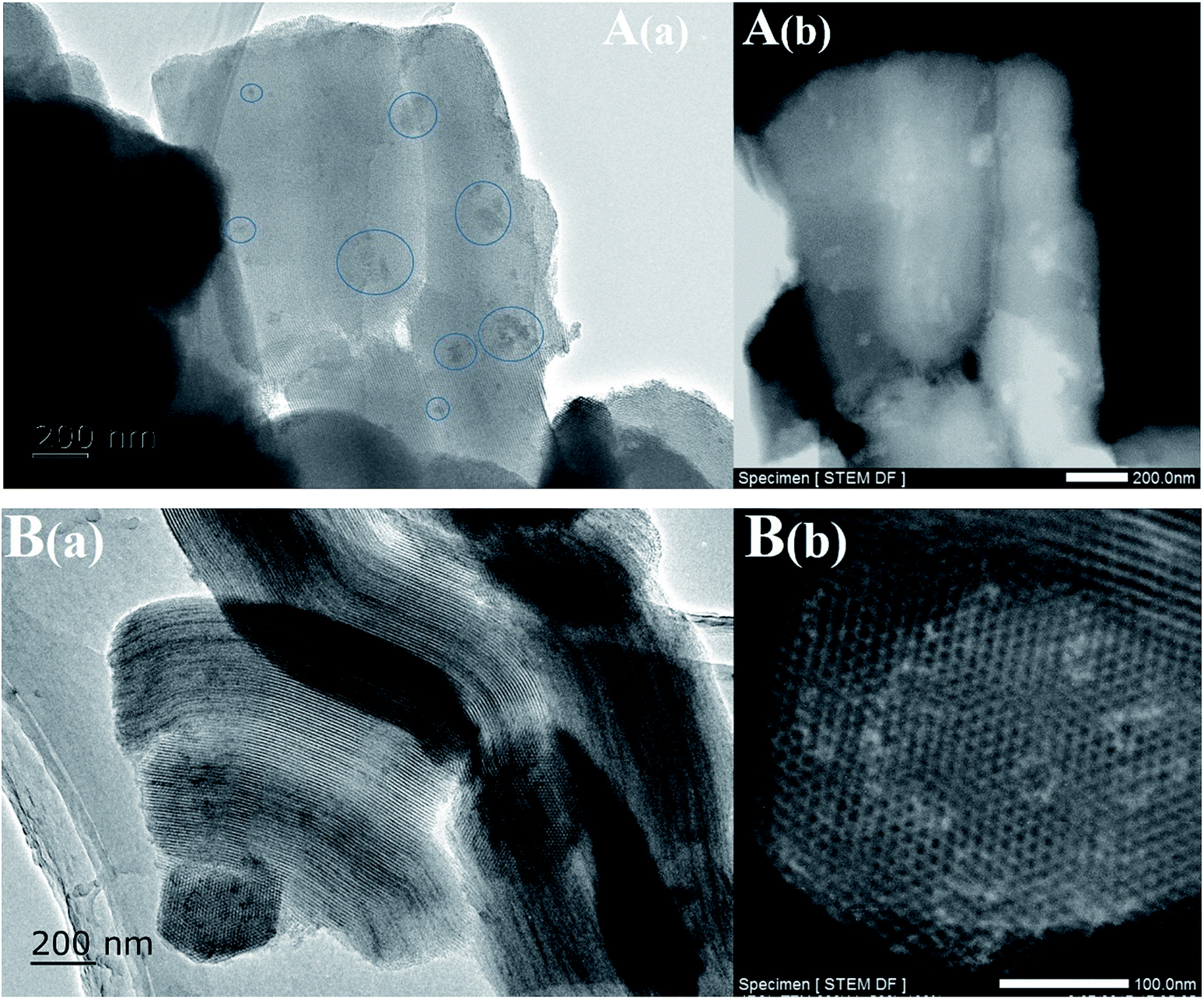 | ||
| Fig. 3 TEM images of the Ni-based catalysts after drying at 100 °C without calcination: (A) Ni/SBA-15 and (B) Ni@SBA-15. | ||
In all high-angle diffractograms, there was one broad and diffuse diffraction signal observed clearly at around 2θ = 15–30°, which could be attributed to amorphous silica (SBA-15). The Ni/SBA-15 sample displayed five strong and sharp diffraction peaks at 2θ = 37.2°, 43.3°, 62.8°, 75.3°, 79.4°, indicating that nickel species were present in the form of cubic NiO (JCPDS no. 44–1159) after calcination.24,25 In contrast, the Ni@SBA-15 sample showed three weak and diffuse diffraction peaks of NiO, confirming that NiO particles were very small and highly dispersed on the SBA-15 support. The average particle size of NiO was evaluated by the Scherrer equation at 2θ angle of 43.3°. The average particle sizes of 15.2 nm and 4.1 nm were obtained for Ni/SBA-15 and Ni@SBA-15 samples, respectively. After H2 reduction at 700 °C for 1 h, the new diffraction peaks of Ni clearly emerged in Fig. 4B. Particularly, for the Ni@SBA-15 sample, the metallic Ni particles retained the approximate crystal sizes and dispersion of NiO, implying the possible existence of the confinement effect.
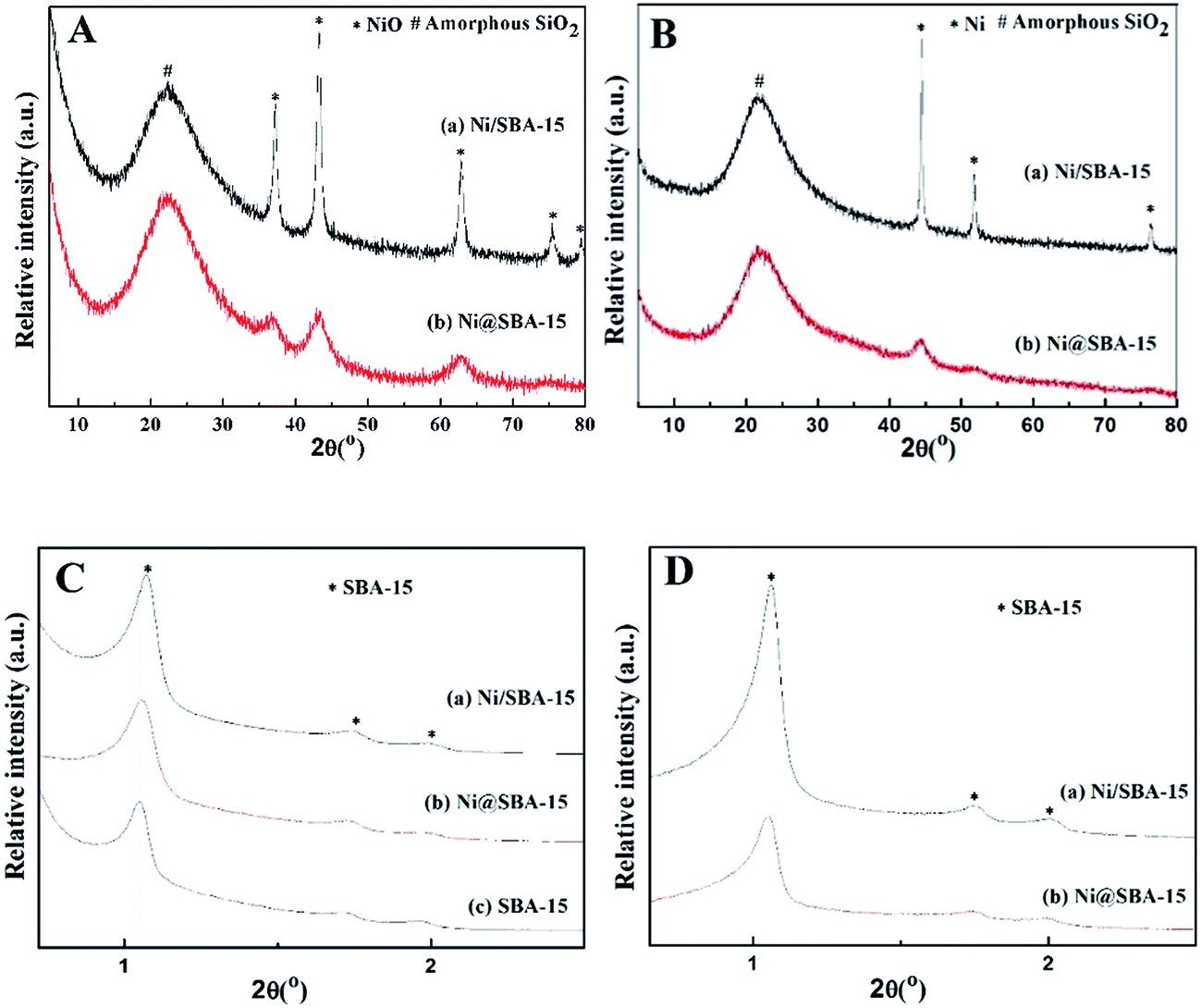 | ||
| Fig. 4 High-angle and low-angle XRD spectra of the Ni-based catalysts: (A) and (C) after calcination; (B) and (D) after reduction. | ||
The representative TEM images of the reduced Ni/SBA-15 catalysts are shown in Fig. 5. All the samples revealed the well-ordered channel structures of mesoporous SBA-15, thus confirming the results obtained by XRD analysis at low angles. In the case of Ni/SBA-15, there were only a small fraction of channels containing the tiny Ni particles, while the dominant large Ni particles were obviously aggregated on the external surface of SBA-15. This demonstrated that water could not sufficiently transport Ni species into the pore channels through conventional impregnation and suppress its redistribution and agglomeration during follow-up drying and calcination. Conversely, for the Ni@SBA-15 catalyst, the TEM observations confirmed that the Ni particles were anchored inside the channels of SBA-15, indicating that the introduction of EG during co-impregnation could confine the location of tiny Ni nanoparticles. Thus, the confinement effect of the channel walls were conducive to the uniform distribution and high dispersion by restricting the growth of the supported metal nanoparticles. The intrinsic characteristics of EG played a key role in the distribution and packaging of Ni nanoparticles, attributing to its high boiling point, viscosity and decomposition features.18,20,26,27
 | ||
| Fig. 5 TEM images of the reduced Ni-based catalysts: (A) Ni/SBA-15 and (B) Ni@SBA-15. | ||
As illustrated in Fig. 6, the reduction performance of various catalysts was measured by H2-TPR. Two samples obtained by different preparation methods exhibited quite different reduction characteristics. The Ni/SBA-15 sample prepared by conventional impregnation is characterized by a sharp main peak centred at 370 °C and a weak shoulder peak located at 460 °C, both being associated with the reduction of Ni2+ to Ni0. The former primary peak could be attributed to the reduction of bulk NiO, while the second small peak was assigned to the difficultly reducible Ni species, such as very small NiO nanoparticles outside or inside the SBA-15 channels, which were in strong interaction with the support.28,29 In comparison, the reduction profile of the sample prepared by co-impregnation was very broad, located in the range of 266 °C to 635 °C. The main reduction peak shifted to higher temperature and occurred at 515 °C, suggesting that the metal-support interaction was apparently strengthened due to the ultra-small particles embedded in the SBA-15 channels.
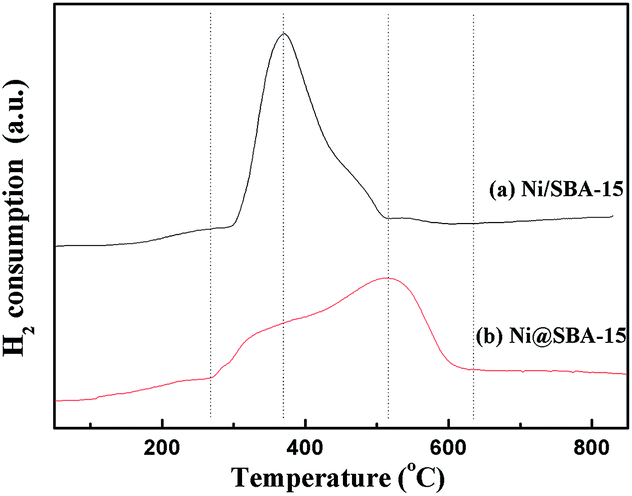 | ||
| Fig. 6 H2-TPR profiles of the Ni-based catalysts. | ||
Moreover, the oxidation states of Ni on Ni-based samples were further investigated by XPS analysis. Fig. 7 presents the XPS spectra of Ni 2p. By comparison of the binding energy of Ni 2p3/2 in the metallic Ni, there was no detection of Ni0 species present in the samples after calcination. The signal of the Ni 2p lines displayed a distinctly intense satellite structure. The difference in binding energies derived from the Ni 2p3/2 satellite separation suggested that no nickel silicate was formed over all the catalysts.19,30 From the XPS results, the Ni 2p3/2 region revealed an asymmetrical signal, indicating the existence of shoulder peaks. The maximum of two Ni 2p3/2 peaks were centred at 854.1 eV and 856.2 eV, respectively. For the Ni@SBA-15 sample, the maximum of this peak slightly shifted towards higher binding energy, corresponding to the strong metal-support interactions, consistent with the above H2-TPR results. Actually, this result can be essentially explained by the electronic interaction occurring between the metal and support, and very small NiO particles embedded in the SBA-15 channels would be more inclined to have charge transfer from NiO clusters to the support, contributing to the formation of the difficultly reducible NiO species and the strong metal-support interaction.31–33
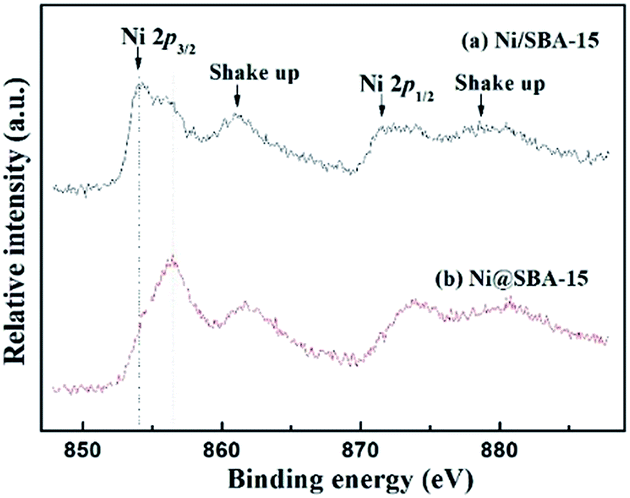 | ||
| Fig. 7 XPS spectra of the Ni 2p region for the calcined Ni-based catalysts. | ||
3.2. Catalytic activity and stability of the DRM reaction
To investigate the catalytic activity and stability, the SBA-15 supported Ni catalysts were applied in the DRM reaction. The reaction was carried out from 600 °C to 800 °C under atmospheric pressure. It is well known that the CO2 reforming of CH4 is a typical endothermic reaction. Therefore, as expected from thermodynamics, the conversions of CH4 and CO2 both increased with the increase of reaction temperature in Fig. 8. Apparently, the Ni@SBA-15 catalyst prepared by co-impregnation showed better catalytic activity by comparing that obtained by conventional impregnation. At 800 °C, for the Ni@SBA-15 catalyst, the CH4 conversion was increased to 92.9% and the CO2 conversion reached 93.2%. Moreover, the conversion of CO2 was appreciably higher than that of CH4, attributed to the occurrence of side reactions such as the reverse water gas shift and the reverse Boudouard reactions favored at high temperature.1–4 It was reported that the number of active sites determined the catalytic activity. Generally, the better metal dispersion of supported metal catalysts could bring out more active sites. Thus, the higher activity of the Ni@SBA-15 catalyst should be ascribed to smaller particle size and higher dispersion of Ni species located inside the SBA-15 channels.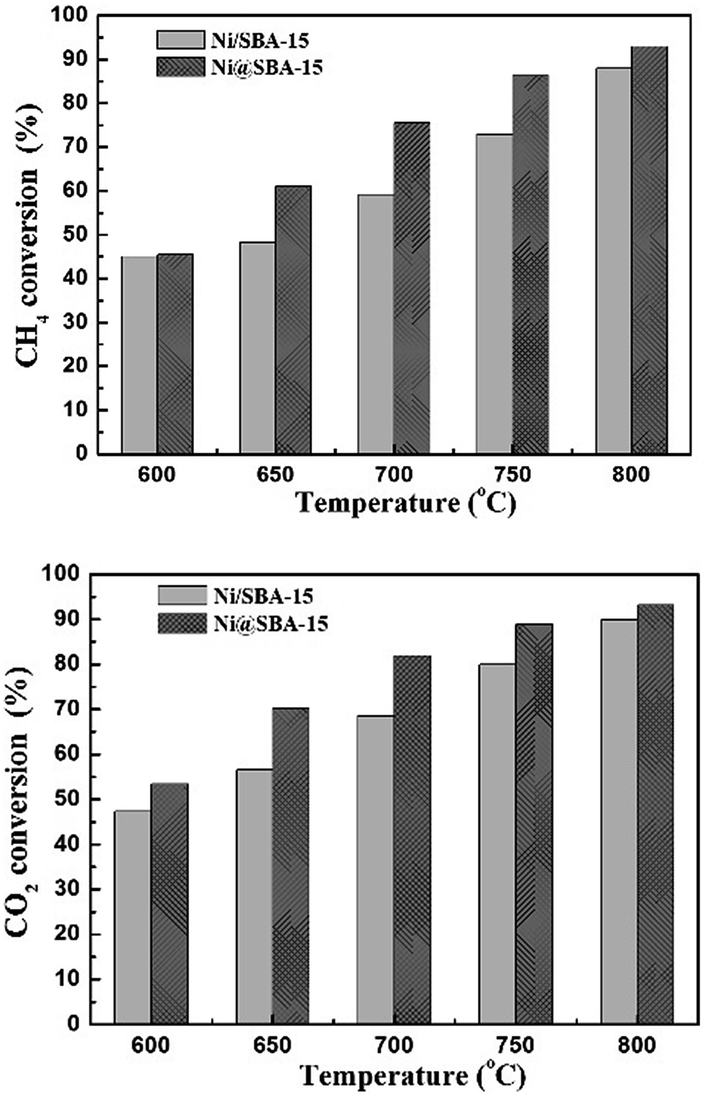 | ||
| Fig. 8 The conversions of CH4 and CO2 in the DRM reaction at different temperatures for the Ni-based catalysts; reaction conditions: P = 1 atm, T = 700 °C, CO2/CH4 = 1:1, GHSV = 36000 cm3 g−1 h−1. | ||
The long-term catalytic stability tests of the catalysts were carried out at 700 °C for 30 h. As reported in Fig. 9, in the case of the Ni/SBA-15 catalyst, the conversion of CH4 and CO2 both decreased monotonously with time on stream. Conversely, the Ni@SBA-15 catalyst expressed the prolonged lifetime in the harsh DRM reaction and the catalytic activity remained stable with no apparent deactivation observed.
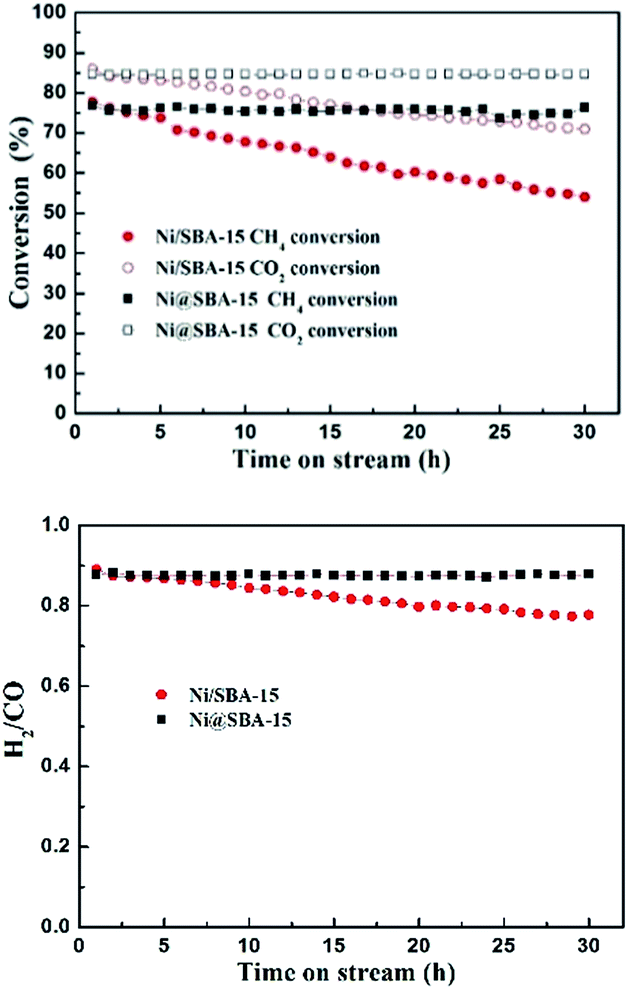 | ||
| Fig. 9 The catalytic stability of the Ni-based catalysts in the DRM reaction; reaction conditions: P = 1 atm, T = 700 °C, CO2/CH4 = 1:1, GHSV = 36000 cm3 g−1 h−1. | ||
In order to compare the impacts of confinement and dispersion on the catalytic performance, we prepared the highly dispersed Ni catalyst supported on nano-SiO2 without typical mesoporous structures. The characterization results (ESI SFig. 5, SFig. 6, SFig. 9 and STable 1†) showed that the samples of Ni@SBA-15 and Ni/nano-SiO2 AE displayed the approximate crystal sizes and dispersion of metallic Ni nanoparticles after reduction. In addition, the Ni/nano-SiO2 AE sample did not have a typical mesoporous structure and the Ni nanoparticles were highly dispersed on the external surface of the nano-SiO2 support. As depicted in ESI SFig. 7 and SFig. 8,† the Ni/nano-SiO2 AE catalyst expressed similar activity to that of the Ni@SBA-15 catalyst, and it showed better catalytic activity compared to the Ni/SBA-15 catalyst obtained by conventional impregnation. This is ascribed to smaller particle size and higher dispersion of Ni species on supports. During the long-term stability testing of the Ni/nano-SiO2 AE catalyst, the conversion of CH4 and CO2 both gradually decreased little by little with time on stream. Conversely, the Ni@SBA-15 catalyst remained stable with no apparent deactivation observed in the harsh DRM reaction.
Generally, the deactivation of Ni-based catalysts is caused by sintering of Ni species and carbon deposition. In order to explore the reason for deactivation, XRD profiles and TEM images of the used Ni-based catalysts, after a long-term stability test, were investigated in Fig. 10 and 11. Obvious diffraction peaks for the quartz crystalline phase were observed (JCPDS no. 47-1144), which were attributed to the addition of the quartz sand as the diluting agent during the DRM reaction. In the low angle XRD patterns, three well-resolved peaks indexed to a hexagonal lattice were distinctly detected. This means that the ordered mesoporous structure of SBA-15 was retained very well after 30 h of long-term reaction at 700 °C, which was further confirmed by the TEM observation. Particularly, it should be noted that the Ni nanoparticles in the Ni@SBA-15 catalyst still maintained the high dispersion even after the severe reaction. This outstanding resistance of Ni nanoparticles to sintering was attributed to the confinement effect of the SBA-15 pores with one given size scope limitation.
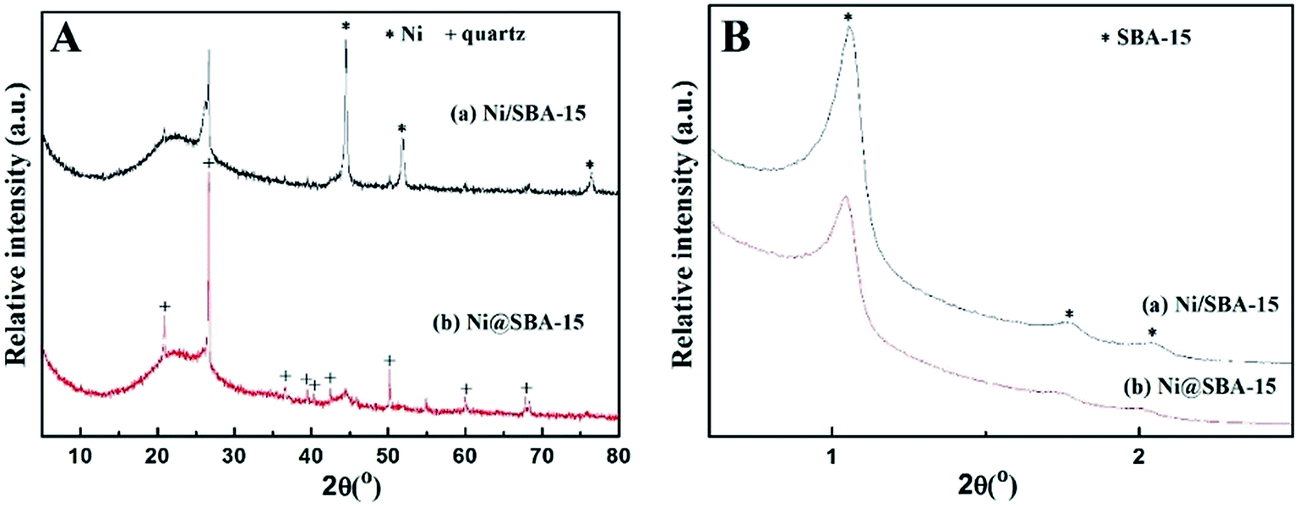 | ||
| Fig. 10 XRD patterns for the Ni-based catalysts after the stability test: (A) high-angle and (B) low-angle. | ||
 | ||
| Fig. 11 TEM images of the Ni-based catalysts after the stability test: (A) Ni/SBA-15 and (B) Ni@SBA-15. | ||
Moreover, one asymmetric diffraction peak appeared at an approximate 2θ value of 26°, indicating the doubtless existence of the shoulder peak ascribed to the graphite,34,35 especially for the used Ni/SBA-15 catalyst. Simultaneously, TEM images clearly displayed the formation of filamentous carbon over the two spent samples. From the results above, it was revealed that at least two different types of carbon, such as graphitic carbon and filamentous carbon (e.g. carbon nanotubes or nanofibers),15,16,19 were deposited on the catalyst surface. During the long-term DRM reaction, the amount of amorphous carbon derived from the CH4 decomposition and CO disproportionation reaction was easily deposited on the surface of large Ni nanoparticles, and was subsequently converted into inert graphite carbon and then gradually covered the active Ni species, which was mainly responsible for the activity loss of the Ni/SBA-15 catalyst. In addition, for both samples, it was verified that no filamentous carbon was observed inside the SBA-15 channels and the carbon deposition was predominantly formed over the edge of the channels or outside the pores. Therefore, the confinement effect essentially hindered the growth of filamentous carbon inside the SBA-15 pores. As a result, the filamentous carbon deposited on the surface of the Ni@SBA-15 catalyst had no serious influence on the diffusion, transport and adsorption of the reactants within the mesopores. Consequently, there was no deactivation. The carbon deposition over the two used catalysts was also quantified by thermogravimetric (TG) and differential scanning calorimetry (DSC) measurements, as depicted in Fig. 12. The weight losses for the used Ni/SBA-15 and Ni@SBA-15 catalysts at high temperatures (500–750 °C) were 15.8% and 10.4%, respectively; the Ni@SBA-15 catalyst realized less carbon deposition.
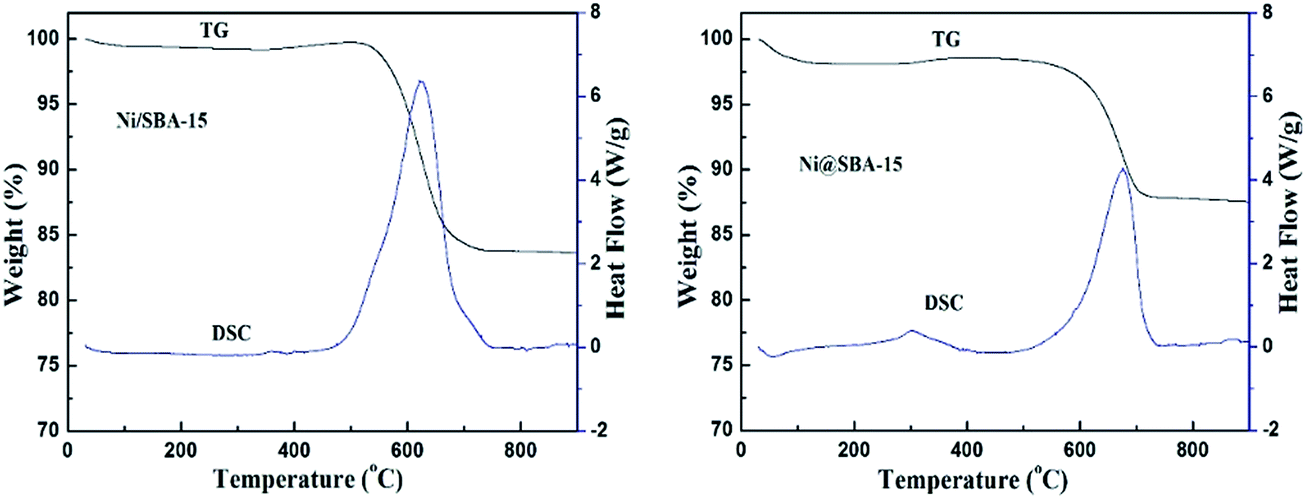 | ||
| Fig. 12 TG and DSC profiles of Ni/SBA-15 and Ni@SBA-15 catalysts after the stability test. | ||
In order to explore the reason for deactivation of the Ni/nano-SiO2 AE catalyst, XRD profiles, TG and DSC profiles and TEM images after a long-term stability test were also investigated, shown in the ESI, SFig. 6, SFig. 9 and SFig. 10.† For the used Ni/nano-SiO2 AE catalyst, the average diameter of Ni nanoparticles increased to 8.6 nm from 4.8 nm, according to calculation by the Scherrer formula, which was further confirmed by the TEM images. Due to the absence of the confinement effect of mesoporous channels and strong metal-support interactions, the highly dispersed Ni/nano-SiO2 AE catalyst still suffered from serious sintering during the harsh DRM reaction. The weight loss of the used Ni/nano-SiO2 AE catalyst at high temperatures (500–750 °C) was 2.6%. The Ni/nano-SiO2 AE catalyst presented the least carbon deposition, which was consistent with the TEM observation. This could be explained by the fact that the small Ni nanoparticles with high dispersion facilitated the improved resistance to coking.
The small Ni nanoparticles stabilized within the SBA-15 porous network were characteristic of higher reactivity than large Ni nanoparticles, and easily activated the coke precursor and eliminated amorphous carbon by the weak oxidizing reagent CO2;15,36–38 consequently, the Ni active sites still remained accessible to the reactants, resulting in not only higher activity but also stability in the DRM reaction. In brief, during the DRM reaction, the rapid deactivation of the Ni/SBA-15 catalyst was mainly attributed to the excessive amount of carbon formation covering the Ni active sites located on the external surface of SBA-15. Sintering of Ni species was mainly responsible for the activity loss of the Ni/nano-SiO2 AE catalyst. In contrast, for the Ni@SBA-15 catalyst, the carbon deposition dominantly took shape outside the mesoporous channels and this could prevent coverage of the Ni active sites by inert deposited carbon. Therefore, the confinement effect of SBA-15 channels had a decisive effect on preventing the aggregation of small Ni nanoparticles, and the stabilized small Ni nanoparticles with strong metal-support interactions facilitated the improved resistance of coking and sintering so that it revealed outstanding performance in the catalytic activity and long-time stability of DRM.
4. Conclusion
In this work, a simple and efficient preparation method of co-impregnation was successfully applied to preparing the Ni@SBA-15 catalyst, which presented higher activity and excellent stability in the dry reforming of methane. Compared to conventional impregnation, the characterization results confirmed that the EG addition during co-impregnation could smoothly transfer and package the nickel species into the SBA-15 channels, and then lead to the homogeneous dispersion of small Ni nanoparticles anchored over the internal surface of mesoporous channels, instead of the disorderly and non-uniform dispersion of large Ni nanoparticles supported over its external surface. During drying, the lower surface tension and high viscosity of the concentrated impregnation solution played a critical role in the distribution of the precursor. Essentially, the confinement effect of mesoporous channels was intensely conducive to inhibiting the sintering of small Ni nanoparticles and preventing the formation of inert carbon deposition, so that the expected catalysts revealed outstanding long-term stability in the severe dry reforming reaction. Therefore, the Ni@SBA-15 catalyst prepared by simple co-impregnation was shown to have potential for commercial application in the dry reforming of methane with CO2 to produce syngas and hydrogen.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (No. 21306195 & 51476176).References
- S. R. Li and J. L. Gong, Chem. Soc. Rev., 2014, 43, 7245 RSC.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813 RSC.
- X. E. Verykios, Int. J. Hydrogen Energy, 2003, 28, 1045 CAS.
- M. Usman and W. M. A. W. Daud, RSC Adv., 2015, 5, 21945 RSC.
- Q. X. Ma, J. Sun, X. H. Gao, J. L. Zhang, T. S. Zhao, Y. Yoneyama and N. Tsubaki, Catal. Sci. Technol., 2016, 6, 6542 CAS.
- H. Tian, X. Y. Li, L. Zeng and J. L. Gong, ACS Catal., 2015, 5, 4959 CrossRef CAS.
- M. M. Nair and S. Kaliaguine, New J. Chem., 2016, 40, 4049 RSC.
- S. Kawi, Y. Kathiraser, J. Ni, U. Oemar, Z. W. Li and E. T. Saw, ChemSusChem, 2015, 8, 3556 CrossRef CAS PubMed.
- S. Arora and R. Prasad, RSC Adv., 2016, 6, 108668 RSC.
- X. Y. Lv, J. F. Chen, Y. S. Tan and Y. Zhang, Catal. Commun., 2012, 20, 6 CrossRef CAS.
- Z. Y. Liu, D. C. Grinter, P. G. Lustemberg, T. D. Nguyen-Phan, Y. H. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola and J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7455 CrossRef CAS PubMed.
- D. P. Liu, X. Y. Quek, W. N. E. Cheo, R. Lau, A. Borgna and Y. H. Yang, J. Catal., 2009, 266, 380 CrossRef CAS.
- P. Maki-Arvela and D. Y. Murzin, Appl. Catal., A, 2012, 451, 251 CrossRef.
- B. W. Lu and K. Kawamoto, RSC Adv., 2012, 2, 6800 RSC.
- N. Wang, W. Chu, T. Zhang and X. S. Zhao, Int. J. Hydrogen Energy, 2012, 37, 19 CrossRef CAS.
- T. Xie, L. Y. Shi, J. P. Zhang and D. S. Zhang, Chem. Commun., 2014, 50, 7250 RSC.
- S. B. Qiu, X. Zhang, Q. Y. Liu, T. J. Wang, Q. Zhang and L. L. Ma, Catal. Commun., 2013, 42, 73 CrossRef CAS.
- A. J. Van Dillen, R. M. Terörde, D. J. Lensveld, J. W. Geus and K. P. de Jong, J. Catal., 2003, 216, 257 CrossRef CAS.
- Q. L. Zhang, M. Z. Wang, T. F. Zhang, Y. R. Wang, X. S. Tang and P. Ning, RSC Adv., 2015, 5, 94016 RSC.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687 CrossRef CAS PubMed.
- J. R. A. Sietsma, J. D. Meeldijk, M. Versluijs-Helder, A. Broersma, J. A. van Dillen, P. E. de Jongh and K. P. de Jong, Chem. Mater., 2008, 20, 2921 CrossRef CAS.
- S. Velu and S. K. Gangwal, Solid State Ionics, 2006, 177, 803 CrossRef CAS.
- A. Ungureanu, B. Dragoi, A. Chirieac, S. Royer, D. Duprez and E. Dumitriu, J. Mater. Chem., 2011, 21, 12529 RSC.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527 CrossRef CAS.
- Y. Q. Chen, L. X. Yuan, T. Q. Ye, S. B. Qiu, X. F. Zhu, Y. Torimoto, M. Yamamoto and Q. X. Li, Int. J. Hydrogen Energy, 2009, 34, 1760 CrossRef CAS.
- M. Wolters, L. J. W. van Grotel, T. M. Eggenhuisen, J. R. A. Sietsma, K. P. de Jong and P. E. de Jongh, Catal. Today, 2011, 163, 27 CrossRef CAS.
- D. Valencia, L. Pena, V. H. Uc and I. Garcia-Cruz, Appl. Catal., A, 2014, 475, 134 CrossRef CAS.
- T. Hou, L. X. Yuan, T. Q. Ye, L. Gong, J. Tu, M. Yamamoto, Y. Torimoto and Q. X. Li, Int. J. Hydrogen Energy, 2009, 34, 9095 CrossRef CAS.
- R. Q. Yang, C. A. Xing, C. X. Lv, L. Shi and N. Tsubaki, Appl. Catal., A, 2010, 385, 92 CrossRef CAS.
- H. P. Ren, Q. Q. Hao, W. Wang, Y. H. Song, J. Cheng, Z. W. Liu, Z. T. Liu, J. Lu and Z. P. Hao, Int. J. Hydrogen Energy, 2014, 39, 11592 CrossRef CAS.
- S. R. Kirumakki, B. G. Shpeizer, G. V. Sagar, K. V. R. Chary and A. Clearfield, J. Catal., 2006, 242, 319 CrossRef CAS.
- K. G. Fang, J. Ren and Y. H. Sun, J. Mol. Catal. A: Chem., 2005, 229, 51 CrossRef CAS.
- A. Y. Stakheev and L. M. Kustov, Appl. Catal., A, 1999, 188, 3 CrossRef CAS.
- Q. X. Ma, D. Wang, M. B. Wu, T. S. Zhao, Y. Yoneyama and N. Tsubaki, Fuel, 2016, 108, 430 CrossRef.
- A. Venugopal, S. N. Kumar, J. Ashok, D. H. Prasad, V. D. Kumari, K. B. S. Prasad and M. Subrahmanyam, Int. J. Hydrogen Energy, 2007, 32, 1782 CrossRef CAS.
- D. Baudouin, U. Rodemerck, F. Krumeich, A. de Mallmann, K. C. Szeto, H. Menard, L. Veyre, J. P. Candy, P. B. Webb and C. Thieuleux, J. Catal., 2013, 297, 27 CrossRef CAS.
- N. Wang, X. P. Yu, K. Shen, W. Chu and W. Z. Qian, Int. J. Hydrogen Energy, 2013, 38, 9718 CrossRef CAS.
- T. Huang, W. Huang, J. Huang and P. Ji, Fuel Process. Technol., 2011, 92, 1868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00149e |
| This journal is © The Royal Society of Chemistry 2017 |