
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cytotoxic trichothecene-type sesquiterpenes from the sponge-derived fungus Stachybotrys chartarum with tyrosine kinase inhibition†
Yong Lia,
Dong Liua,
Zhongbin Chenga,
Peter Prokschb and
Wenhan Lin *a
*a
aState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing, 100191, P. R. China. E-mail: whlin@bjmu.edu.cn; Fax: +86-10-82806188
bInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine University, 40225 Duesseldorf, Germany
First published on 23rd January 2017
Abstract
Bioassay-guided fractionation of the sponge (Niphates recondite)-associated fungus Stachybotrys chartarum revealed that the EtOAc fraction shows inhibitory effects against tumor cell lines. Chromatographic separation of the active fraction resulted in the isolation of 15 trichothecene-based sesquiterpenes, including four new compounds, namely chartarenes A–D (1–4). The structures of the new compounds were determined on the basis of extensive spectroscopic analysis and chemical conversion. Compounds 1–15 exerted potent or selective inhibition against a panel of tumor cell lines including HCT-116, HepG2, BGC-823, NCI-H1650, and A2780, with IC50 values ranging from 10−2 to 10 μM. In addition, these compounds showed potent inhibition against tumor-related kinases FGFR3, IGF1R, PDGFRb, and TRKB.
1. Introduction
Trichothecenes are a group of sesquiterpene-based mycotoxins,1–6 which are markedly cytotoxic toward eukaryotic organisms,7 including high phytotoxicity.8–10 The mechanism of toxicity is attributed to the inhibition of protein and DNA synthesis, and the mitochondrial electron transport system.11,12 Biogenetically, the mevalonate pathway is involved in the generation of this typical scaffold and chemically related derivatives such as apotrichothecene-type and sambucinol-type analogues.13,14 Apart from the phytotoxicity and mammalian intoxications, trichothecene-related compounds exhibited potent inhibitory effects for tumors,4 and diacetoxyscirpenol (anguidine) has even been developed for clinical trials as an anticancer agent.15 The structure–activity relationships revealed that the structural variety directly affects the cytotoxic activity, while destruction of the epoxide moiety and rearrangement of the ring system resulted in the virtual elimination of cytotoxicity.16 It was noted that a slight change in trichothecenes could dramatically reduce the toxicity but still retain the activity of apoptosis induction in human cancer cells, as exemplified by 2′,3′-epoxymyrothecine A and myrothecine A inducing the apoptosis of tumor cell lines through the induction of PARP cleavage and JNK phosphorylation with low cytotoxicity.3 The derivatives of roridins A and H and verrucarins A and J exhibited extraordinary activities in vivo against P388 mouse leukemia with high therapeutic index,17,18 and trichothecinol A strongly inhibited TPA-induced tumor promotion for cancer preventive activity.19 These findings suggested that trichothecenes are promising leads which are applicable for cancer chemotherapy. During our study on antitumor-active metabolites from marine-derived fungi, we found that the EtOAc extract of a rice solid cultured fungus, Stachybotrys chartarum, isolated from a sponge (Niphates recondite), showed significant cytotoxicity against a panel of tumor cell lines including HCT-116 and HepG2 with IC50 values <10 μg mL−1. This fungus was then subjected to large-scale fermentation, while chromatographic separation of the EtOAc fraction resulted in the isolation of 15 trichothecene-based sesquiterpenes (1–15) (Fig. 1).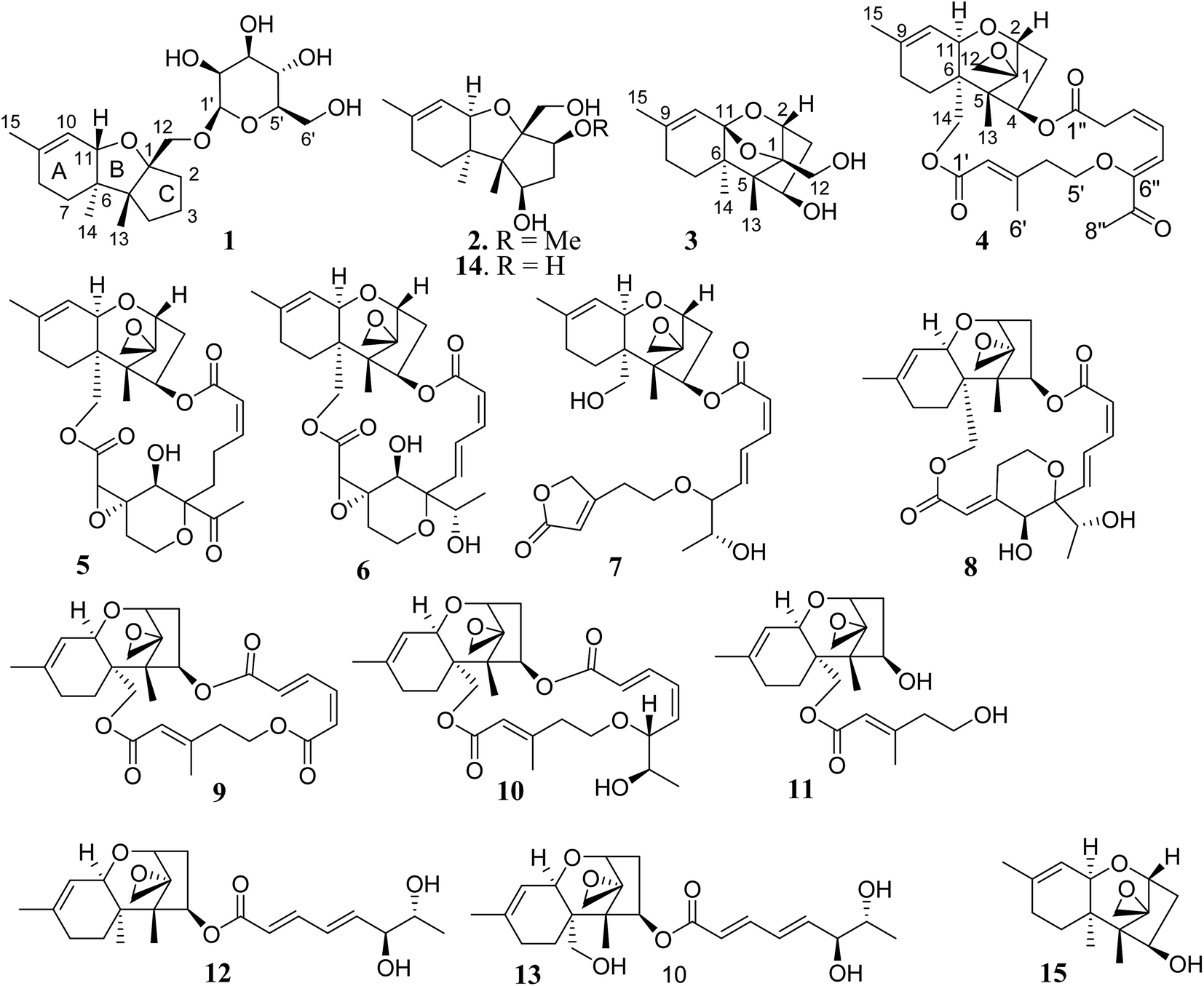 | ||
| Fig. 1 Structures of 1–15. | ||
2. Experimental section
2.1. General procedures
Optical rotations were measured with a Rudolph Autopol III automatic polarimeter. IR spectra were obtained with a Thermo Nicolet Nexus 470 FT-IR spectrometer. 1H and 13C NMR, and 2D NMR spectra were recorded on Bruker Avance 400 NMR and Bruker Avance 500 NMR spectrometers (400/500 MHz for 1H NMR and 100/125 MHz for 13C NMR, respectively). ESIMS and HRESIMS spectra were obtained with a Thermo Scientific LTQ Orbitrap XL instrument. Silica gels (160–200 and 200–300 mesh, Qingdao Marine Chemistry Co. Ltd) and ODS (50 μm, YMC) were used for column chromatography. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) were used for TLC analyses. HPLC was performed with an Alltech instrument (426-HPLC pump) equipped with an Alltech uvis-200 detector at 210 nm and semipreparative reversed-phased columns (YMC-packed C8, 5 μm, 250 mm × 10 mm).2.2. Microorganism
The fungal strain WGC-25C-6 was isolated from the sponge Niphates recondite, which was collected from the inner coral reef at a depth of 10 m in Beibuwan Bay, Guangxi Province of P. R. China, in August 2010. The fungal strain was identified by comparing the morphological character and 18S rDNA sequence with those of standard records. Specifically, the morphological examination was performed by scrutinizing the fungal culture, the mechanism of spore production, and the characteristics of the spores. For inducing sporulation, the fungal strain was separately inoculated on PDA. All experiments and observations were repeated at least twice leading to the identification of the strain as Stachybotrys chartarum. The fungal strain WGC-25C-6 was deposited at the State Key Laboratory of Natural and Biomimetic Drugs, Peking University, China.2.3. Fermentation
Large-scale fermentation was carried out in 30 Fernbach flasks (500 mL) each containing 80 g of rice. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.2.4. Extraction and isolation
The fermented material was extracted successively with EtOAc (3 × 500 mL). The EtOAc layer was evaporated to dryness under vacuum to afford a crude extract (30 g).The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay results indicated that the EtOAc extract had significant inhibitory effects against the tumor cell lines HCT-116 (IC50 < 10 μg mL−1) and HepG2 (IC50 < 10 μg mL−1). The EtOAc fraction (10 g) was subjected to silica gel vacuum liquid chromatography, eluting with CH2Cl2–MeOH (gradient from 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:0) to obtain 10 fractions (FA–FJ). Additional MTT assay indicated that the fractions FA and FC–FJ showed weak cytotoxicity toward HCT-116 and HepG2 cell lines with IC50 > 10 μg mL−1, whereas the fraction FB exhibited inhibitory effects against HCT-116 and HepG2 cell lines with IC50 < 5 μg mL−1. FB (2.2 g) was fractionated on a silica gel column eluting with CH2Cl2–MeOH (from 100:0 to 100:5) to afford three subfractions (SB1–SB3). SB3 (412.2 mg) was separated by column chromatography on ODS gel eluting with H2O–MeOH (from 1:1 to 0:1) to yield 2 (183.7 mg) and 3 (100.1 mg). Compounds 5 (199 mg), 4 (2.8 mg), 9 (5.3 mg), and 10 (9.0 mg) were purified from SB2 (325.5 mg) by semipreparative HPLC (C18) with a mobile phase of MeOH–H2O (4:1). FB2 (800 mg) was separated by column chromatography on silica gel (300 mesh) eluting with CH2Cl2–MeOH (50:1) to afford four subfractions (FB2-1 to FB2-4). FB2-3 (344.6 mg) was purified by Sephadex LH-20 with CHCl3–MeOH (1:1) to yield 14 (8.9 mg), 8 (16.9 mg), 13 (20.7 mg), 15 (90.2 mg), 11 (19.5 mg), and 7 (13.9 mg). FB2-4 (142.4 mg) was chromatographed on an ODS column eluting with MeOH–H2O (gradient from 1:1 to 1:0) to afford 12 (3.9 mg) and 6 (2.3 mg). FB2-2 (12.7 mg) was purified by semipreparative HPLC eluting with MeOH–H2O (3:2) to yield 1 (4.2 mg).
:1 to 1:0) to obtain 10 fractions (FA–FJ). Additional MTT assay indicated that the fractions FA and FC–FJ showed weak cytotoxicity toward HCT-116 and HepG2 cell lines with IC50 > 10 μg mL−1, whereas the fraction FB exhibited inhibitory effects against HCT-116 and HepG2 cell lines with IC50 < 5 μg mL−1. FB (2.2 g) was fractionated on a silica gel column eluting with CH2Cl2–MeOH (from 100:0 to 100:5) to afford three subfractions (SB1–SB3). SB3 (412.2 mg) was separated by column chromatography on ODS gel eluting with H2O–MeOH (from 1:1 to 0:1) to yield 2 (183.7 mg) and 3 (100.1 mg). Compounds 5 (199 mg), 4 (2.8 mg), 9 (5.3 mg), and 10 (9.0 mg) were purified from SB2 (325.5 mg) by semipreparative HPLC (C18) with a mobile phase of MeOH–H2O (4:1). FB2 (800 mg) was separated by column chromatography on silica gel (300 mesh) eluting with CH2Cl2–MeOH (50:1) to afford four subfractions (FB2-1 to FB2-4). FB2-3 (344.6 mg) was purified by Sephadex LH-20 with CHCl3–MeOH (1:1) to yield 14 (8.9 mg), 8 (16.9 mg), 13 (20.7 mg), 15 (90.2 mg), 11 (19.5 mg), and 7 (13.9 mg). FB2-4 (142.4 mg) was chromatographed on an ODS column eluting with MeOH–H2O (gradient from 1:1 to 1:0) to afford 12 (3.9 mg) and 6 (2.3 mg). FB2-2 (12.7 mg) was purified by semipreparative HPLC eluting with MeOH–H2O (3:2) to yield 1 (4.2 mg).
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| 1 | 95.2 C | 91.2 C | 95.8 C | |||
| 2 | 37.0 CH2 | 1.95, m; 2.00, m | 85.7 CH | 3.48, dd (J = 5.0, 8.8 Hz) | 80.2 CH | 4.06, dd (J = 2.0, 4.8 Hz) |
| 3 | 25.9 CH2 | 1.68, m; 1.65, m | 36.9 CH2 | 1.47, m; 2.08, m | 41.7 CH2 | 1.68, ddd (J = 4.8, 6.0, 16.0 Hz); 2.37, ddd (J = 2.0, 8.9, 16.0 Hz) |
| 4 | 36.9 CH2 | 1.30, m; 2.00, m | 69.2 CH | 3.89, ddd (J = 4.8, 5.0, 8.0 Hz) | 74.0 CH | 3.91, ddd (J = 6.0, 6.4, 8.9 Hz) |
| 5 | 56.1 C | 57.6 C | 54.9 C | |||
| 6 | 45.2 C | 42.7 C | 47.3 C | |||
| 7 | 27.5 CH2 | 1.36, ddd (J = 4.4, 5.0, 12.4 Hz); 1.52, ddd (J = 3.0, 12.0, 12.4 Hz) | 25.0 CH2 | 1.06, m; 1.85, m | 30.5 CH2 | 1.36, ddd (J = 4.8, 5.0, 12.8 Hz), 1.75, m |
| 8 | 29.3 CH2 | 1.96, m | 28.1 CH2 | 1.82, m; 1.96, m | 28.8 CH2 | 1.92, ddd (J = 3.0, 4.8, 14.0 Hz); 2.10, m |
| 9 | 134.7 C | 139.4 C | 143.9 C | |||
| 10 | 122.1 CH | 5.47, brs | 119.2 CH | 5.44, d (J = 3.2 Hz) | 117.5 CH | 5.26, s |
| 11 | 81.1 CH | 4.12, brs | 76.3 CH | 3.62, d (J = 3.2 Hz) | 107.1 C | |
| 12 | 71.8 CH2 | 3.31, d (J = 10.4 Hz); 3.88, d (J = 10.4 Hz) | 62.4 CH2 | 3.41, dd (J = 6.0, 12.0 Hz); 3.52, dd (J = 6.0, 12.0 Hz) | 58.1 CH2 | 3.69, d (J = 6.0 Hz) |
| 13 | 22.9 CH3 | 0.81, s | 10.1 CH3 | 0.93, s | 10.7 CH3 | 0.85, s |
| 14 | 15.8 CH3 | 0.99, s | 14.0 CH3 | 1.06, s | 14.5 CH3 | 0.75, s |
| 15 | 20.2 CH3 | 1.61, s | 23.5 CH3 | 1.65, s | 23.0 CH3 | 1.71, s |
| 1′ | 101.9 CH | 4.37, d (J = 2.5 Hz) | ||||
| 2′ | 71.0 CH | 3.62, dd (J = 2.5, 5.2 Hz) | ||||
| 3′ | 74.2 CH | 3.27, dd (J = 5.2, 7.0 Hz) | ||||
| 4′ | 67.6 CH | 3.23, t (J = 7.0 Hz) | ||||
| 5′ | 78.0 CH | 3.02, ddd (J = 6.0, 7.0, 10.0 Hz) | ||||
| 6′ | 61.9 CH2 | 3.66, dd (J = 6.0, 12.0 Hz) 3.44, dd (J = 10.0, 12.0 Hz) | ||||
| OH-4 | 4.50, d (J = 4.8 Hz) | 4.72, d (J = 6.4 Hz) | ||||
| OH-12 | 3.90, t (J = 6.0 Hz) | 4.98, t (J = 6.0 Hz) | ||||
| OMe | 58.2 CH3 | 3.26, s | ||||
2.5. Acidic hydrolysis of 1
Compound 1 (2 mg) was dissolved in acetone (2 mL) and then was treated with 6 N HCl (1 mL) and heated at 90 °C for 12 h. The mixture was dried under vacuum, and then dissolved in 5 mL H2O. After extraction by EtOAc, the monosaccharide was treated with (+)-2-octanol (0.2 mL) in the presence of trifluoroacetic acid (1 drop) in a stoppered reaction vial at 130 °C overnight. The mixture was evaporated to dryness and acetylated with Ac2O in pyridine. The acetylated (+)-2-octyl glycoside was analyzed by gas chromatography using corresponding authentic samples prepared from sugars. Comparison of the retention time of the sugar derivative derived from 1 (TR = 19.18 min) with those of standard samples indicated the sugar of 1 to be in agreement with D-mannose.2.6. X-ray crystallographic analysis
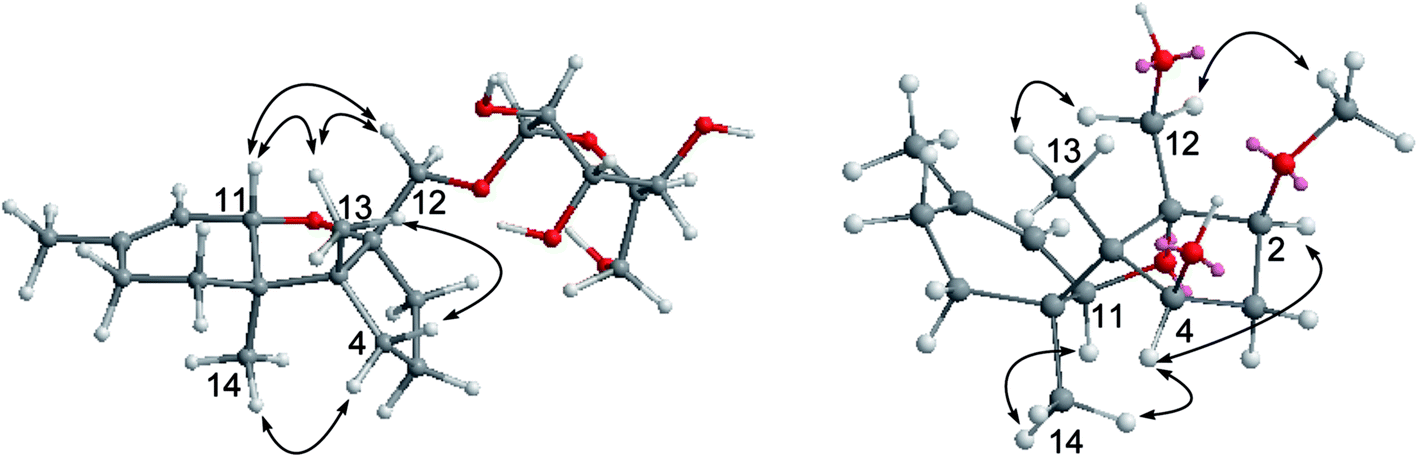
Crystal data were obtained with a Rigaku MicroMax 002+ single-crystal X-ray diffractometer. Cell parameter measurements and data collection were performed with a Bruker APEX2 CCD diffractometer using the wavelength for CuKα (λ = 1.5418 Å) radiation. Compounds were crystallized in polar space groups. The crystal structures of 3, 6, and 14 were solved by direct methods (SHELXS-97), and subsequent Fourier difference techniques (SHELEX-97, version 6.10, Bruker AXS Inc.). The crystallographic data for the structures of 3, 6, and 14 have been deposited with the Cambridge Crystallographic Data Centre (CCDC numbers: 1516457 for 3, 1516459 for 6, 1516458 for 14).†Colorless crystals of 3 were obtained from CH2Cl2/MeOH (1:1) using the vapor diffusion method. C15H22O4 (M = 266.34 g mol−1), orthorhombic, space group P212121, a = 6.4486(3) Å, b = 11.3707(10) Å, c = 17.7648(15) Å, α = 90°, β = 90°, γ = 90°, V = 1302.61(17) Å3, Z = 4, T = 98.8 K, μ(CuKα) = 0.792 mm−1, Dcalc = 1.3580 g cm−3, reflections measured (9.24° ≤ 2θ ≤ 143.6°), 2490 unique (Rint = 0.0191, Rsigma = 0.0324) which were used in all calculations. The final R1 was 0.0404 (I > 2σ(I)) and wR2 was 0.1049 (all data). Flack parameter = 0.2(2).
Colorless crystals of 6 were obtained from MeOH. C29H38.5O11.25·H2O (M = 567.10 g mol−1), monoclinic (0.40 × 0.35 × 0.35), space group P212121 (no. 4), a = 10.4204(3) Å, b = 13.0839(3) Å, c = 20.0354(7) Å, α = 90°, β = 90°, γ = 90°, V = 2731.60(14) Å3, Z = 4, T = 103(2) K, μ(CuKα) = 0.886 mm−1, Dcalc = 1.307 g cm−3, reflections measured (8.08° ≤ 2θ ≤ 143.52°), 5222 unique (Rint = 0.0176, Rsigma = N/A) which were used in all calculations. The final R1 was 0.0351 (I > 2σ(I)) and wR2 was 0.0925 (all data). Flack parameter = −0.03(13).
Colorless crystals of 14 were obtained from acetone/MeOH (1:1). C15H24O4 (M = 268.34 g mol−1), orthorhombic (0.40 × 0.20 × 0.15), space group P212121, a = 6.96010(16) Å, b = 7.10939(15) Å, c = 27.6935(7) Å, α = 90°, β = 90°, γ = 90°, V = 1370.33(5) Å3, Z = 4, T = 101.5 K, μ(CuKα) = 0.753 mm−1, Dcalc = 1.301 g cm−3, reflections measured (12.78° ≤ 2θ ≤ 143.78°), 2610 unique (Rint = 0.0170, Rsigma = 0.02601) which were used in all calculations. The final R1 was 0.0381 (I > 2σ(I)) and wR2 was 0.0980 (all data). Flack parameter = 0.01(16).
2.7. Cytotoxicity assay
Test samples were dissolved in dimethyl sulfoxide (DMSO) to make stock solutions and further diluted in culture medium upon assay. The cancer cell lines were cultured in RPMI 1640 medium, containing 10% fetal bovine serum. The cell lines were maintained at 37 °C in a humidified environment containing 5% CO2. To determine the effects of the compounds on cell viability, the cell number was quantified using a standard colorimetric MTT assay. Cells were plated in a 96-well plate (5 × 103 cells per well) and allowed to attach overnight. Cells were treated with 1, 2, 4, 8, and 10 μM of each compound in culture medium for 72 h. Then, new culture medium was added with 20 μL of MTT (5 mg mL−1 stock in PBS) per well and incubated for 4 h at 37 °C. Finally, the culture medium was discarded, and 150 μL of DMSO was added per well to dissolve the purple formazan crystals. Absorbance of the solution was measured using a microplate reader spectrophotometer (Bio-Rad Laboratories, Inc., Hercules, CA, USA), at a wavelength of 490 nm. The absorbance of untreated cells in the medium (negative control) was 100%. Paclitaxel was used as a positive control and showed cytotoxic activity with IC50 values of 0.03, 0.01, 0.01, 0.04, and 0.01 μM against the HCT-116, HepG2, BGC-823, NCI-H1650, and A2780 tumor cell lines, respectively. The compound with IC50 < 1 μM was further screened by the diluted concentrations of 0.1, 0.2, 0.4, and 0.8 μM, while the compound with IC50 < 0.1 μM was diluted to 0.01 μM for additional assay. The human malignant cell lines (colon carcinoma HCT-116, hepatocellular carcinoma HepG2, gastric carcinoma BGC-823, non-small cell lung carcinoma NCI-H1655, and ovarian carcinoma A2780) used in this study were purchased from the China Infrastructure of Cell Line Resources.2.8. Anti-kinase assay
Assay for protein kinase inhibition was performed in 96-well Flash Plates from PerkinElmer/NEN (Boston, MA) in a 50 μL reaction volume. The reaction cocktail contained 20 μL of assay buffer, 5 μL of ATP solution (in H2O), 5 μL of test compound (in 10% DMSO), 10 μL of substrate, and 10 μL of purified recombinant protein kinase. The assay for all enzymes contained 60 mM HEPES–NaOH (pH 7.5), 3 mM MgCl2, 3 mM MnCl2, 3 μM Na-orthovanadate, 1.2 mM DTT, 50 μg mL−1 PEG 20000, and 1 μM [γ-33P]-ATP (ca. 5 × 105 cpm per well). The human kinases FGFR3 (CD333), IGF1R (CD221), PDGFRb (CD140b), and TRKB (NTRK2) were obtained from Sino Biological Inc.3. Results and discussion
3.1. Structure elucidation of new compounds
Chartarene A (1) has a molecular formula of C21H34O7 as determined by HRESIMS and NMR data, containing five degrees of unsaturation. The 1H NMR spectrum exhibited the resonances of three methyl singlets (δH 0.81, 0.99, 1.61), an olefinic proton at δH 5.47 (brs, H-10), as well as a number of alkyl protons and hydroxylated methine and methylene protons. The 13C NMR spectrum showed a total of 21 carbon resonances, including two olefinic carbons at δC 134.7 (C-9) and 122.1 (C-10) for a double bond. Based on the 2D NMR data analyses, an apotrichothecene nucleus20 structurally related to apotrichodiol21 was established. This assignment was evidenced by the COSY correlations between H2-7 (δH 1.36, 1.52)/H2-8 (δH 1.96), H2-3 (δH 1.65, 1.68) to H2-2 (δH 1.95, 2.00) and H2-4 (δH 1.30, 2.00), in addition to the HMBC interactions from H3-15 (δH 1.61) to C-8 (δC 29.3), C-9 and C-10, H3-14 (δH 0.99) to C-5 (δC 56.1), C-6 (δC 45.2), C-7 (δC 27.5), and C-11 (δC 81.1), H3-13 (δH 0.81) to C-1 (δC 95.2), C-4 (δC 36.9), C-5 and C-6, and H2-12 (δH 3.31, 3.88) to C-1, C-2 (δC 37.0), and C-5. The HMBC interaction between H-11 (δH 4.12) and C-1 clarified an ether bridge between C-1 and C-11 (Fig. 2). Thus, a tricyclic core with methyl substitution at C-5, C-6 and C-9 and an oxymethylene at C-1, as well as a double bond at C-9 and C-10 was deduced. The remaining six oxygenated carbons (δC 101.9, 78.0, 74.2, 71.0, 67.6, 61.9) were attributed to a sugar moiety. The J values (JH-3′/H-4′ = 7.0 Hz, JH-4′/H-5′ = 7.0 Hz, JH-3′/H-2′ = 2.5 Hz) in association with the NOE interactions from H-1′ (δH 4.37) to H-3′ (δH 3.27) and H-5′ (δH 3.02) indicated axial orientation of H-1′, H-3′, H-4′, and H-5′, while H-2′ was in equatorial orientation. Acidic hydrolysis22 of 1 resulted in the generation of a sugar, which was identical to D-mannose based on the comparison of its HPLC retention time, specific rotation and NMR data with those of authentic sample. The linkage of sugar to C-12 of the tricyclic nucleus through an ether bond was determined by the HMBC interaction between H2-12 and C-1′ (δC 101.9). The NOE interactions from H3-13 to H-11, H2-12 and Hb-7, in association with the NOE relationship between H3-14 and Ha-7 (Fig. 3) indicated the trans junction of rings A and B and cis junction of rings B and C, while H3-13 and H3-14 were in opposite faces.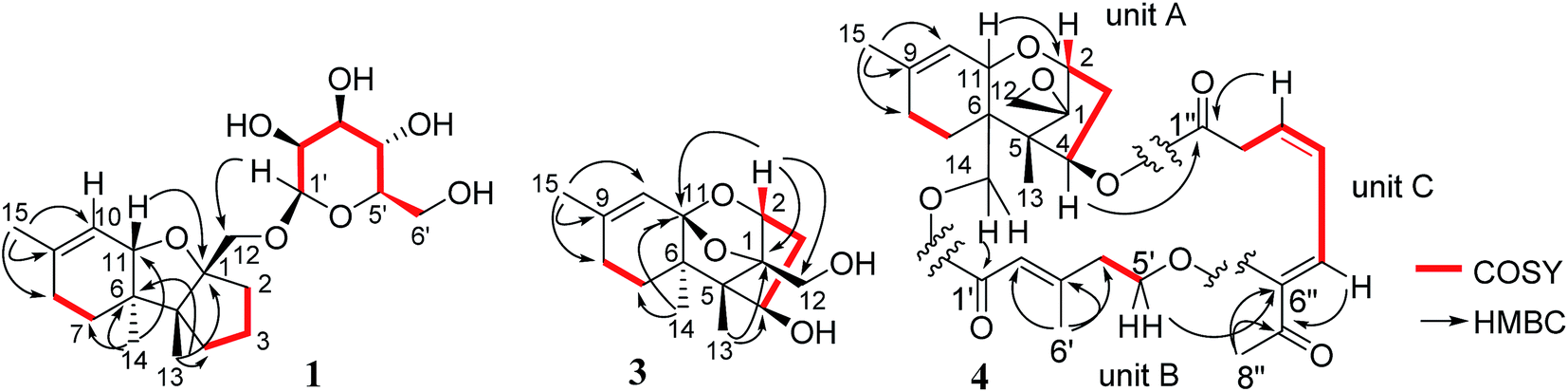 | ||
| Fig. 2 Key COSY and HMBC correlations of 1, 3 and 4. | ||
 | ||
| Fig. 3 Key NOE correlations of 1 and 2. | ||
The molecular formula of chartarene B (2) was determined as C16H26O4 based on the HRESIMS and NMR data (Table 1). Analyses of 2D NMR data (COSY, HMQC, and HMBC) established the planer structure of 2 possessing a tricyclic nucleus, which was the same as that of 1 with the exception of lack of sugar moiety and the presence of a methoxy group at ring C. The COSY correlations from H2-3 (δH 1.47, 2.08) to H-2 (δH 3.48) and H-4 (δH 3.89) in association with the HMBC correlations from H2-12 (δH 3.41, 3.52) to C-1 (δC 91.2), C-2 (δC 85.7) and C-5 (δC 57.6) and the HMQC correlations between H-2/C-2 and H-4/C-4 (δC 69.2) indicated the substitution of oxygen atom at C-2 and C-4. In addition, the HMBC correlation between H-2 and a methoxy carbon at δC 58.2 clarified a methoxy substitution at C-2. Thus, C-4 was located with a hydroxy group, and this assignment was supported by the COSY relationship between H-4 and OH-4 (δH 4.50). The NOE interactions between H-2/H-4 and H3-14/H-11, and from H2-12 to OMe and H3-13 (Fig. 3), determined the cis fusion of rings A/B and B/C, while H-2 and H-4 were oriented in the same face as H3-14.
The HRESIMS and NMR data indicated the molecular formula of chartarene C (3) to be C15H22O4, requiring five degrees of unsaturation. The 1D and 2D NMR data established a sambucinol-type analogue with the planer structure of 3 to be related to loukacinol A.5 The distinction was attributed to C-8, where a methylene group of 3 replacing a ketone of loukacinol A was evident from H2-8 (δH 1.92, 2.10) correlated to the olefinic carbons C-9 (δC 143.9) and C-10 (δC 117.5) in the HMBC spectrum. The similar NOE interactions between 3 and loukacinol A indicated both compounds possessing the same configurations. These assignments were confirmed by the analyses of the X-ray diffraction data using the Flack parameter (Fig. 4).
 | ||
| Fig. 4 X-ray structures of 3, 6, and 14. | ||
The molecular formula of chartarene D (4) was determined as C29H36O8 by HRESIMS (m/z 535.23084 [M + Na]+) and NMR data. Analyses of the 2D NMR data indicated the structure of 4 as consisting of three building blocks (units A–C). Unit A was identical to verrucarol23 based on comparable NMR data (Table 2) in association with the HMBC interactions from H3-15 (δH 1.66, s) to C-8 (δC 27.6) and both olefinic carbons C-9 (δC 139.2) and C-10 (δC 119.6), H3-13 (δH 0.69, s) to C-1 (δC 65.8), C-4 (δC 75.6), C-5 (δC 48.2), and C-6 (δC 42.8), as well as the epoxy protons H2-12 (δH 2.80, 3.07) to C-1, C-2 (δC 78.4) and C-5. Unit B was determined as a 5-oxy-3-methyl-2-pentenoic unit, based on the HMBC interactions from the methyl protons H3-6′ (δH 2.13, s) to C-2′ (δC 117.3), C-3′ (δC 157.6), and C-4′ (δC 41.1), olefinic proton H-2′ (δH 5.79, s) to the carbonyl carbons C-1′ (δC 165.7) and C-4′, in addition to the COSY relationship between H2-4′ and the oxymethylene H2-5′ (δH 3.77, 4.17). Subsequently, unit C was determined as a 6-oxygenated 7-oxo-3,5-octadienoic acid, based on the COSY correlations from H2-2′′ (δH 3.16, 3.35) to H-5′′ (δH 6.75, d, J = 10.0 Hz), in association with the HMBC interactions from H3-8′′ (δH 2.27, s) to a ketone C-7′′ (δC 195.3) and an olefinic carbon C-6′′ (δC 150.5) and from H-3′′ and H2-2′′ to a carbonyl carbon C-1′′. The linkage of units A and B by an ester bond across C-14 and C-1′ was deduced by the HMBC interaction between H2-14 (δH 3.83, 4.28) and C-1′, while the conjunction of units B and C through an ester bond across C-4 and C-1′′ was indicated by the HMBC correlation between H-4 (δH 5.98) and C-1′′ (δC 170.1) (Fig. 2). The connection of units B and C with an ether bond between C-5′ and C-6′′ was deduced by the HMBC relationship between H2-5′ (δH 3.77, 4.17) and C-6′′. Thus, 4 was established as a roridoid-type analogue, structurally related to roridin E with the migration of the olefinic bonds to C-3′′/C-4′′ and C-5′′/C-6′′ and the substitution of a ketone at C-7′′. The NOE interaction between H-2′ and H2-4′ in association with the chemical shift of the methyl carbon C-6′ (δC 18.3, <20 ppm) determined 2′E geometry. The JH-3′′/H-4′′ value (12.0 Hz) and the NOE interaction between H3-8′′ and H-5′′ confirmed 3′′Z and 5′′Z geometries. Acidic hydrolysis of 4 produced a product which was identical to verrucarol, confirming the stereogenic centers in moiety A of 4 to be the same as those of verrucarol.
| 4 | HMBC | ||
|---|---|---|---|
| δC | δH | ||
| 1 | 65.8 C | ||
| 2 | 78.4 CH | 3.68, d (J = 4.0 Hz) | C-1, C-4, C-5, C-11 |
| 3 | 42.6 CH2 | 1.88, m; 2.50 m | |
| 4 | 75.6 CH | 5.98, dd (J = 2.4, 6.4 Hz) | C-3, C-5, C-6, C-1′′ |
| 5 | 48.2 C | ||
| 6 | 42.8 C | ||
| 7 | 21.2 CH2 | 1.91, m; 1.94, m | |
| 8 | 27.6 CH2 | 1.98, m; 2.00, m | |
| 9 | 139.2 C | ||
| 10 | 119.6 CH | 5.37, d (J = 3.2 Hz) | C-6, C-8, C-11 |
| 11 | 66.6 CH | 3.87, d (J = 3.2 Hz) | C-6, C-9, C-10, C-2 |
| 12 | 47.7 CH2 | 2.80, d (J = 3.6 Hz); 3.07, d (J = 3.6 Hz) | C-1, C-2, C-5 |
| 13 | 7.25 CH3 | 0.69, s | C-1, C-3, C-4, C-5 |
| 14 | 63.7 CH2 | 3.83, d (J = 12.0 Hz); 4.28, d (J = 12.0 Hz) | C-5, C-6, C-7, C-11, C-1′ |
| 15 | 23.3 CH3 | 1.66, s | C-8, C-9, C-10 |
| 1′ | 165.7 C | ||
| 2′ | 117.3 CH | 5.79, s | C-1′, C-4′, C-6′ |
| 3′ | 157.6 C | ||
| 4′ | 41.1 CH2 | 2.40, m; 2.50, m | |
| 5′ | 68.5 CH2 | 3.77, m; 4.17, m | C-3′, C-4′, C-6′′ |
| 6′ | 18.3 CH3 | 2.13, s | C-3′, C-4′, C-5 |
| 1′′ | 170.1 C | ||
| 2′′ | 36.3 CH2 | 3.16, dd (J = 3.0, 12.0 Hz), 3.35, dd (J = 3.0, 12.0 Hz) | C-1′′, C-4′′ |
| 3′′ | 132.9 CH | 6.18, dt (J = 3.0, 16.0 Hz) | C-1′′, C-5′′ |
| 4′′ | 124.9 CH | 6.56, dd (J = 10.0, 16.0 Hz) | C-2′′, C-6′′ |
| 5′′ | 126.5 CH | 6.75, d (J = 10.0 Hz) | C-3′′, C-7′′ |
| 6′′ | 150.5 C | ||
| 7′′ | 195.3 C | ||
| 8′′ | 26.6 CH3 | 2.27, s | C-6′′, C-7′′ |
Moreover, the known compounds were identical to mytoxin A (5),24 satratoxin G (6),25 roridin L-2 (7),26 satratoxin H (8),27 muconomycin B (9),28 roridin E (10),29 verrol (11),30 trichodermadienediol B (12),31 isotrichoverrol B (13),32 2,4,12-trihydroxyapotrichothecene (14), and trichodermol (15),33 based on the comparison of their NMR and MS data in association with specific rotation with those reported in the literature. 2,4,12-Trihydroxyapotrichothecene (14) was isolated from a microorganism for the first time, while the absolute configurations of 14 and satratoxin G (6) were uncertain in the literature. In the present work, the absolute configurations of satratoxin G (6) and 2,4,12-trihydroxyapotrichothecene (14) were established by the analyses of the X-ray diffraction data using Flack parameters (Fig. 4).
3.2. Bioassay
Compounds 1–15 were tested against a panel of human tumor cell lines, including human colon carcinoma (HCT-116), human hepatocellular carcinoma (HepG2), gastric cancer (BGC-823), non-small-cell lung adenocarcinoma (NCI-H1655), and human ovarian cancer (A2780), by the MTT method.34 As shown in Table 3, all compounds exhibited selective inhibition against the tumor cell lines, while compounds 5, 6, 8, and 10 showed potent cytotoxic activities against the panel of tumor cell lines with IC50 values less than 10 nM. Preliminary analyses of the structure–activity relationship revealed that the cytotoxic effects are related to the scaffolds and the substitution. Apotrichothecene-based analogues with a sugar moiety at C-12 and without hydroxy group at C-2 and C-4 (1) or with a methoxy group at C-2 (2) showed weaker cytotoxicity in comparison with the 2,4,12-trihydroxy analogue (14). For the trichodermol-type analogues, those with a macrocyclic ring along with a tetrahydropyran ring (5, 6, 8) exerted remarkable effects, whereas the analogues without a tetrahydropyran ring in the macrocyclic moiety (4, 9) showed less cytotoxicity. However, compound 10 is an exception which showed IC50 within 10 nM, while the structural distinction of 4 and 10 was attributed to the olefinic rearrangement and the substitution at C-7′′ of the macrocyclic ring. Compounds 7 and 11–13 with a linear moiety at C-4 or C-14 had decreased inhibitory effects in comparison with 5.| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| HCT-116 | HepG2 | BGC-823 | NCI-H1650 | A2780 | |
| 1 | 3.39 ± 0.01 | 3.95 ± 0.01 | 2.87 ± 0.02 | >10 | 2.38 ± 0.02 |
| 2 | 5.58 ± 0.03 | >10 | >10 | >10 | >10 |
| 3 | 0.74 ± 0.02 | 2.09 ± 0.01 | >10 | 2.58 ± 0.03 | 2.07 ± 0.02 |
| 4 | 1.48 ± 0.01 | 0.90 ± 0.02 | 0.68 ± 0.04 | 1.23 ± 0.02 | 0.69 ± 0.03 |
| 5 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| 6 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| 7 | 1.73 ± 0.03 | 1.2 ± 0.02 | 1.81 ± 0.04 | 2.36 ± 0.03 | 1.61 ± 0.01 |
| 8 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| 9 | 1.32 ± 0.03 | 0.81 ± 0.03 | 0.84 ± 0.02 | 0.86 ± 0.01 | 0.68 ± 0.02 |
| 10 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| 11 | 2.77 ± 0.04 | 1.45 ± 0.02 | 2.33 ± 0.01 | 2.68 ± 0.01 | 2.00 ± 0.03 |
| 12 | 1.65 ± 0.03 | 0.86 ± 0.05 | 0.81 ± 0.03 | 1.31 ± 0.01 | 0.68 ± 0.04 |
| 13 | 3.48 ± 0.06 | 2.62 ± 0.03 | 2.64 ± 0.04 | 2.36 ± 0.02 | 2.12 ± 0.01 |
| 14 | 0.87 ± 0.05 | 0.69 ± 0.04 | 0.65 ± 0.04 | 0.84 ± 0.07 | 0.69 ± 0.03 |
| 15 | 7.22 ± 0.04 | 3.69 ± 0.04 | 2.55 ± 0.05 | 2.68 ± 0.06 | 2.68 ± 0.03 |
| Taxol | 0.03 | 0.01 | 0.01 | 0.04 | 0.01 |
In order to gain an insight into the mechanism of the antitumor-active compounds, a panel of tumor growth-related tyrosine kinases including FGFR3 (fibroblast growth factor receptor 3), IGF1R (insulin-like growth factor 1 receptor), PDGFRb (β-type platelet-derived growth factor receptor), and TRKB (tropomyosin receptor kinase B) were selected for testing. The bioassay results revealed that most compounds exerted potent inhibitory activities (Table 4), except for 1 and 13 showing weak activity with IC50 values of more than 20 μM.
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| FGFR3 | IGF1R | PDGFRb | TRKB | WT | |
| 1 | >25 | >25 | >25 | >25 | >25 |
| 2 | 2.4 | 6.9 | 10.4 | 7.0 | 12.9 |
| 3 | 1.1 | 3.0 | 5.3 | 2.7 | 6.3 |
| 4 | 0.1 | 0.1 | 0.8 | 0.7 | 0.7 |
| 5 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| 6 | 0.1 | 0.1 | 0.5 | 0.2 | 0.9 |
| 7 | 0.4 | 0.5 | 2.3 | 1.9 | 1.7 |
| 8 | <0.1 | <0.1 | <0.1 | <0.1 | 0.1 |
| 9 | 0.3 | 0.4 | 1.2 | 0.7 | 1.5 |
| 10 | 0.4 | 0.4 | 1.4 | 1.0 | 2.1 |
| 11 | 0.1 | 0.2 | 0.7 | 0.4 | 0.9 |
| 12 | 0.5 | 0.7 | 1.9 | 1.0 | 1.9 |
| 13 | 24.0 | >25 | >25 | >25 | >25 |
| 14 | 0.5 | 1.0 | 2.1 | 1.0 | 2.2 |
| 15 | 0.9 | 0.8 | 3.1 | 1.8 | 3.0 |
FGF signaling plays a role in various aspects of cancer biology, involving proliferation, anti-apoptosis, drug resistance, angiogenesis, EMT, and invasion; thus FGFR-targeted therapeutics is an active topic in the field of clinical oncology.35,36 IGFIR regulates the tumor microenvironment to promote cancer progression and metastasis,37,38 while PDGFR inhibition is an antiangiogenic approach potentially affecting all solid tumors due to tumor vasculature having a pericyte coverage.39,40 Meanwhile, high levels of TrkB are correlated with poor patient prognosis in neuroblastomas and ovarian, pancreatic, colon, prostate, and gastric cancers, while TrkB is overexpressed in ovarian adenocarcinoma metastases compared with primary lesions.41,42 The potent inhibition toward the spectrum of tyrosine kinases suggested that the antitumor effects of the trichothecene-type derivatives were mediated by multiple targets.
4. Conclusion
Trichothecene-type sesquiterpenoids are frequently found in Stachybotrys species in different terrestrial localities, while the unique scaffold of trichothecenes may be considered as chemotaxonomic markers. This work firstly reported the sponge-derived Stachybotrys chartarum to be a new fungal strain of functionalized trichothecenes. Trichothecene with a sugar linkage such as 1 was found from nature for the first time. The potent cytotoxic effects of the isolated compounds against a panel of tumor cell lines are induced by the inhibition of the multiple kinase targets FGFR3, IGF1R, PDGFRb, and TRKB. These findings provided additional data indicating that trichothecenes are promising kinase inhibitors for the development of antitumor leads.Acknowledgements
This work was supported by grants of National High Technology and Science 973 program (2015CB755906), NSFC (81630089, 41376127) and NSFC-Shangdong Joint Fund for Marine Science (U1406402).References
- K. F. Nielsen, T. Grafenhan, D. Zafari and U. Thrane, J. Agric. Food Chem., 2005, 53, 8190–8196 CrossRef CAS PubMed.
- M. Namikoshi, K. Akano, S. Meguro, I. Kasuga, Y. Mine, T. Takahashi and H. Kobayashi, J. Nat. Prod., 2001, 64, 396–398 CrossRef CAS.
- T. Lin, G. Wang, Y. Zhou, D. Zeng, X. Liu, R. Ding, X. Jiang, D. Zhu, W. Shan and H. Chen, J. Agric. Food Chem., 2014, 62, 5993–6000 CrossRef CAS PubMed.
- M. P. Carvalho, H. Weich and W. R. Abraham, Curr. Med. Chem., 2016, 23, 23–35 CrossRef PubMed.
- A. Loukaci, O. Kayser, K. U. Bindseil, K. Siems, J. Frevert and P. M. Abreu, J. Nat. Prod., 2000, 63, 52–56 CrossRef CAS.
- W. O. Godtfredsen and S. Vangedal, Acta Chem. Scand., 1965, 19, 1088–1102 CrossRef CAS PubMed.
- L. H. Senter, D. R. Sanson, D. G. Corley, M. S. Tempesta, A. A. Rottinghaus and G. E. Rottinghaus, Mycopathologia, 1991, 113, 127–131 CrossRef CAS PubMed.
- S. P. McCormick, T. Kato, C. M. Maragos, M. Busman, V. M. Lattanzio, G. Galaverna, C. Dall-Asta, D. Crich, N. P. Price and C. P. Kurtzman, J. Agric. Food Chem., 2015, 63, 731–738 CrossRef CAS PubMed.
- T. Suzuki and Y. Iwahashi, Toxins, 2014, 6, 453–463 CrossRef PubMed.
- H. K. Abbas, T. Yoshizawa and W. T. Shier, Toxicon, 2013, 74, 68–75 CrossRef CAS PubMed.
- J. W. He, G. S. Bondy, T. Zhou, D. Caldwell, G. J. Boland and P. M. Scott, Food Chem. Toxicol., 2015, 84, 250–259 CrossRef CAS PubMed.
- C. Arunachalam and F. M. Doohan, Toxicol. Lett., 2013, 217, 149–158 CrossRef CAS PubMed.
- L. O. Zamir, A. Nikolakakis, F. Sauriol and O. Mamer, J. Agric. Food Chem., 1999, 47, 1823–1835 CrossRef CAS PubMed.
- J. Merhej, F. Richard-Forget and C. Barreau, Appl. Microbiol. Biotechnol., 2011, 91, 519–528 CrossRef CAS PubMed.
- C. H. Diggs, M. J. Scoltock and P. H. Wiernik, Cancer Clin. Trials, 1978, 1, 297–299 CAS.
- D. W. Anderson, R. M. Black, C. G. Lee, C. Pottage, R. L. Rickard, M. S. Sandford, T. D. Webber and N. E. Williams, J. Med. Chem., 1989, 32, 555–562 CrossRef CAS PubMed.
- W. Ye, Y. Chen, H. Li, W. Zhang, H. Liu, Z. Sun, T. Liu and S. Li, Molecules, 2016, 21(781), 1–11 Search PubMed.
- B. B. Jarvis, J. O. Midiwo and E. P. Mazzola, J. Med. Chem., 1984, 27, 239–244 CrossRef CAS PubMed.
- K. Konishi, A. Iida, M. Kaneko, K. Tomioka, H. Tokuda, H. Nishino and Y. Kumeda, Bioorg. Med. Chem., 2003, 11, 2511–2518 CrossRef CAS PubMed.
- L. O. Zamir, K. A. Devor, A. Nikolakakis, Y. Nadeau and F. Sauriol, Tetrahedron Lett., 1992, 33, 5181–5184 CrossRef CAS.
- L. O. Zamir, K. A. Devor, Y. Nadeau and F. Sauriol, J. Biol. Chem., 1987, 262, 15354–15358 CAS.
- S. G. Ma, Y. C. Hu, S. S. Yu, Y. Zhang, X. G. Chen, J. Liu and Y. X. Liu, J. Nat. Prod., 2008, 71, 41–46 CrossRef CAS PubMed.
- J. Gutzwiller and C. Tamm, Helv. Chim. Acta, 1963, 46, 1786–1790 CrossRef CAS.
- B. B. Jarvis, F. T. Comezoglu, Y. W. Lee, J. L. Flippen-Anderson, R. D. Gilardi and C. F. George, Bull. Soc. Chim. Belg., 1986, 95, 681–697 CrossRef CAS.
- R. M. Eppley, E. P. Mazzola, M. E. Stack and P. A. Dreifuss, J. Org. Chem., 1980, 45, 2522–2523 CrossRef CAS.
- R. J. Bloem, T. A. Smitka, R. H. Bunge, J. C. French and E. P. Mazzola, Tetrahedron Lett., 1983, 24, 249–252 CrossRef CAS.
- R. M. Eppley, E. P. Mazzola, R. J. Highet and W. J. Bailey, J. Org. Chem., 1977, 42, 240–243 CrossRef CAS PubMed.
- J. S. Vittimberga and B. M. Vittimberga, J. Org. Chem., 1965, 30, 746–748 CrossRef CAS PubMed.
- P. Traxler, W. Zuercher and C. Tamm, Helv. Chim. Acta, 1970, 53, 2071–2085 CrossRef CAS PubMed.
- B. B. Jarvis, V. M. Vrudhula, J. O. Midiwo and E. P. Mazzola, J. Org. Chem., 1983, 48, 2576–2578 CrossRef CAS.
- R. Ren, C. Chen, X. Liu, H. Ge, R. Tan and R. Jiao, Helv. Chim. Acta, 2015, 98, 1435–1441 CrossRef CAS.
- B. B. Jarvis, S. Wang and H. L. Ammon, J. Nat. Prod., 1996, 59, 254–261 CrossRef CAS PubMed.
- T. A. Smitka, R. H. Bunge, R. J. Bloem and J. C. French, J. Antibiot., 1984, 37, 823–828 CrossRef CAS PubMed.
- J. B. Burton, Methods Mol. Med., 2005, 110, 69–78 CAS.
- M. Katoh and H. Nakagama, Med. Res. Rev., 2014, 34, 280–300 CrossRef CAS PubMed.
- P. S. Lee and A. A. Secord, Cancer Treat. Rev., 2014, 40, 507–512 CrossRef PubMed.
- H. Werner and C. T. Roberts Jr, Genes, Chromosomes Cancer, 2003, 36, 113–120 CrossRef CAS PubMed.
- D. C. Yang, R. L. Elliott and J. F. Head, Expert Opin. Ther. Targets, 2002, 6, 375–385 CrossRef CAS PubMed.
- J. S. Huang, S. S. Huang, B. Kennedy and T. F. Deuel, J. Biol. Chem., 1982, 257, 8130–8136 CAS.
- H. N. Antoniades, Baillieres Clin. Endocrinol. Metabol., 1991, 5, 595–613 CrossRef CAS.
- H. Dawson, S. Grundmann, V. H. Koelzer, J. A. Galván, R. Kirsch, E. Karamitopoulou, A. Lugli, D. Inderbitzin and I. Zlobec, Histopathology, 2015, 66, 715–725 CrossRef PubMed.
- K. W. Sinkevicius, C. Kriegel, K. J. Bellaria, J. Lee, A. N. Lau, K. T. Leeman, P. Zhou, A. M. Beede, C. M. Fillmore, D. Caswell, J. Barrios, K. K. Wong, L. M. Sholl, T. M. Schlaeger, R. T. Bronson, L. R. Chirieac, M. M. Winslow, M. C. Haigis and C. F. Kim, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10299–10304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1516457–1516459. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26956g |
| This journal is © The Royal Society of Chemistry 2017 |