
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoplastic starch–polyethylene blends homogenised using deep eutectic solvents†
Andrew P. Abbott *a,
Tariq Z. Abolibdaa,
Wanwan Qua,
William R. Wiseb and
Luka A. Wrighta
*a,
Tariq Z. Abolibdaa,
Wanwan Qua,
William R. Wiseb and
Luka A. Wrighta
aDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK. E-mail: apa1@le.ac.uk
bInstitute for Creative Leather Technologies, University of Northampton, Northampton, NN2 7AL, UK
First published on 23rd January 2017
Abstract
Polyolefin based plastics are extensively used for packaging applications and as such they tend to have a short service life but they have a long environmental persistence. One strategy to accelerate the mechanical degradation of polyolefin plastics in the environment is to blend them with carbohydrate based polymers. Unfortunately polyolefins are hydrophobic whereas carbohydrates tend to be hydrophilic so the two do not blend without chemical modification of the carbohydrate. In this study high density polyethylene, HDPE and thermoplastic starch, TPS are used as the polymers with deep eutectic solvents, DESs as the modifiers. Both TPS and DESs are biodegradable and the DESs are water miscible and biocompatible ensuring that the composite plastic contains a biodegradable flaw which should enable mechanical and chemical degradation. It is shown that DESs enable facile mixing of the two polymers. The composite has a strength similar to TPS but a ductility greater than either of the two components. The glass transition temperature of the composite plastic shows that they are homogeneously mixed and data suggests that the DESs act as lubricants rather than plasticisers.
Introduction
An ideal material needs to be stable during its service life but biodegrade rapidly when it is emitted into the environment. In the last few decades the fate of plastics in the environment has become a significant issue. The discovery that ocean currents are concentrating water-borne plastics into areas the size of continents is headline news. The so-called Great Pacific Garbage Patch and North Atlantic Garbage Patch has a plastic content of about 5 kg km−2 of assorted fragments which are a hazard to all forms of aquatic life.1,2 In addition to environmental concerns the use of plastic is using about 6% of the annual oil produced which is naturally not sustainable.Much of the plastic which has been discarded into the environment originates from packaging applications and as such requires a relatively short service life but a rapid environmental breakdown rate. Polyolefin plastics e.g. polyethylene, polypropylene etc. are extremely stable as they are difficult to degrade through oxidation, reduction or photolysis. Some polymers will biodegrade more rapidly than polyolefins e.g. polylactic acid and polycaprolactam however these are prohibitively expensive for high-volume/low value applications such as packaging. An alternative method has been to blend polyolefin plastics with carbohydrate based polymers which can degrade more rapidly decreasing the mechanical strength of the polymer and allowing it to fragment in the environment. Starch is the ideal material due to its ease of degradation. Unmodified starch has been added as a filler for PE but it is immiscible and so stiffens the plastic and makes it more brittle.3 Gelatinised starch has been blended with more polar polymers such as ethylene–acrylic acid copolymer in the presence of other additives such as polyvinyl alcohol.4 LDPE:TPS has been produced and while it has interesting mechanical properties it has been shown to be highly heterogeneous.5
Several studies have modified the starch to make it less hydrophillic. Kim modified potato starch through hydroxypropylation and found that it could be blended with polyethylene.6 Esterification of starch with octanoyl chloride resulted in octanoated starch which could be blended with LDPE to produce a water resistant material.7 The area of starch polyolefin blends has been reviewed by Hamad et al.8
Deep eutectic solvents are mixtures of quaternary ammonium salts and hydrogen bond donors.9,10 The liquids have properties which are similar to ionic liquids and the strong hydrogen bonding between the two components can be used as a modifier for carbohydrates. Recently, several groups have shown that DESs, can be used as efficient plasticisers of starch and starch composites.11–14 A significant decrease in the energy needed for melt processing was observed. The materials displayed improved tensile strength and did not recrystallise on storage.15 The starch plastics had reduced water sensitivity and acted like true thermoplastics which could be recycled without significant loss of mechanical properties.9 The thermoplastic starch was also demonstrated as a binder for wood particles to make a thermoplastic material, which could be recycled.10 Mixtures of glycerol and quaternary ammonium salts were also used to plasticise starch and this DES had the advantage that it can be made with a Sheldon E-factor of zero.16 The optimization of the plastic was made and it was shown that the mechanical properties could be made similar to those for HDPE. In the current study DESs will be used to modify HDPE and attempts are made to homogenise HDPE and TPS. The ability of water to modify the mechanical properties is also investigated.
Experimental
Corn flour (Weikfields Foods Ltd.) was used as received with no additional drying or modification steps performed, unless otherwise stipulated. Glycerol, urea, ethylene glycol and choline chloride were obtained from Sigma Aldrich (>99% purity) and used as received. High density polyethylene was obtained from Artenius Tech Polymers, and was used as received. The preparation of the deep eutectic solvents has previously been described elsewhere.9,17 Pelletised thermoplastic starch samples were prepared by mixing corn flower with the pre-prepared glyceline, in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt ratio, respectively and the resultant mixture subjected to extrusion in a Prism TSE 24 TC self-wiping extruder with a Prism volumetric feeder and an air swept face-cut pelletising system. The internal barrel temperature was maintained at 150 °C. Pelletised HDPE:DES blends were obtained by mixing the desired ratios of pre-prepared DESs and virgin HDPE prior to extrusion at a controlled barrel temperature of 150 °C, across all five heating zones. The screw profile for the extruder was as follows: 4.5 L/D conveying screw, 0.75 L/D 30° kneading, 0.75 L/D 60° kneading, 1.25 L/D 90° kneading, 7 L/D conveying screw, 0.5 L/D 60° kneading, 0.75 L/D kneading, 3 L/D conveying screw, 1.5 L/D single lead discharge screws.
:1 wt ratio, respectively and the resultant mixture subjected to extrusion in a Prism TSE 24 TC self-wiping extruder with a Prism volumetric feeder and an air swept face-cut pelletising system. The internal barrel temperature was maintained at 150 °C. Pelletised HDPE:DES blends were obtained by mixing the desired ratios of pre-prepared DESs and virgin HDPE prior to extrusion at a controlled barrel temperature of 150 °C, across all five heating zones. The screw profile for the extruder was as follows: 4.5 L/D conveying screw, 0.75 L/D 30° kneading, 0.75 L/D 60° kneading, 1.25 L/D 90° kneading, 7 L/D conveying screw, 0.5 L/D 60° kneading, 0.75 L/D kneading, 3 L/D conveying screw, 1.5 L/D single lead discharge screws.
The pelletised or powdered samples were placed between two copper plates lined with non-stick silicone sheets and equipped with a 1 mm copper separating unit with a 100 mm2 aperture. The resultant sandwich was then placed in a hydraulic press (Fontune Grotnes Laboratory Press TH400) and a force of 120 kN was applied to the sample at 150 °C for 10 min. The sample was subsequently cooled to ambient temperature whilst remaining under pressure prior to removal from the hydraulic press. The cooled sample was subjected to a mechanical ‘dog-bone’ press (Ceast Hollow Die Punch, Type 6051) to cut test shapes (test area size: L = 30 mm, W = 4 mm, D = 0.7–1.2 mm).
Tensile testing was performed on the dog-bone samples using an Instron 3343 tensile apparatus (Instron Ltd., Assembles, USA) with a load cell of 500 N. The material stress and strain was controlled by Instrom Bluehill 2 software and average numerical values were taken from eight or more samples. In each case, the thickness of the sample was measured using a micrometer and subjected to a strain rate between 2–10 mm min−1.
Differential Scanning Calorimetry (DSC) analysis was performed to analyse thermal events with the samples. A Mettler Toledo DSC1 was used to determine glass transition temperatures and the results analysed with STARe software. Samples (5–10 mg) were placed on in a standard aluminium DSC pan before the lid of the pan was pierced. The sample was placed within the furnace next to an, otherwise identical, empty reference pan. The samples were heated from −160 °C to 150 °C at 10 °C min−1 and held at 150 °C for 5 min before cooling to 25 °C at 20 °C min−1.
Results and discussion
Plasticisation of HDPE with various DESs: mechanical and thermal analysis
The first aim of this study was to determine whether DESs would mix with a non-polar polymer such as HDPE. Ionic liquids have been used as plasticisers with polyolefinic polymers such as polymethylmethacrylate and polyvinylchloride but these are relatively polar and it is less surprising that they could act as a plasticiser.18,19 In this section, the blending of HDPE in the presence of three DESs is evaluated at 1 and 3 wt% DES. Attempts to increase the amount of DES to 5 wt% resulted in the formation of non-homogeneous samples and as such no further analysis of these samples was performed. The samples were all produced by twin screw extrusion of mixtures of HDPE:DES at 150 °C (see Experimental section) and pelletised prior to formation of 1 × 100 × 100 mm sheets by compression moulding at 150 °C for 10 minutes (Fig. 1). Under these processing conditions, it was found that largely homogeneous samples were obtained. | ||
| Fig. 1 Samples of extruded HDPE:DES blends. | ||
To understand how the DESs modify HDPE it is necessary to determine if the two components are uniformly distributed throughout the plastic. If the DES formed a continuous phase then the samples should be electrically conducting; the fact that they are electrical insulators shows that the DES is not continuous and therefore must be evenly distributed. The density of the DES-modified HDPE (m-HDPE) is lower than that for un-modified HDPE (0.948 g cm−3 versus 0.994 g cm−3, respectively); thus implying that addition of DES to these samples increases the free volume of the HDPE. This is surprising given the density if the DESs is higher than that of HDPE (1.1 to 1.3 g cm−3).
Fig. 1 shows that when 3 wt% DES is incorporated mottling of the samples is evident. It was found that both altering the DES employed as well as varying the DES content had a marked impact upon the qualitative appearance of the samples. The degree of homogeneity is shown to decrease as the weight percentage of DES increases and is attributed to increased agglomeration of particles within the extruder due to the increased liquid volume.
In an attempt to quantify the efficiency of homogenisation, mechanical analysis was performed on the samples produced. The ultimate tensile strength (UTS) of the six blended materials was determined (Fig. 2a) and compared against the percent elongation at break (Fig. 2b). These data clearly demonstrate that that whilst the UTS of HDPE:DES mixtures remain largely unaffected by the presence of DES, the elongation at break increases significantly (Fig. 2b). The increase in elongation at break for samples containing 1% DES by weight is indicative of significant modification of HDPE by deep eutectic solvents. However, increasing the weight content of DES to 3% by weight, all samples exhibit a significant reduction in elongation at break, despite a largely unaffected ultimate tensile strength. Indeed, it can be seen that all three DESs aid the ductility of the HDPE, with approximately double the elongation at break when compared with un-modified HDPE. However, upon addition of 3% by weight DES to the sample, the ductility is significantly reduced to levels comparable to unmodified HDPE; suggestive of non-homogeneous, non-advantageous mixing. An effective plasticiser should decrease the ultimate tensile strength but increase the elongation at break.
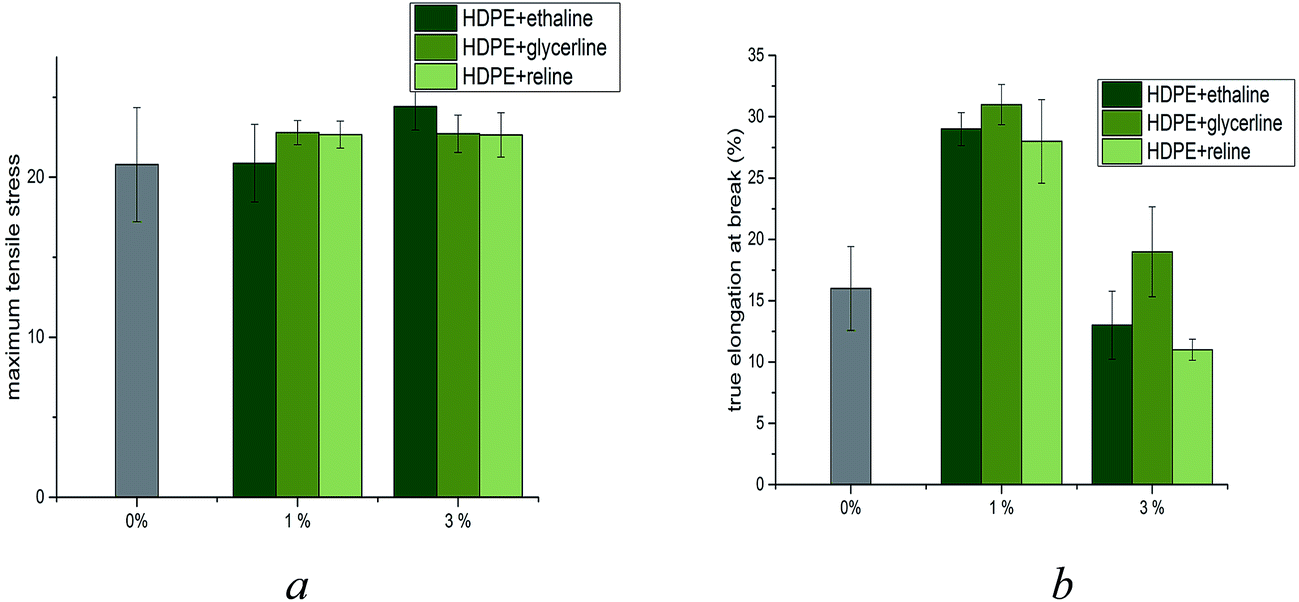 | ||
| Fig. 2 Mechanical analysis of HDPE:DES blends (a) UTS (b) percent true elongation at break. | ||
In an attempt to further investigate this on a molecular level, Differential Scanning Calorimetry (DSC) was performed to elucidate the effects of DESs on the glass transition temperature (Tg) of DES modified HDPE.
DSC results for HDPE:DES blends containing 1% modifier show a slight reduction in Tg (Table 1). The Tg for unmodified HDPE is −81 °C and a reduction in Tg upon incorporation of a DES is only 1 to 4 °C indicating minimal reduction of intramolecular bonding interactions between individual polymer chains, thus showing limited plasticising ability. It can be concluded that DESs act more as a modifier or lubricant than a true plasticiser.
| Tg/°C ethaline | Tg/°C glyceline | Tg/°C reline | |
|---|---|---|---|
| a HDPE:TPS blends. | |||
| 0% | −81.3 | −81.3 | −81.3 |
| 1% | −85.4 | −84.0 | −82.7 |
It has recently reported that DESs act as efficient plasticisers for starch. In this section, the use of starch plasticised with glyceline (TPS) as a composite with glyceline-modified HDPE (m-HDPE) blends is investigated together with the use of DESs as effective homogenising agents. In the first instance, pre-modified thermoplastic starch pellets (TPS) (75% starch and 25% glyceline, by weight, see Experimental section) was mixed with modified HDPE pellets (m-HDPE) (99% HDPE and 1% glyceline, by weight) in two compositions (30 and 40 wt% TPS) and subjected to extrusion at 150 °C and pelletised.
Using a similar methodology to that employed in the preparation of m-HDPE blends, the formation of 1 × 100 × 100 mm sheets was achieved by compression moulding of the pelletised composite material. In this instance, the visual appearances of the samples were the same, regardless of the increased starch content (Fig. 3). This shows for the first time that the carbohydrate polymer can be homogeneously mixed with a polyolefin without chemical modification of carbohydrate. This makes for more facile synthesis of the plastic using less polyolefin and incorporating a larger sustainable component content. The DESs are simple to handle and they have already been applied to tonne scale processes masking this a practical approach to material modification.
 | ||
| Fig. 3 Representative 7:3 m-HDPE:TPS composite. | ||
To evaluate the mechanical properties of the m-HDPE:TPS blended composites, stress–strain data were collected and compared against independently prepared samples of thermoplastic starch and m-HDPE (Fig. 4).
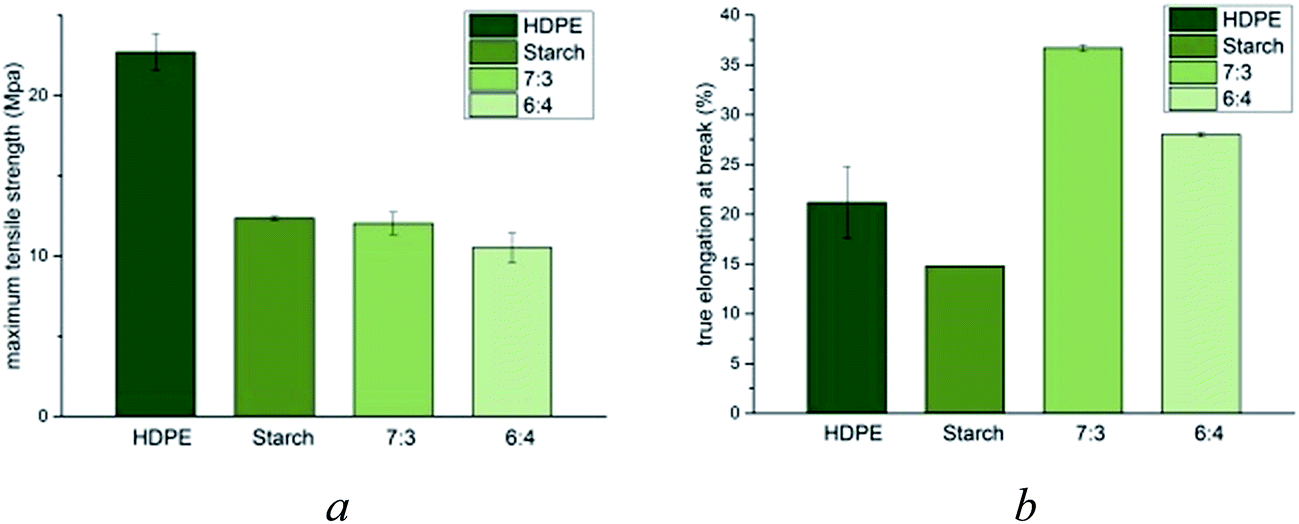 | ||
| Fig. 4 Mechanical analysis of m-HDPE:TPS blends (a) UTS (b) percent true elongation at break. | ||
These data show that upon addition of thermoplastic starch to m-HDPE, the ultimate tensile strength is reduced to that exhibited by TPS (Fig. 4a). Analysis of the elongation at break shows a significant increase upon addition of TPS to m-HDPE (Fig. 4b). Increasing the weight percentage of TPS to the m-HDPE:TPS blend to 40% results in an observed decrease in percent true elongation at break when compared to mixtures containing 70% m-HDPE:30% TPS. Nevertheless, the value remains greater than either m-HDPE or TPS. The DES probably has a role in homogenising the two polymers; in HDPS it fills the void spaces in the polymer matrix and in starch is disrupts the hydrogen bonding. The common miscibility of the DESs in both polymers must enable mutual mixing.
This observation is in contrast to the data reported in the literature, wherein an increase in starch content to HDPE mixtures (in the absence of a compatibiliser) which results in decreased ductility of the resultant composite material.20 This shows that having a molecular (ionic) modifier behaves in a completely different way to a macromolecular modifier confirming the above assumption that the DES acts as a lubricant.
The strongest and yet most ductile sample was achieved when the m-HDPE:TPS composition was 70:30 wt%. The results obtained in this section suggest that in order to efficiently plasticise HDPE:TPS mixtures, DESs provide a low-cost, environmentally benign solution, without the need to chemically modify the backbone of the starch sub-structure.
Differential scanning calorimetry was performed on the m-HDPE:TPS composites in an attempt to identify the degree of homogenisation present in the samples. Quite coincidentally the Tg values for TPS (−84.8 °C) and m-HDPE (−84 °C) (entries 1 and 2) are extremely close to each other. Interestingly, for both blends, no Tg at ca. −84 °C was observed at all. Instead, the 7:3 and 6:4 composites exhibit unique Tg at −113.4 and −73.2 °C, respectively (entries 3 and 4, Table 2).
| Entry | Composition | Tg (°C) | Wt loss on boiling (%) | Wt% DES |
|---|---|---|---|---|
| 1 | TPS | −84.8 | — | |
| 2 | m-HDPE | −84.0 | 0.3 | 1.0 |
| 3 | HDPE:TPS (7:3) |
−73.2 | 10.5 | 8.2 |
| 4 | HDPE:TPS (6:4) |
−113.4 | 19.8 | 10.6 |
The presence of a single glass transition temperature, distinct from those expected for m-HDPE or TPS, reveals that the effect of blending starch and HDPE with a deep eutectic solvent is to modify both materials; generating a unique blend with thermal characteristics not attributable to being HDPE-like or TPS-like in origin. This is an important observation because it implies that the material will behave in a unique fashion and not as a heterogeneous mixture of HDPE and thermoplastic starch. Most interestingly, the presence of a single Tg proves that the material is completely homogeneous and discounts the possibility of localised thermoplastic starch pockets existing within the composite material. This feature implies that the starch has been evenly distributed throughout the m-HDPE:TPS composite and that both the starch and HDPE components are acting as concomitant structural disruptors; enabling access to a valuable decomposition pathway of the composite material by exposure to boiling water (vide infra). The incorporation of large quantities of a hydrophilic material (starch) into a non-polar material such as HDPE must be facilitated by the formation of strong intermolecular interactions between the starch polymer chains and the deep eutectic solvent, which is acting as a homogenising agent. Indeed, the absence of a Tg at −84.8 °C specifically for the TPS (Table 2, entry 2) or one relating to m-HDPE at −84.0 °C indicates that the DES must be interacting strongly with both components in order to generate a homogeneous material.
Effect of treatment of m-HDPE-TPS composites in boiling water
Blending starch with HDPE incorporates a breakdown mechanism that while not acutely affecting the material it should produce a chronic weakness following aqueous immersion on the timescale of months. In cold water negligible changes in mass or appearance were observed over a week showing that it should be serviceable under normal operating conditions. To accelerate degradation samples of m-HDPE-TPS blends were immersed in boiling water for 3 hours. The weight of these samples was recorded before and after boiling, following desiccation over dehydrated silica for 48 hours.Table 2 shows that the 7:3 m-HDPE:TPS composites lose ca. 10 wt% post-boiling when compared to its original mass. This is more than the 8.2% DES which is in the composite showing that it is not just the water soluble component which is removed during boiling. As would be expected, boiling the 6:4 m-HDPE:TPS composite results in an even larger loss in mass with almost 20% of the sample being lost. While a small amount of the starch is water soluble it is evident from the appearance of the solution that the starch component is leached from the composite and forms a colloidal distribution. This can also be seen from the density of the films before and after boiling in water for 3 h (Table 3). Note the density of HDPE is about 0.91 g cm−3 and TPS is 1.40 g cm−3. The density of the boiled materials approach that of pure HDPE but appear lower due to internal voids.
 |
Visually, the boiled samples have a significantly altered appearance with extensive fragmentation of the surface observed (Fig. 5). This can be seen as macroscopic flakes but also as microscopic voids in the surface (Table 3). Under tension the material is still strong (Fig. 6) but the surface can be readily abraded with just a fingernail into fragments which are generally 1–5 mm in the x and y coordinates and 1–5 μm in the z direction.
 | ||
| Fig. 5 Low resolution microscopy image (24× magnification) showing significant fragmentation of 7:3 composite following treatment in boiling water. | ||
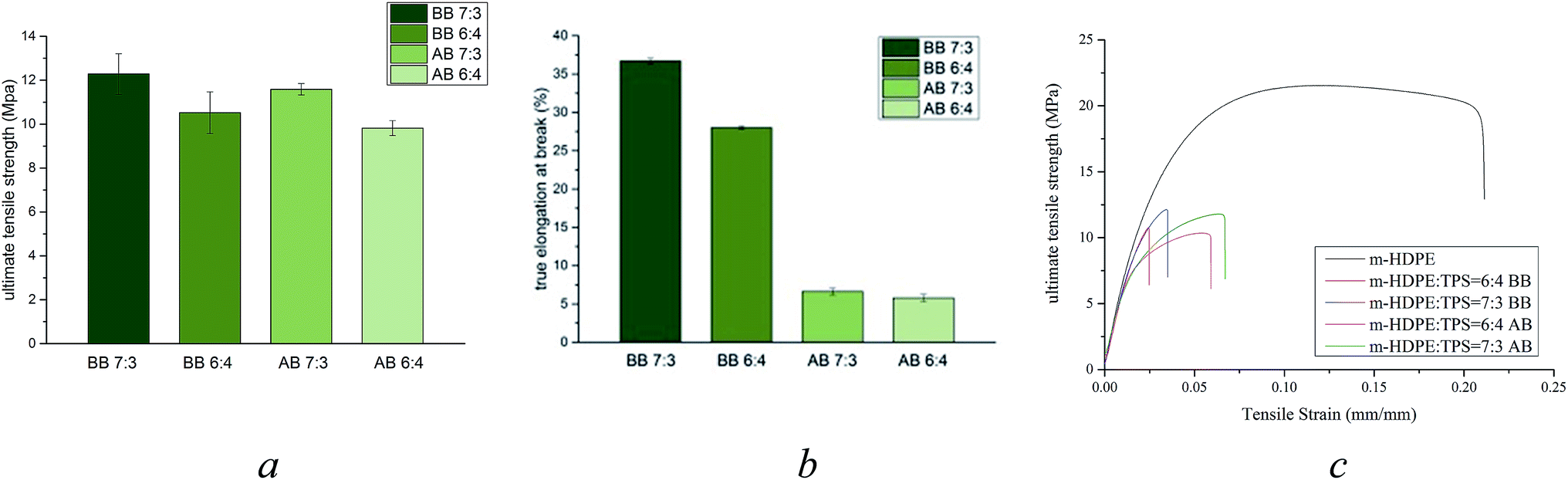 | ||
| Fig. 6 Mechanical analysis of m-HDPE:TPS blends (BB – before boiling and AB – after boiling) (a) UTS (b) elongation at break (c) stress–strain curves. | ||
Fig. 6 shows the mechanical analysis data for the 6:4 and 7:3 m-HDPE:TPS samples before and after boiling. These data show that the UTS of the samples is largely unaffected by treatment in boiling water for both blends (Fig. 6a) whereas the percent elongation at break has been significantly reduced for both composites after treatment in boiling water; (Fig. 6b). This shows that the materials are more brittle and in general less flexible.
Conclusions
This study has shown that DESs can be used to modify the properties of polyolefins such as HDPE. They do not significantly affect the strength or the glass transition temperature of the material but they do increase the ductility of the samples suggesting that they may act as lubricants rather than plasticisers. At a loading of 1 wt% the DES is homogeneously distributed but at 3 wt% the mottled appearance shows an uneven distribution of the two components. The incorporation of a DES into HDPE enables it to be blended with thermoplastic starch which has also been modified using a DES. This is the first time that non-chemically modified carbohydrates have been able to be blended with polyolefins. The study shows that HDPE:TPS blends are stable in water over the timescale of weeks at ambient temperature but when boiled the starch and DES components leach from the plastic and enable it to be mechanically degraded. This shows that starch can easily be blended with HDPE using DESs as modifiers and this not only increases the amount of sustainable material included in plastic but also improves the mechanical degradation once immersed in water.Acknowledgements
The authors would like to thank the Royal Society for funding the work through the Mercer Award for Innovation MI130014.References
- R. Yamashita and A. Tanimura, Mar. Pollut. Bull., 2007, 54, 485–488 CrossRef CAS PubMed.
- O. J. Dameron, M. Parke, M. A. Albins and R. Brainard, Mar. Pollut. Bull., 2007, 54, 423–433 CrossRef CAS PubMed.
- J. L. Willett, J. Appl. Polym. Sci., 1994, 54, 1685–1695 CrossRef CAS.
- F. H. Otey, R. P. Westhoff and W. M. Doane, Ind. Eng. Chem. Prod. Res. Dev., 1980, 19, 592–595 CrossRef CAS.
- F. J. Rodriguez-Gonzalez, B. A. Ramsay and B. D. Favis, Polymer, 2003, 44, 1517–1526 CrossRef CAS.
- M. Kim, Carbohydr. Polym., 2003, 54, 173–181 CrossRef CAS.
- J. Aburto, S. Thiebaud, I. Alric, E. Borredon, D. Bikiaris, J. Prinos and C. Panayiotou, Carbohydr. Polym., 1997, 34, 101–112 CrossRef CAS.
- K. Hamad, M. Kaseem, Y. G. Ko and F. Deri, Polym. Sci., Ser. A, 2014, 56, 812–829 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, A. D. Ballantyne, J. Palenzuela Conde, K. S. Ryder and W. R. Wise, Green Chem., 2012, 14, 1302 RSC.
- A. P. Abbott, J. Palenzuela Conde, S. Davis and W. R. Wise, Green Chem., 2012, 14, 3067–3070 RSC.
- E. Leroy, P. Decaen, P. Jacquet, G. Coativy, B. Pontoire, A.-L. Reguerre and D. Lourdin, Green Chem., 2012, 14, 3063–3066 RSC.
- M. Zdanowicz, T. Spychaj and H. Mąka, Carbohydr. Polym., 2016, 140, 416–423 CrossRef CAS PubMed.
- A. P. Abbott, T. Z. Abolibda, S. J. Davis, F. Emmerling, D. Lourdin, E. Leroy and W. R. Wise, RSC Adv., 2014, 4, 40421–40427 RSC.
- A. P. Abbott, R. C. Harris, K. S. Ryder, C. D'Agostino, L. F. Gladden and M. D. Mantle, Green Chem., 2011, 13, 82–90 RSC.
- A. P. Abbott, G. Capper, D. L. Davies, R. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- M. Rahman and C. S. Brazel, Polym. Degrad. Stab., 2006, 91, 3371–3382 CrossRef CAS.
- S. Y. Choi, H. Rodríguez, A. Mirjafari, D. F. Gilpin, S. McGrath, K. R. Malcolm, M. M. Tunney, R. D. Rogers and T. McNally, Green Chem., 2011, 13, 1527–1535 RSC.
- M. L. Méndez-Hernández, C. S. Tena-Salcido, Z. Sandoval-Arellano, M. C. González-Cantú, M. Mondragón and F. J. Rodríguez-González, Polym. Bull., 2011, 67, 903–914 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00135e |
| This journal is © The Royal Society of Chemistry 2017 |