
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of the in vitro effects of gallic acid-grafted-chitooligosaccharides in the suppression of AGS human gastric cancer cell proliferation
BoMi Ryu†
 a,
So-Yeon Kim†b,
Thanh-Sang Voc,
Won-Suk Kimd,
Dong Gyu Kim*ef and
Se-Kwon Kim*be
a,
So-Yeon Kim†b,
Thanh-Sang Voc,
Won-Suk Kimd,
Dong Gyu Kim*ef and
Se-Kwon Kim*be
aDepartment of Marine Life Science, Jeju National University, Jeju, Republic of Korea
bMarine Bioprocess Research Center, Pukyong National University, Busan 608-739, Republic of Korea. E-mail: sknkim@pknu.ac.kr; Fax: +82-51-629-6865; Tel: +82-51-629-6870
cNTT Institute of Hi-Technology, Nguyen Tat Thanh University, Ho Chi Minh City, Vietnam
dMajor in Pharaceutical Engineering Division of Bio-Industry, Silla University, Busan, Korea
eSpecialized Graduate School Science and Technology Convergence, Department of Marine Bio Convergence Science, Pukyong National University, Busan 608-737, Republic of Korea. E-mail: dgkim@pknu.ac.kr
fDepartment of Mechanical & Shipbuilding Convergence Engineering, College of Lifelong Education, Pukyong National University, Republic of Korea
First published on 5th May 2017
Abstract
Gastric cancer is the second most common cause of cancer-related deaths in the world. In this study, a bioactive derivative of chitooligosaccharide, named gallic acid-grafted-chitooligosaccharide (G-COS), was evaluated for its capabilities against the proliferation of AGS human gastric cancer cells. It was found that G-COS treatment caused significant inhibition of gastric cancer cell growth at concentrations of 200 and 400 μg mL−1. The anti-growth effect of G-COS in AGS cells was characterized using flow cytometry, fluorescence microscopy, DNA fragmentation and evaluation of protein expression. Notably, G-COS-induced apoptosis was related to the increase in the expression of p53, p21, Bax, and caspases (-9 and -3) and the decrease in the activation of Bcl-2, p-IκB-α and NF-κB (p50 and p65). These findings indicate that G-COS has potential to be applied in the treatment of gastric cancer as a cancer chemopreventative agent.
Introduction
Gastric cancer is the second leading cause of cancer-related death as one of the most common malignancies with poor prognosis.1,2 The geographic and ethnic differences in gastric cancer incidence and mortality suggest that gastric cancer is closely related to diet such as ingestion of salt-preserved food, nitrates, and smoked or pickled food.2 Despite the advanced targeted therapy and improved understanding of the biology and development of the malignancy, progress in the treatment of gastric cancer has been limited.3 The most efficacious remedy for gastric cancer is surgery, however many patients have inoperable cancers. Therefore, chemotherapy regimens have remained an integral part of primary treatment until now.4Most cancer cells block apoptosis via anti-apoptotic signalling pathways in order to survive despite undergoing genetic and morphologic transformations. Therefore, drugs promoting apoptosis may be effective against many cancers and are an important strategy to counteract cancer.5 Apoptotic cells are characterized by certain morphological features, including condensation of the cytoplasm and nucleus, cell surface expression of phosphatidylserine, and internucleosomal cleavage of DNA.6 Further, the translocation of phosphatidylserine to the exterior surfaces of the plasma membrane is a distinguishing feature of early apoptosis.7 The tumor suppressor gene p53 whose product can act as a suppressor of transformation inhibits cell growth through activation of cell cycle arrest and apoptosis in more than half of human cancers.8 The important component of apoptosis occurs primarily through a p53-dependent pathway and involves its target proteins of the Bcl-2 family (i.e., Bax, Bcl-2) and activation of caspase family.
Phenolic acids, especially hydroxycinnamic acids such as gallic acid, caffeic, and sinapinic acids, and benzoic acids derivatives such as p-hydroxybenzoic, protocatechuic, vanillic and syringic acids are natural plant hydrophilic antioxidants.9 The antioxidant activity of these compounds is due to the ability to quench the free radicals, terminate the chain reactions in lipid oxidation and chelate metal ions.9,10 Hydroxyl groups, present on their structure are believed to be associated with their antioxidant activity. However, those derivatives metabolize in the body within short time and exhibit relatively low solubility in aprotic media. The hydrophilic nature of these compounds reduces their effectiveness in stabilizing lipophilic systems, such as fats and oil and become a serious disadvantage. The hydrophilicity of phenolic acids can be reduced up to some extent by reacting them with slow digestible compounds such as chitooligosaccharides (COSs) to enhance their slow releasing ability while increasing the antioxidant effect of COSs.6,11–13 A COS conjugated with gallic acid (G-COS) showed no significance effect in viability up to 200 μg mL−1 in macrophage and mast cells,14,15 and a substitute of G-COS, gallic acid showed toxicity in AGS cells at IC50 of 170.12 μg mL−1.16
The objective of this study was to characterize the anti-tumor activity of G-COS and the related mechanistic pathway following treatment with G-COS in AGS gastric cancer cells.
Results and discussion
Structural characterization of G-COS
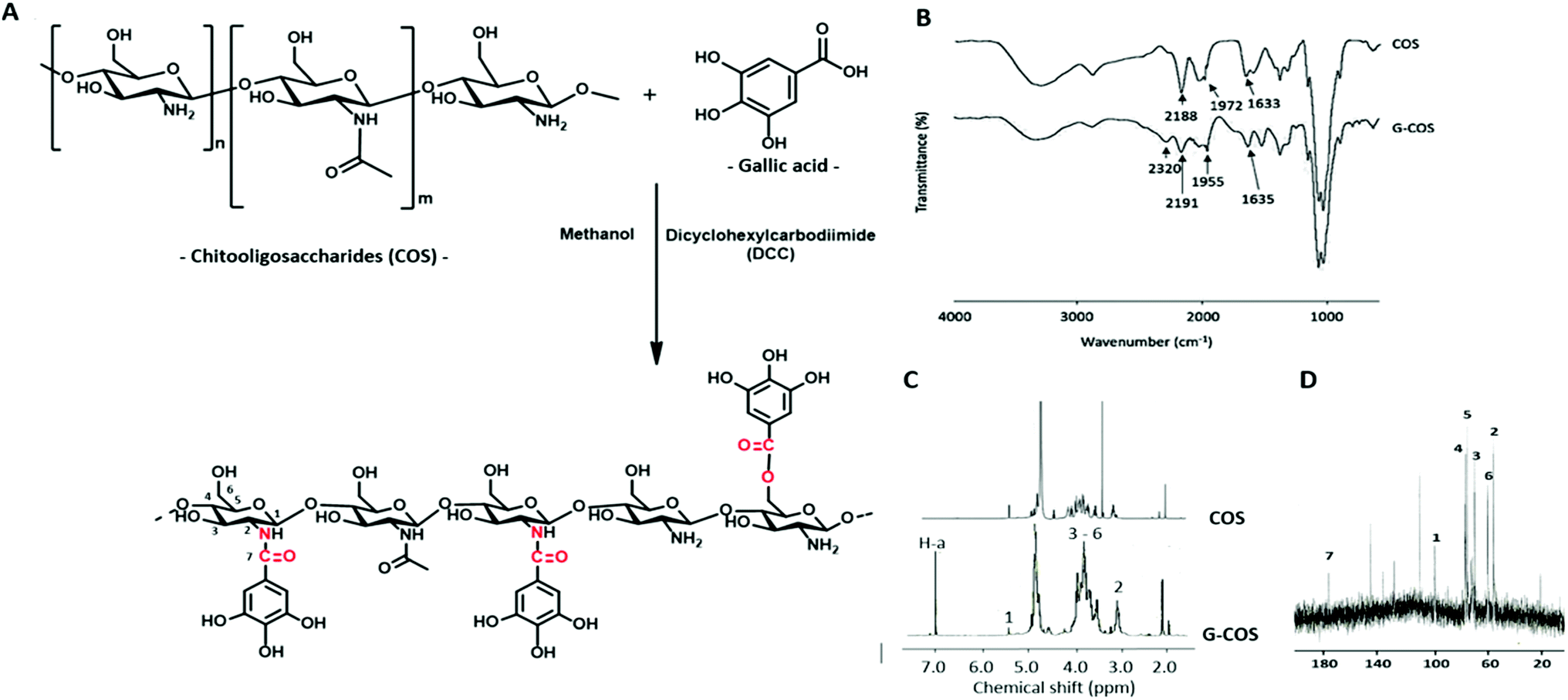
In the synthesis mechanism of G-COS (Fig. 1A), dicyclohexyl carbodiimide (DCC) is used as a dehydration agent and for retaining of the amine group, and initiated the reaction of carboxyl groups with gallic acid. The reaction includes formation of the intermediate active ester that further reacts with the amine function to yield the amide bond. The FT-IR spectra of G-COS and COS are shown in Fig. 1B to identify their molecular interactions. Several typical peaks of amide bonds were observed in the COS spectrum, i.e., the absorbance bands at 2188, 1972, and 1633 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (amide I), N–H bending (amide II), and C–N stretching (amide III), respectively.17 In comparison, G-COS, G-COS showed the characteristic peaks at 2320 cm−1 (ester) or 1955 cm−1 (amide), suggesting the nucleophilic reaction of covalent linkage of gallic acid with COS occurs at either C-6 (via the formation of ester linkage) or C-2 (via the formation of amide linkage).18 In addition, a new peak at 1731 cm−1 was observed in G-COS due to the CC stretching in esters, representing the formation of ester bond between the carboxyl groups of gallic acid and hydroxyl groups of COS.
O stretching (amide I), N–H bending (amide II), and C–N stretching (amide III), respectively.17 In comparison, G-COS, G-COS showed the characteristic peaks at 2320 cm−1 (ester) or 1955 cm−1 (amide), suggesting the nucleophilic reaction of covalent linkage of gallic acid with COS occurs at either C-6 (via the formation of ester linkage) or C-2 (via the formation of amide linkage).18 In addition, a new peak at 1731 cm−1 was observed in G-COS due to the CC stretching in esters, representing the formation of ester bond between the carboxyl groups of gallic acid and hydroxyl groups of COS.
 | ||
| Fig. 1 Synthetic pathway of G-COS (A). FT-IR spectra (B) and 1H NMR (C) for COS and G-COS and 13C NMR (D) for G-COS. | ||
Further characterization of GCOS was performed by 1H NMR. As shown in Fig. 1C, gallic acid showed only one peak at 7.1 ppm (H-a of benzene ring). Chemical shift at 3.1 ppm (quartet, H-2), 3.3–4.1 ppm (multiplet, H-3 to H-6), 5.3 ppm (doublet, H-1) and a single peak at 2.0 ppm were representing the protons on amino and the hydroxyl groups of COS.11 In detail, splitting of H-1 is observed as doublet with a weak coupling constant (J = 0.14 Hz) due to the spin–spin coupling of H-2. And, doublet of doublet observed for H-2 is due to spin–spin coupling of nearby H-1 and H-3 which are at different chemical environment. The spectrum of G-COS contained all the peaks of COS, and a new peak at around 7.1 ppm originated from gallic acid, confirming the successful grafting of gallic acid onto COS. This result corresponded to 13C NMR spectra (Fig. 1D) that the aromatic carbon of gallyl group for G-COS observed at 109.2, 127.8, 136.7, and 145.2 ppm (CC) and 173.6 ppm (CO).
Evaluation of the antitumor activity of G-COS using cytotoxicity assays
We evaluated the anti-growth effect of G-COS in comparison with COS on AGS human gastric adenocarcinoma cells. In order to evaluate the effect of G-COS and COS on AGS cells, cells were treated with the different concentrations of G-COS or COS for 24 h and viability was then measured by MTT assay (Fig. 2A). G-COS showed significantly higher growth inhibitory effects in a dose-dependent manner (p < 0.001), compared to the COS. In addition, the fold change of cell viability upon treatment with 200 and 400 μg mL−1 of G-COS were 0.68 and 0.43 compared to the blank, respectively. Direct observation by inverted microscopy revealed that numerous morphological changes occurred in cells treated with G-COS or COS (Fig. 2B, upper panel). Additional experiments were conducted to determine if the inhibitory effect of G-COS on cell growth was the result of apoptotic cell death. Hoechst staining (Fig. 2, lower panel) revealed that nuclei with chromatin condensation and apoptotic bodies with significant loss of cell volume and widespread membrane bleeding were formed in cells that were cultured with G-COS (as indicated by yellow arrows in Fig. 2), and that this phenomenon occurred in a concentration-dependent manner; however, these features were present in only a few cells in the 400 μg mL−1 treatment of COS (Fig. 2B, bottom panel).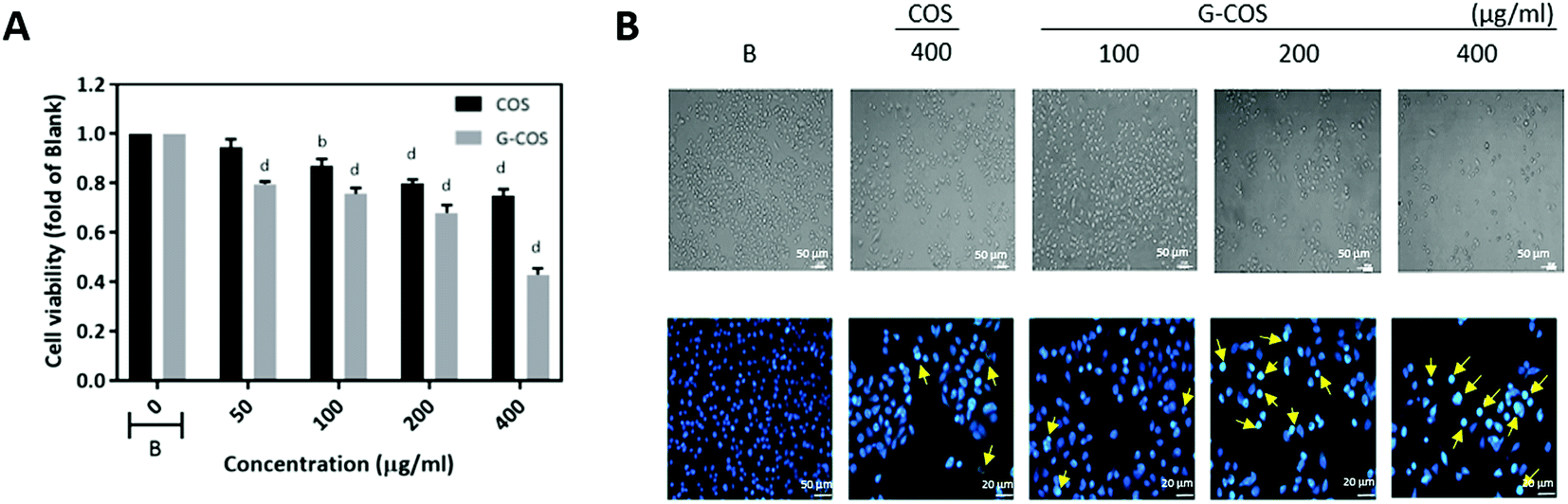 | ||
| Fig. 2 Effect of G-COS and COS on the viability of AGS cells. AGS cells were incubated with the indicated concentrations of G-COS and COS for 24 h. Cell viability was determined by an MTT assay (A). Morphological changes were determined by phase contrast microscopy (viewed at magnification of ×100) (upper panel) or fixed and stained with Hoechst 33258 (lower panel) (B). Arrows indicate apoptotic cells. The data are shown as means ± SD of three independent experiments. ap < 0.05, bp < 0.01, cp < 0.005, dp < 0.001 compared with the Blank (B). | ||
Despite the cytotoxic effect of gallic acid and COS in various cancer cells,16,19,20 its conjugations with COS (G-COS) did not show any significant effect on the viability against leukemia mast cell or macrophages cells in tested concentrations (up to 200 μg mL−1).14,15 However, interestingly, we observed that G-COS treatment induced strong anti-growth effect in AGS cells compared to COS.
Quantification of apoptotic cell death using flow cytometry
The agarose gel electrophoresis showed that G-COS treatment induced the progressive accumulation of fragmented DNA, which appeared as a typical ladder pattern of DNA fragmentation due to internucleosomal cleavage associated with apoptosis, and that this accumulation occurred in a concentration-dependent manner (Fig. 3A).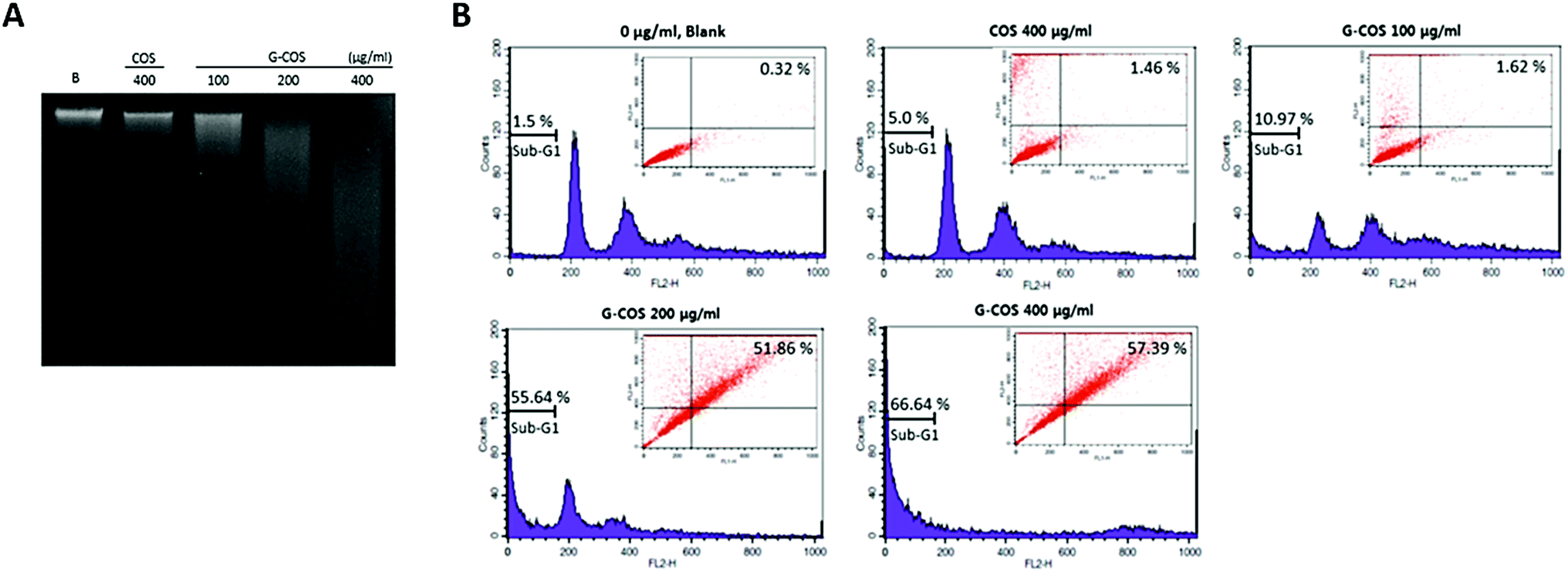 | ||
| Fig. 3 Induction of apoptosis by G-COS and COS treatment of AGS cells. Total DNA was extracted from the AGS cells and the DNA fragmentation assay was analysed (A). Sub-G1 DNA content and apoptotic AGS cells were measured by flow cytometry analysis. The percentages of gates (apoptosis) are represented in each panel (B). The data are shown as means ± SD of three independent experiments. | ||
Apoptosis and cell cycle dysfunction are closely associated biochemical processes and any disturbance in cell cycle progression may lead to apoptotic cell death.21 The degree of apoptosis was determined by analyzing the amount of sub-G1 DNA and apoptotic cells in AGS cells that were treated with G-COS or COS using flow cytometry. As shown in Fig. 3B, the treatment of AGS cells with G-COS resulted in an increased accumulation of cells in the sub-G1 phase at concentrations of 200 and 400 μg mL−1 to 55.64% and 66.64%, respectively. Also, fragmented and damaged DNA was detected by annexin V-FITC and in the cells with 200 and 400 μg mL−1 of G-COS, a 51.86 and 57.39% of annexin V-positive cells were detected, however, COS treatment showed only 1.46% (inner panel of Fig. 3B). This result suggest that cells can undergo early apoptosis after being exposed to G-COS.
Effects of G-COS on the levels of genes associated with apoptosis
To investigate the apoptotic pathways activated by G-COS, we used Western blotting to measure the expression of the tumor suppressor p53 and its downstream p21 and pro-apoptotic proteins like Bcl-2 family members (Bax/Bcl-2) as well as the expression of the caspases (caspase-9 and caspase-3).Tumor suppressor genes p53 is involved in the control of apoptosis, cell cycle and maintenance of genomic stability to repair the damaged DNA, in relation with regulation of downstream effectors p21.22 We monitored the response of p53 and its downstream p21 in AGS cells in the presence G-COS to understand whether G-COS has any effect on the expression of this apoptosis related signaling pathway in AGS cells. Result in Fig. 4A showed that upon G-COS treatment (100, 200 and 400 μg mL−1), p53 was significantly increased (p < 0.001) while COS (400 μg mL−1) although showed a slight increase in p53, did not significantly increase its levels. Meanwhile, its downstream activator p21 showed a significant expression at 400 μg mL−1 of G-COS treatment, whereas COS did not affect the expression of p21 compared to the blank.
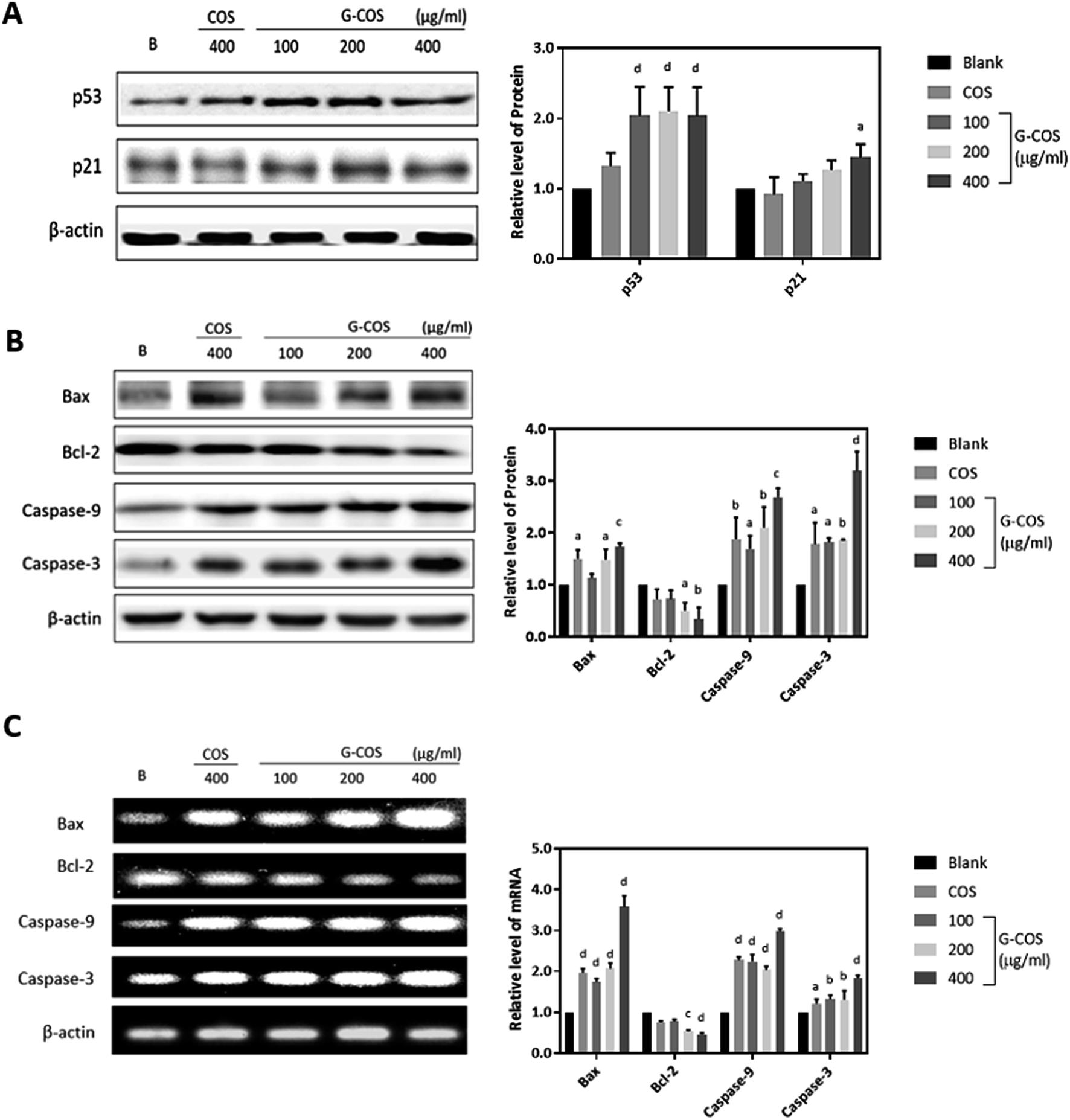 | ||
| Fig. 4 The effect of G-COS and COS on the levels of the apoptosis related genes. After incubation with G-COS and COS for 24 h, the protein level for p53 and 21 (A) and Bax, Bcl-2, caspase 9 and caspase 3 (B, upper panel) were analysed by Western blot. mRNA levels for Bax, Bcl-2, caspase 9 and caspase 3 were analysed by RT-PCR (C, bottom panel). 400 μg mL−1 of COS were used to compare with G-COS in this experiments. Quantitative results of the band intensity were estimated using AlphaEase® gel image analysis software program and normalized to the β-actin group. Similar results were obtained from three independent experiments, and representative blots or gels are shown. ap < 0.05, bp < 0.01, cp < 0.005, dp < 0.001 compared with the Blank. | ||
We further examined the effect of G-COS on the apoptosis related proteins. As shown in Fig. 4B, the protein levels of both Bax and Bcl-2 were significantly altered at 200 and 400 μg mL−1 of G-COS treatment whereas COS treatment resulted in a significant difference on only Bax (p < 0.05) and not Bcl-2. This change at protein level corresponded to the result from mRNA level change (Fig. 4C). Furthermore, addition of G-COS resulted in an increase in caspase-9, and -3 at both protein and mRNA levels compared to the blank (Fig. 4B and C). The pro-apoptotic protein Bax and anti-apoptotic gene Bcl-2 which is related to p53 dependent apoptosis then causes proteolytic activation of the caspases, leading to the characteristic morphological and biochemical features of apoptosis.23
Our results suggested that G-COS treatment led to the activation of caspases-9 and -3 and resulted in DNA fragmentation of AGS cells, as well as in a significant increase in the expression of pro-apoptotic proteins Bax and p21 and tumor suppressor p53.
Effects of G-COS on the levels of NF-κB dependent responsive genes
NF-κB is a transcription factor that can modulate the apoptotic response, known an unexpected pro-apoptotic activity of NF-κB in the p53-induced apoptotic response.24,25 Following stimulation with carcinogens, IκB is phosphorylated and degraded, and free NF-κB (p50 and p65) is translocated into the nucleus.13,26Treatment with G-COS substantially reduced the protein level of phospho-IκB-α (p-IκB-α) at 200 and 400 μg mL−1 compared to the blank, meanwhile COS of 400 μg mL−1 exhibited similar expression of p-IκB-α with 200 μg mL−1 of G-COS (p < 0.05, Fig. 5). Also, the increased expression of p50 and p65 at protein levels were both decreased follows by addition of G-COS and COS. The result suggests that both G-COS and COS has modulated NF-κB signalling pathway in AGS cells.
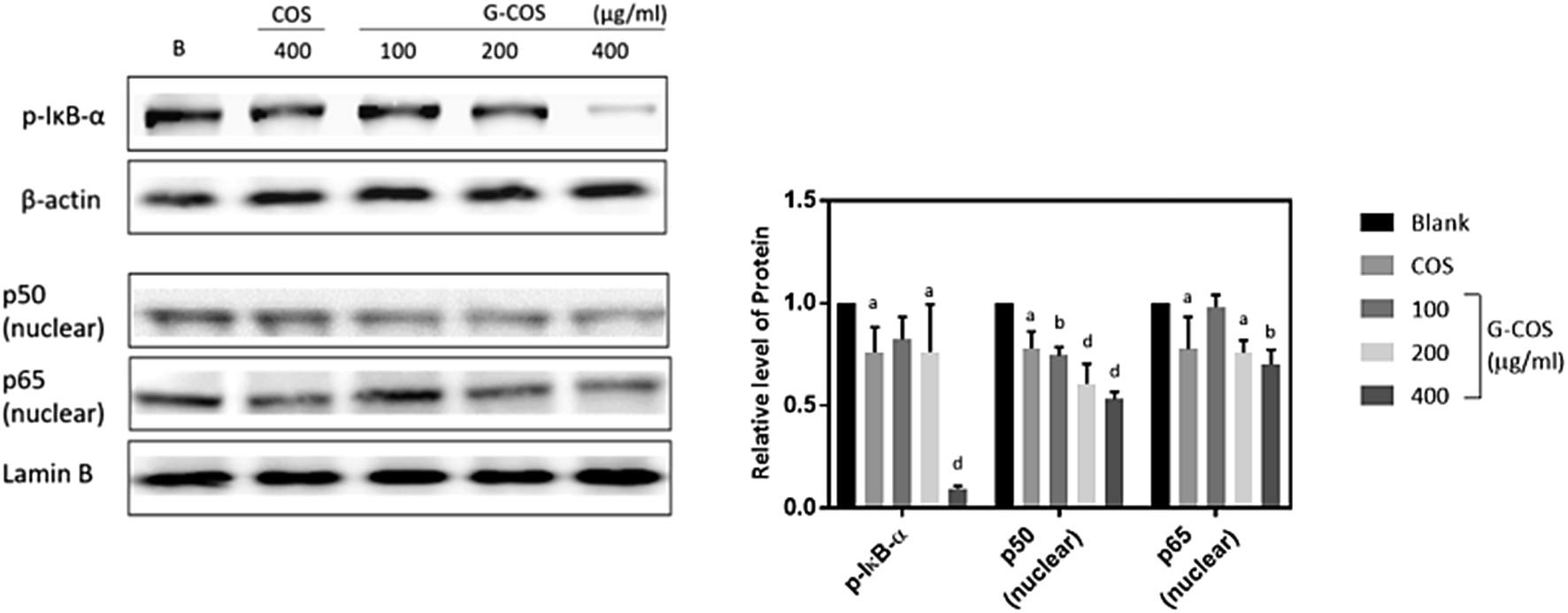 | ||
| Fig. 5 The effect of G-COS and COS on NF-κB activation. Whole and nuclear cell extract from AGS with cells were analysed by Western blot. 400 μg mL−1 of COS were used to compare with G-COS in this experiments. Quantitative results of the band intensity were estimated using AlphaEase® gel image analysis software program. β-Actin and lamin B were used as loading controls for the whole cell and nuclear fractions, respectively. Similar results were obtained from three independent experiments, and representative blots are shown. ap < 0.05, bp < 0.01, cp < 0.005, dp < 0.001 compared with the Blank. | ||
Conclusion
This study identified that conjugation of gallic acid with COS (G-COS), enhances the apoptosis properties of COS in human gastric cancer AGS cells. Inhibition of growth and progression via apoptosis were successfully induced in AGS cells by treatment with G-COS. The possible mechanisms of action and involved metabolic pathways were characterised in Fig. 6.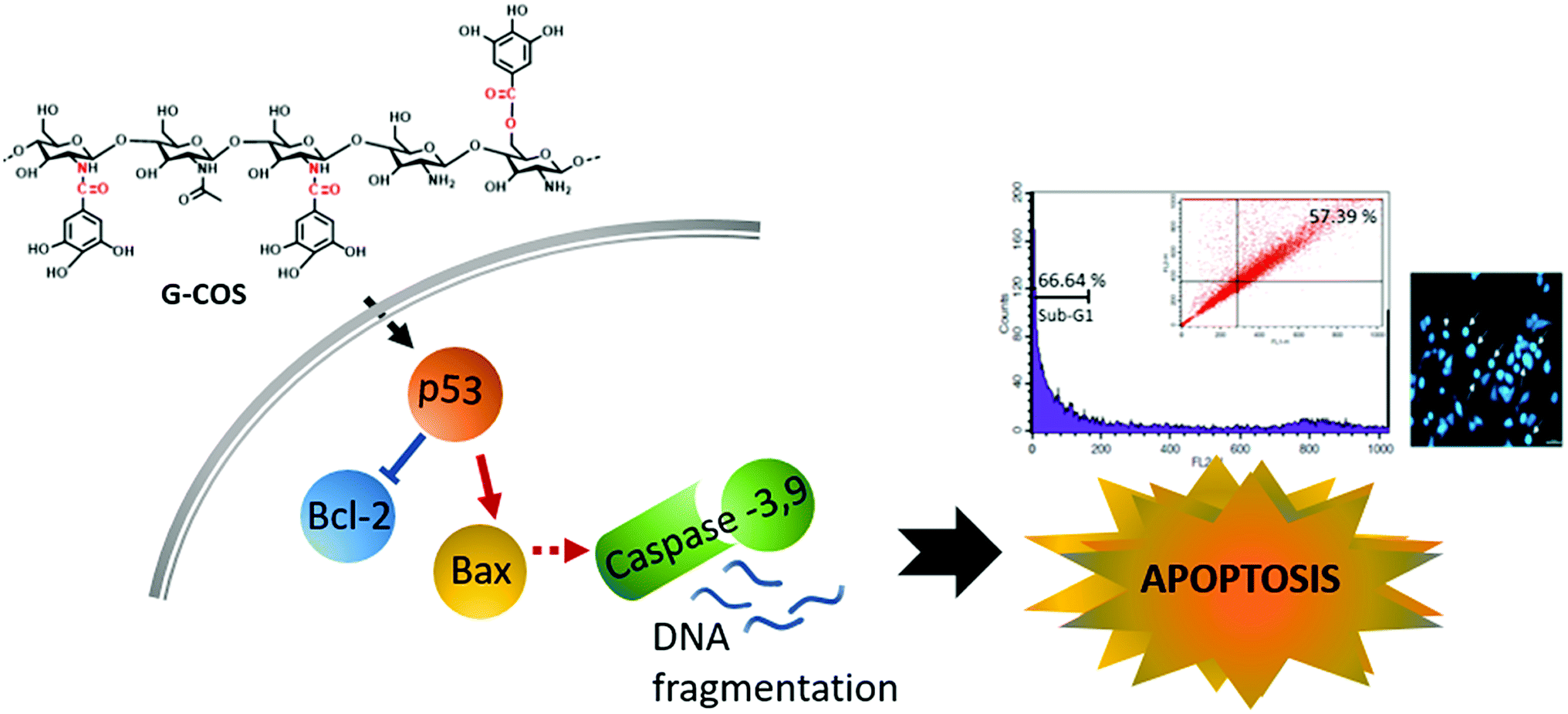 | ||
| Fig. 6 The proposed signaling pathways of G-COS-induced apoptosis of AGS cells. | ||
G-COS treatment was resulted in increased expression of p53 level as follows: the expression of Bax increased whereas the expression of Bcl-2 decreased at the protein level, and reduced the protein levels of NF-κB family (p-IκB-α, p65/p50) associated with apoptosis mechanistic pathway. These results contribute to the understanding of the anti-cancer activity of G-COS by proposing the role of G-COS in regulating the balance between apoptotic factors, and provide insight for G-COS as a potential chemotherapeutic agent for gastric cancer.
Materials and methods
Material
Chitooligosaccharides (3–5.0 kDa) was provided by Kitto Life Co. (Seoul, Korea). Gallic acid (98%), triethylamine (99%), and dicyclohexylcarbodiimide (99%) were purchased from the Acros Organics (New Jersey, USA). Human gastric adenocarcinoma (AGS) cells were obtained by American Type Culture Collection (ATCC, Manassas, VA, USA). Cell culture media (DMEM), Trypsin–EDTA, penicillin/streptomycin were purchased from Lonza Co. (Walkersville, MD, USA). Dimethyl sulfoxide (DMSO), 3-(4,5-dimethyl-2yl)-2,5-diphenyltetrazolium bromide (MTT) and paraformaldehyde, PBS, bisbenzimide Hoechst 33342, propidium iodide (PI) and RIPA buffer were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Annexin V-FITC apoptosis detection kit was purchased from Becton Dickinson (San Diego, CA). Primary and secondary antibodies used for Western blot analysis were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). All other chemicals and reagents used in this study were of analytical grade.Preparation of gallic acid-grafted-chitooligosaccharides (G-COS)
The gallic acid-grafted-chitooligosaccharides (G-COS) were obtained as per a procedure described in the literature with some modifications.14 Briefly, chitooligosaccharides (COS, 2.5 g) with the degree of polymerization in the range of 3–5, were suspended in 60 mL of 66.7% methanol (v/v) and pH adjusted to 6.8 with triethylamine. The resulting slurry was stirred in a 500 mL flask at room temperature. Gallic acid (0.55 M) in methanol was reacted with dicyclohexylcarbodiimide (DCC, 0.5 M in methanol) and was then added dropwise into the COS solution under constant stirring for 5 h at room temperature. The formed dicyclohexyl urea was filtered over and the filtrate was kept at 4 °C overnight. After being dissolved in 90 mL of diethyl ether and precipitated again, the precipitate was added into 20 mL of distilled water. The solution was thoroughly dialyzed (cut off below 1 kDa) against distilled water with changes of water every 8 h to remove unreacted gallic acid (GA). Finally, the resulting solution was lyophilized to obtain water-soluble G-COS (1.5 g).FT-IR and NMR analyses
Fourier transformed infrared spectroscopy (FT-IR) and proton nuclear magnetic resonance spectroscopy (1H NMR) were respectively used to confirm the synthesis of COS and G-COS conjugates. The obtained G-COS conjugates or COS used for the FT-IR analysis was first dried and ground into a powder form. The IR spectrum was recorded as power dispersions in KBr in a frequency range of 4000–400 cm−1 using the FT-IR spectrometer (Perkin Elmer Spectrum GX, Beaconsfield Bucks, England). 1H NMR and 13C NMR spectra were recorded on a JEOL JNM-ECP-400 spectrometer (JEOL, Tokyo, Japan) under a static magnetic field of 400 MHz and 100 MHz. Deuterium oxide (D2O, 4.8 ppm) used as internal standard for 1H.Cell culture and viability assay
AGS cells were cultured in DMEM supplemented with heat-inactivated 5% fetal bovine serum and 100 μg mL−1 penicillin/streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. The viability levels of AGS cells were measured by the ability of mitochondria to convert MTT to insoluble formazan product.27 The cells were seeded in 96-well plates at a density of 5 × 103 cells per well for 24 h, and subjected to different concentration of G-COS (50, 100, 200, 400 μg mL−1) or COS for another 24 h. Subsequently, MTT solution (1 mg mL−1, final concentration) was added and incubated for 4 h. After removing the staining solution, the resulting purple formazan was dissolved with DMSO and the absorbance at 540 nm was read on a GENios microplate reader (Tecan Austria GmbH, Grodig, Austria). Viability of the cells was normalized to the cells with no-treatment (Blank).Cell morphology and Hoechst staining
To study the morphological changes of the cells following GCOS or COS treatments, the cells were incubated with different concentrations of GCOS (100, 200, 400 μg mL−1) or 400 μg mL−1 of COS for 24 h. After being washed with PBS, cells were fixed with 4% paraformaldehyde (pH 7.4) solution for 1 h at room temperature and stained with 1 μg mL−1 of Hoechst 33258 for another 1 h at room temperature in the dark. Morphological changes were observed by using a fluorescence microscope. The fluorescence intensity of Hoechst 33258 was analysed by a Leica CTR 6000 microscope (Wetzlar, Germany).DNA fragmentation assay
The cells were incubated with GCOS or COS and lysed in a buffer containing 10 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA and 0.5% Triton X-100 for 20 min. The lysates were vortexed and cleared by centrifugation at 10000g for 10 min. The fragmented DNA in the supernatant was extracted using an equal volume of neutral phenol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :chloroform:isoamylalcohol (25:24:1, v/v/v) and analysed electrophoretically on 1.0% agarose gel containing ethidium bromide (EtBr, Sigma).
:chloroform:isoamylalcohol (25:24:1, v/v/v) and analysed electrophoretically on 1.0% agarose gel containing ethidium bromide (EtBr, Sigma).
Cell cycle assay
The cells were harvested and washed once with PBS, fixed in ice-cold 70% ethanol and stored at 4 °C. Prior to analysis, the cells were washed once again with PBS, suspended in 1 mL of a cold PI solution containing 100 μg mL−1 RNase A, 50 μg mL−1 PI, 0.1% (w/v) sodium citrate, and 0.1% (v/v) NP-40, and further incubated on ice for 30 min in the dark. Flow cytometric analyses were carried out using a flow cytometer (BD Biosciences, CA, USA), and CellQuest software was used to determine the relative DNA content based on the presence of a fluorescence.Annexin V-FITC and propidium iodide staining
Annexin V-FITC apoptosis detection kit was used for the detection of apoptotic cells as per manufacturer's protocol. Briefly, cells were harvested by trypsinization and washed with PBS, centrifuged at 1500 rpm for 5 min at 4 °C and re-suspended in 1× binding buffer (10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2). Then AnnexinV FITC and PI were added for 15 min in the dark and finally 400 μL of 1× binding buffer was added. Fluorescence of the cells was determined immediately using flow cytometry (Cell Quest software; BD Biosciences, CA, USA). Acquisition gates of the cells and a minimum of 10000 events were collected for each sample.
Western blot analysis
After 24 h incubation with G-COS or COS, the cells were harvested and lysed in RIPA buffer. Lysates were centrifuged at 14000g for 20 min at 4 °C. Protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific, Grand Island, NY, USA). Twelve micrograms of protein from each sample was applied to 10% SDS-PAGE gels and was electrophoretically transferred to nitrocellulose membrane. The nonspecific binding sites on the membranes were blocked at 4 °C overnight with TBS-T buffer (20 mM Tris, pH 7.6, 0.1% Tween 20) containing 5% (w/v) bovine serum albumin. The blots were then incubated with primary antibodies for 1 h at room temperature and were subsequently incubated with a secondary antibody conjugated to horseradish peroxidase which was used to detect the respective proteins. The bands were detected by using an enhanced chemiluminescent ECL assay kit (Amersham Pharmacia Biosciences) and quantified by a LAS3000® Luminescent image analyzer (Fujifilm Life Science, Tokyo, Japan). The relative amounts of proteins associated with each specific antibody were normalized to the respective β-actin and lamin B bands.Reverse transcriptase (RT) PCR detection
Cells were treated with G-COS and COS for 24 h, then, harvested and total cellular RNA was isolated using TRIzol reagent (Invitrogen Co., CA, USA). The RNA was reverse transcribed into cDNA using oligo-(dT) primer and amplified to target gene using each primers; Bax, Bcl-2, caspase-3,-8,-9. The PCR reaction was performed as follow: 95 °C for 45 min, then 30 cycles at 60 °C, 50 s; 72 °C, 60 s; and 72 °C, 5 min. PCR products were visualized on gel and analyzed by UV illumination using AlphaEase® gel image analysis software (Alpha Innotech., San Leandro, CA, USA). Each mRNA expression level was normalized to the mRNA expression level of housekeeping gene, β-actin.Statistical analysis
Data were displayed as mean ± SD from three independent experiments. Statistical analyses were calculated by Microsoft Excel or GraphPad Prism. Data were analyzed statistically using two-way ANOVA and significant differences between treatment means were determined using Dunnett's multiple range tests at the P < 0.05 levels.Acknowledgements
This research was supported by a grant from Marine Bioprocess Research Center of the Marine Biotechnology Program (2004-6002) funded by the Ministry of Oceans and Fisheries, Republic of Korea.References
- J. D. Liu, S. Y. Lin, Y. S. Ho, S. Pan, L. F. Hung, S. H. Tsai, J. K. Lin and Y. C. Liang, Mol. Carcinog., 2003, 37, 16–24 CrossRef CAS PubMed.
- L. P. Carcas, J. Carcinog., 2014, 13, 14 CrossRef PubMed.
- M. A. Shah and D. P. Kelsen, J. Natl. Compr. Cancer Network, 2010, 8, 437–447 CrossRef CAS.
- X. Y. Xiao, M. Hao, X. Y. Yang, Q. Ba, M. Li, S. J. Ni, L. S. Wang and X. Du, Cancer Lett., 2011, 302, 69–75 CrossRef CAS PubMed.
- S. W. Fesik, Nat. Rev. Cancer, 2005, 5, 876–885 CrossRef CAS PubMed.
- S.-H. Lee, B. Ryu, J.-Y. Je and S.-K. Kim, Carbohydr. Polym., 2011, 84, 571–578 CrossRef CAS.
- M. van Engeland, F. C. Ramaekers, B. Schutte and C. P. Reutelingsperger, Cytometry, 1996, 24, 131–139 CrossRef CAS PubMed.
- M. Hollstein, D. Sidransky, B. Vogelstein and C. C. Harris, Science, 1991, 253, 49–53 CAS.
- M.-C. Figueroa-Espinoza and P. Villeneuve, J. Agric. Food Chem., 2005, 53, 2779–2787 CrossRef CAS PubMed.
- F. A. Silva, F. Borges, C. Guimarães, J. L. Lima, C. Matos and S. Reis, J. Agric. Food Chem., 2000, 48, 2122–2126 CrossRef CAS PubMed.
- D. N. Ngo, M. M. Kim and S. K. Kim, Int. J. Biol. Macromol., 2012, 50, 624–631 CrossRef CAS PubMed.
- M. Z. Karagozlu, J.-A. Kim, F. Karadeniz, C.-S. Kong and S.-K. Kim, Process Biochem., 2010, 45, 1523–1528 CrossRef CAS.
- B. Ryu, S. W. Himaya, R. J. Napitupulu, T. K. Eom and S. K. Kim, Carbohydr. Res., 2012, 350, 55–61 CrossRef CAS PubMed.
- D. H. Ngo, Z. J. Qian, T. S. Vo, B. Ryu, D. N. Ngo and S. K. Kim, Carbohydr. Polym., 2011, 84, 1282–1288 CrossRef CAS.
- T. S. Vo, D. H. Ngo and S. K. Kim, Eur. J. Pharm. Sci., 2012, 47, 527–533 CrossRef CAS PubMed.
- H. M. Chen, Y. C. Wu, Y. C. Chia, F. R. Chang, H. K. Hsu, Y. C. Hsieh, C. C. Chen and S. S. Yuan, Cancer Lett., 2009, 286, 161–171 CrossRef CAS PubMed.
- J. Zhang, J. Han, X. Zhang, J. Jiang, M. Xu, D. Zhang and J. Han, Carbohydr. Polym., 2015, 129, 25–34 CrossRef CAS PubMed.
- Q. Hu, T. Wang, M. Zhou, J. Xue and Y. Luo, J. Agric. Food Chem., 2016, 64, 5893–5900 CrossRef CAS PubMed.
- H. H. Ho, C. S. Chang, W. C. Ho, S. Y. Liao, C. H. Wu and C. J. Wang, Food Chem. Toxicol., 2010, 48, 2508–2516 CrossRef CAS PubMed.
- B. R. You and W. H. Park, Toxicol. in Vitro, 2010, 24, 1356–1362 CrossRef CAS PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- W. K. Ng, L. S. Yazan and M. Ismail, Toxicol. In Vitro, 2011, 25, 1392–1398 CrossRef CAS PubMed.
- H. Shin, Y. S. Lee and Y. C. Lee, Oncol. Rep., 2012, 27, 1111–1115 CAS.
- L. Verstrepen and R. Beyaert, Biochem. Pharmacol., 2014, 92, 519–529 CrossRef CAS PubMed.
- K. M. Ryan, M. K. Ernst, N. R. Rice and K. H. Vousden, Nature, 2000, 404, 892–897 CrossRef CAS PubMed.
- A. Ahmad, B. Biersack, Y. Li, D. Kong, B. Bao, R. Schobert, S. B. Padhye and F. H. Sarkar, Adv. Anticancer Agents Med. Chem., 2013, 13, 1002–1013 CrossRef CAS.
- M. B. Hansen, S. E. Nielsen and K. Berg, J. Immunol. Methods, 1989, 119, 203–210 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |