
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel valproic aminophenol amides with enhanced glial cell viability effect†
Andrea Alpuche-García,
Xochitl Dávila-González,
Leticia Arregui * and
Hiram I. Beltrán*
* and
Hiram I. Beltrán*
Departamento de Ciencias Naturales, DCNI, UAM Cuajimalpa, 05300, Ciudad de México, Mexico. E-mail: arregui@correo.cua.uam.mx; hbeltran@correo.cua.uam.mx
First published on 21st February 2017
Abstract
New valproic acid derivatives were synthesized by coupling valproyl chloride with ortho-aminophenols, resulting in seven N-(ortho-hydroxyphenyl)valproamides. These amides share similar structural characteristics and exhibit tuneable electronic and steric contributions either without particular substituents, or through the inclusion of electro-donating (–Me), electro-withdrawing (–NO2) or pi electro-donating/sigma electro-withdrawing (–Cl) substituents at the aromatic ring. The identity of such derivatives was evidenced through spectroscopic characterization using FTIR, 1H, 13C and HETCOR NMR, as well as by analyzing their melting points. In particular, for three derivatives it was feasible to determine their chemical structures in the crystal phase; all three behaved in a similar fashion and exhibited very similar conformations independent of the attached substituents. The base compound was found to exhibit 15.8 times more activity in C6 cells and 4.4 times more activity in U373 cells compared with VPA. In general, the parent compound, or those having –Me as a substituent, presented a greater effect on C6 cells than U373 cells. However, those with –NO2 and –Cl substituents, as well as VPA, required similar doses for the IC50 in both cell lines. Modification of the base compound with a –Me or –NO2 substituent increased the effect on cell viability to ca. 20 times that of VPA in both C6 and U373, indicating that a larger structure causes an important enhancement in the inhibition of cell viability. In both cell lines, –Cl containing derivatives were the most active compounds. For these derivatives, an activity increase of ca. 59 and 47 times that of VPA was observed for C6 and U373 cells, respectively. An important perspective is that VPA analogues possessing an aromatic ring with a –Cl substituent may become central structures in the search for more potent pharmaceutical prototypes.
Introduction
Valproic acid (VPA) is a fatty acid with anticonvulsant properties used in the treatment of epilepsy, and this drug also has antitumor activity due to its effects as a histone deacetylase inhibitor.1 The occurrence of many types of cancer are generally accompanied by histone hypoacetylation.2 Because of this, many new strategies to fight cancer are focused on the restoration of de-acetylation balance through the use of histone deacetylase inhibitors (iHDAC) such as VPA.3–6 Nevertheless, despite the promising effects offered by VPA against cancer, it causes side effects mainly related to blood and bone marrow toxicity, and high concentrations are required to achieve the desired pharmacological activity, mostly in the mM range.7Glioblastoma is the most common and devastating primary tumour in adults. Epigenetic changes together with genetic modifications have been associated with its generation and progression. VPA and other iHDAC have been explored for glioblastoma treatment alone or with chemotherapy or radiotherapy, and promising effects have been found. However, additional efforts to find more effective drugs are required, in particular those involving the testing of drugs in glioblastoma cell lines, given the previously reported evidence.8
In order to diminish or avoid the side effects associated with VPA, as well as to enhance its activity, a common and useful strategy has been the chemical modification of the parent compound. For instance, in 2005, Eyal et al.9 evaluated and compared the iHDAC of VPA and its constituent isomers, including valnoctic acid (VCA), propylisopropylacetic acid (PIA), diisopropylacetic acid (DIA), 2,2,3,3-tetramethylcyclopropylcarboxylic acid (TMCA), and VPA metabolites: 2-en-AVP and 4-en-AVP. The authors found that almost any chemical modification of the VPA structure results in the decay of its activity as iHDAC. Another strategy, developed in 2006 by Deubzer and co-workers,10 was the employment of VPA analogues with longer side chains, as well as the inclusion of alkenyl fragments in the structure. These changes caused multifunctional properties of such derivatives as iHDAC, showing interesting cell cycle modulation due to the induction of p21 expression, as well as low toxicity on CD34+ bone marrow cells.10 Another recent effort to enhance the beneficial effects of VPA derivatives was carried out using phosphovalproic acid, employed as iHDAC, which involved tracking its dominant molecular target STAT3. This compound includes ester functionality to anchor the VPA fragment to the diethyl (4-hydroxybenzyl) phosphate parent compound. This compound has shown synergistic behaviour in enhanced pancreatic cancer inhibition, compared with VPA.11 Lastly, in 2009, the compound N-9-(2-hydroxy)ethoxymethylguanine disubstituted with VPA was patented12 and in 2014 it was evaluated by Tarasenko and co-workers13 as an anticancer agent in various cell lines with promising results. In particular, ester and amide groups link this compound to the VPA moiety, indicating that these functionalities should work to generate new promising derivatives of VPA. Some other trials to find potent VPA analogues with enhanced iHDAC activity have also emerged lately.14
Thus, the current work aims to obtain and characterize seven new VPA derivatives, wherein the molecular design strategy is not to vary the valproic fragment, but instead the carboxylate moiety, through the formation of amidic functionalities. This change is accompanied by the inclusion of an aromatic ring containing different electro-withdrawing or electro-releasing substituents in order to modulate bioactivities.15 Finally, the new structures recover the OH fragment present in VPA, through the inclusion of a phenolic group. The results of this contribution are that the seven valproic amides exhibit an enhanced effect on cell viability compared with VPA in two glioma cell lines.
Experimental section
General information
Available solvents and reagents were used as received. 2-Propylpentanoic acid (VPA, CAT P6273-100ML), 2-amino-p-cresol (APh2, CAT 144908-50G), 2-amino-5-methyl-phenol (APh3, CAT 144916-10G), 2-amino-4-nitro-phenol (APh4, CAT A70402-5G), 2-amino-5-nitro-phenol (APh5, CAT 303585-25G), 2-amino-4-chloro-phenol (APh6, CAT C44400-100G) and 2-amino-5-chloro-phenol (APh7, CAT 552224-25G) were acquired from Sigma-Aldrich; 2-aminophenol (APh1, CAT 09110-100G) was obtained from Fluka. The solvents employed were acetonitrile (CH3CN, CAT 9011-03-4L), tetrahydrofuran (THF, CAT 9450-03-4L), ethyl acetate (AcOEt, CAT 75-05-8-2L) and hexane (Hex, CAT 930L) and were acquired from Reasol.1D & 2D nuclear magnetic resonance spectroscopy (1D & 2D NMR)
NMR experiments were performed on a VARIAN Mercury 200-BB spectrometer and on an Anasazi spectrometer EFT-60. The 1H and 13C chemical shifts [δ = ppm] were taken as relative to internal SiMe4 (TMS, δ(1H) = 0.00 ppm, δ(13C) = 0.00 ppm), and the coupling constants were quoted in Hz. The employed solvent was CDCl3, and just for the compounds AV4 and AV5, 0.1 mL of DMSO-d6 was added to achieve full dissolution due to the presence of nitro groups.Fourier transform infrared (FT-IR) spectra
The FT-IR spectra were recorded in the range of 4000–400 cm−1 on a Bruker Tensor-27 FT-IR spectrophotometer, using KBr pellets or KBr mixtures of each of the isolated samples. The samples were measured using the ATR technique.Single crystal X-ray diffraction (X-ray)
X-ray diffraction data were acquired on an Enraf Nonius diffractometer FR590 Kappa-CCD (λMoKa = 0.71073 Å, graphite monochromator, T = 293 K, using rotating image scan with CCD). Monocrystals were mounted in LINDEMAN tubes. Reflections were corrected for Lorentz and polarization effects. The SHELX-S and SHELX-L 2014 program suite16 was used for the first structural solution, as well as for the refinement and output data. These programs, as well as the molecular perspectives, were employed and obtained in the OLEX2 environment.17 All heavy atoms were found through electronic density differences in the Fourier maps and refined anisotropically. Some of the hydrogen atoms were also found through Fourier maps and refined isotropically, and the remnants were geometrically calculated and added for the final refinement.Thin layer chromatography (TLC)
Thin layer chromatography (TLC) aluminium sheet plates of silica 60 F254 of 0.2 mm width (Merck, KGaA) were employed to track the course of each reaction. The TLC separation of each compound was developed using a hexane/ethyl acetate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture and the plates were revealed using a UV-vis lamp. In all obtained VA products, the appearance of single spots was indicative of their purity and also confirmed that the desired reaction had been achieved.
:1 solvent mixture and the plates were revealed using a UV-vis lamp. In all obtained VA products, the appearance of single spots was indicative of their purity and also confirmed that the desired reaction had been achieved.
Synthetic methodology for compounds VA1–7
:4 solvent mixture. In this way it was feasible to obtain compound VA1 in a yield of 64.8%. Similar to VA1, the other six valproic amides (VA2–7) were prepared by just varying the corresponding ortho-aminophenol source. As stated in a very recent contribution, this synthetic strategy solely provided N-acylation of the carboxylate moiety, instead of O-acylation.19![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N–), 1545, 1497 (–C(O)N–), 1387, 1313, 1264, 772, 750, 722, 693. 1H NMR (δ = ppm, CDCl3, J = Hz): 8.95, 8.73 (OH, sb, 1H & NH, s, 1H), 7.44 (H3, d, Jo = 7.25, 1H), 7.02 (H5, t, Jo = 7.25, 1H), 6.95 (H4, t, Jo = 7.25, 1H), 6.83 (H6, d, Jo = 7.25, 1H), 2.46 (H8, q, J = 6.42, 1H), 1.49 (H9, m, 4H), 1.23 (H10, m, 4H), 0.92 (H11, t, J = 6.42, 6H). 13C NMR (δ = ppm, CDCl3): 177.3 (C7, 1C), 148.2 (C1, 1C), 126.5 (C2, 1C), 126.0 (C5, 1C), 122.4 (C4, 1C), 120.4 (C3, 1C), 118.4 (C6, 1C), 48.0 (C8, 1C), 35.4 (C9, 2C), 20.8 (C10, 2C), 14.1 (C11, 2C).O)–N–) 1541, 1507 (–C(O)N–), 1358, 1317, 1267, 1259, 1034, 1015, 949, 868, 821, 780, 766, 727, 713. NMR 1H (δ = ppm, CDCl3, J = Hz): 9.20, 8.94 (OH, sb, 1H & NH, s, 1H), 7.37 (H3, s, 1H), 6.83–6.81 (H5, H6, m, 2H), 2.48 (H8, q, J = 7.3, 1H), 2.24 (H12, s, 3H), 1.55 (H9, m, 4H), 1.36 (H10, m, 4H), 0.92 (H11, t, J = 7.3, 6H). NMR 13C (δ = ppm, CDCl3): 175.9 (C7, 1C), 145.5 (C1, 1C), 128.8 (C4, 1C), 125.8 (C2, 1C), 125.8 (C5, 1C), 122.0 (C3, 1C), 117.2 (C6, 1C), 47.3 (C8, 1C), 35.2 (C9, 2C), 20.5 (C10, 2C), 20.4 (C12, 1C), 15.9 (C11, 2C). Suitable monocrystals for the X-ray diffraction analysis were obtained in a 4:1 hexane/ethyl acetate solvent mixture.O)–N–), 1598, 1546, 1510 (–C(O)N–), 1469, 1459, 1322, 943, 897, 872, 815, 796, 771, 760, 733. NMR 1H (δ = ppm, CDCl3, J = Hz): 8.90, 8.45 (OH, sb, 1H & NH, s, 1H), 7.16 (H3, d, Jo = 7.02, 1H), 6.82 (H6, s, 1H), 6.65 (H4, d, Jo = 7.02, 1H), 2.49 (H8, q, J = 6.72, 1H), 2.27 (H12, s, 3H), 1.48 (H9, m, 4H), 1.29 (H10, m, 4H), 0.92 (H11, t, J = 6.72, 6H). NMR 13C (δ = ppm, CDCl3): 177.0 (C7, 1C), 148.4 (C1, 1C), 136.9 (C5, 1C), 123.4 (C2, 1C), 122.3 (C3, 1C), 121.1 (C4, 1C), 119.4 (C6, 1C), 47.9 (C8, 1C), 35.4 (C9, 2C), 20.8 (C12, 1C), 20.0 (C10, 2C), 14.2 (C11, 2C). Suitable monocrystals for the X-ray diffraction analysis were obtained in a 4:1 hexane/ethyl acetate solvent mixture.O)–N–), 1539, 1503 (–C(O)N–), 1465, 1461, 1427, 1400, 1341 (–N(O)–O), 1327, 1268, 1244, 1197, 1167, 1153, 993, 878, 819, 781, 743. NMR 1H (δ = ppm, CDCl3, J = Hz): 9.22, 8.86 (OH, sb, 1H & H3, d, Jm = 2.74, 1H), 8.82 (NH, s, 1H), 7.88 (H5, dd, Jo = 8.96, Jm = 2.74, 1H), 6.98 (H6, d, Jo = 8.96, 1H), 2.52 (H8, q, J = 7.14, 1H), 1.50 (H9, m, 4H), 1.38 (H10, m, 4H), 0.92 (H11, t, J = 7.14, 6H). NMR 13C (δ = ppm, CDCl3): 176.3 (C7, 1C), 153.4 (C1, 1C), 140.2 (C4, 1C), 126.7 (C2, 1C), 120.8 (C5, 1C), 116.8 (C3, 1C), 116.0 (C6, 1C), 47.8 (C8, 1C), 35.1 (C9, 2C), 20.6 (C10, C2), 14.0 (C11, 2C).O)–N–), 1592, 1506 (–C(O)N–), 1465, 1424, 1400, 1346 (–N(O)–O), 1327, 1268, 1247, 1194, 896, 875, 825, 880, 743. NMR 1H (δ = ppm, CDCl3, J = Hz): 9.07, 8.35 (OH, sb, 1H & NH, s, 1H), 8.20 (H3, d, Jo = 8.88, 1H), 7.76 (H4, d, Jo = 8.88, 1H), 7.63 (H6, s, 1H), 2.43 (H8, q, J = 7.14, 1H), 1.48 (H9, m, 4H), 1.36 (H2, m, 4H), 0.92 (H11, t, J = 7.14, 6H). NMR 13C (δ = ppm, CDCl3): 175.0 (C7, 1C), 146.1 (C1, 1C), 142.5 (C5, 1C), 132.6 (C2, 1C), 118.7 (C3, 1C), 114.7 (C4, 1C), 109.5 (C6, 1C), 47.1 (C8, 1C), 34.5 (C9, 2C), 19.9 (C10, C2), 13.4 (C11, 2C).O)–N–), 1541, 1524 (–C(O)N–), 1291, 867, 821 (C–Cl). NMR 1H (δ = ppm, CDCl3): 8.17, 7.86 (OH, s, 1H & NH, s, 1H), 7.40 (H3, s, 1H), 6.95 (H5, d, 1H), 6.95 (H6, d, 1H), 2.35 (H8, q, J = 6.60, 1H), 1.45 (H9, m, 4H), 1.20 (H10, m, 4H), 0.92 (H11, t, J = 6.60, 6H). NMR 13C (δ = ppm, CDCl3): 177.3 (C7, 1C), 147.0 (C1, 1C), 127.0 (C5, 1C), 126.3 (C2, 1C), 125.0 (C4, 1C), 121.9 (C3, 1C), 119.5 (C6, 1C), 48.3 (C8, 1C), 35.5 (C9, 2C), 21.0 (C10, C2), 14.2 (C11, 2C). Suitable monocrystals for the X-ray diffraction analysis were obtained in a 4:1 hexane/ethyl acetate solvent mixture.O)–N–), 1531, 1495 (–C(O)N–) 919, 896, 807 (C–Cl). NMR 1H (δ = ppm, CDCl3, J = Hz): 8.50, 7.95 (OH, s, 1H & NH, s, 1H), 7.18 (H3, d, Jo = 8.40, 1H), 7.00 (H6, d, Jm = 2.04, 1H), 6.80 (H4, dd, Jo = 8.40, Jm = 2.04, 1H), 2.36 (H8, q, J = 6.60, 1H), 1.47 (H9, m, 4H), 1.37 (H10, m, 4H), 0.92 (H11, t, J = 6.60, 6H). NMR 13C (δ = ppm, CDCl3): 177.4 (C7, 1C), 149.1 (C1, 1C), 131.9 (C5, 1C), 124.8 (C2, 1C), 123.1 (C3, 1C), 120.6 (C4, 1C), 119.3 (C6, 1C), 48.4 (C8, 1C), 35.5 (C9, 2C), 21.0 (C10, C2), 14.3 (C11, 2C).
O)–N–), 1545, 1497 (–C(O)N–), 1387, 1313, 1264, 772, 750, 722, 693. 1H NMR (δ = ppm, CDCl3, J = Hz): 8.95, 8.73 (OH, sb, 1H & NH, s, 1H), 7.44 (H3, d, Jo = 7.25, 1H), 7.02 (H5, t, Jo = 7.25, 1H), 6.95 (H4, t, Jo = 7.25, 1H), 6.83 (H6, d, Jo = 7.25, 1H), 2.46 (H8, q, J = 6.42, 1H), 1.49 (H9, m, 4H), 1.23 (H10, m, 4H), 0.92 (H11, t, J = 6.42, 6H). 13C NMR (δ = ppm, CDCl3): 177.3 (C7, 1C), 148.2 (C1, 1C), 126.5 (C2, 1C), 126.0 (C5, 1C), 122.4 (C4, 1C), 120.4 (C3, 1C), 118.4 (C6, 1C), 48.0 (C8, 1C), 35.4 (C9, 2C), 20.8 (C10, 2C), 14.1 (C11, 2C).O)–N–) 1541, 1507 (–C(O)N–), 1358, 1317, 1267, 1259, 1034, 1015, 949, 868, 821, 780, 766, 727, 713. NMR 1H (δ = ppm, CDCl3, J = Hz): 9.20, 8.94 (OH, sb, 1H & NH, s, 1H), 7.37 (H3, s, 1H), 6.83–6.81 (H5, H6, m, 2H), 2.48 (H8, q, J = 7.3, 1H), 2.24 (H12, s, 3H), 1.55 (H9, m, 4H), 1.36 (H10, m, 4H), 0.92 (H11, t, J = 7.3, 6H). NMR 13C (δ = ppm, CDCl3): 175.9 (C7, 1C), 145.5 (C1, 1C), 128.8 (C4, 1C), 125.8 (C2, 1C), 125.8 (C5, 1C), 122.0 (C3, 1C), 117.2 (C6, 1C), 47.3 (C8, 1C), 35.2 (C9, 2C), 20.5 (C10, 2C), 20.4 (C12, 1C), 15.9 (C11, 2C). Suitable monocrystals for the X-ray diffraction analysis were obtained in a 4:1 hexane/ethyl acetate solvent mixture.O)–N–), 1598, 1546, 1510 (–C(O)N–), 1469, 1459, 1322, 943, 897, 872, 815, 796, 771, 760, 733. NMR 1H (δ = ppm, CDCl3, J = Hz): 8.90, 8.45 (OH, sb, 1H & NH, s, 1H), 7.16 (H3, d, Jo = 7.02, 1H), 6.82 (H6, s, 1H), 6.65 (H4, d, Jo = 7.02, 1H), 2.49 (H8, q, J = 6.72, 1H), 2.27 (H12, s, 3H), 1.48 (H9, m, 4H), 1.29 (H10, m, 4H), 0.92 (H11, t, J = 6.72, 6H). NMR 13C (δ = ppm, CDCl3): 177.0 (C7, 1C), 148.4 (C1, 1C), 136.9 (C5, 1C), 123.4 (C2, 1C), 122.3 (C3, 1C), 121.1 (C4, 1C), 119.4 (C6, 1C), 47.9 (C8, 1C), 35.4 (C9, 2C), 20.8 (C12, 1C), 20.0 (C10, 2C), 14.2 (C11, 2C). Suitable monocrystals for the X-ray diffraction analysis were obtained in a 4:1 hexane/ethyl acetate solvent mixture.O)–N–), 1539, 1503 (–C(O)N–), 1465, 1461, 1427, 1400, 1341 (–N(O)–O), 1327, 1268, 1244, 1197, 1167, 1153, 993, 878, 819, 781, 743. NMR 1H (δ = ppm, CDCl3, J = Hz): 9.22, 8.86 (OH, sb, 1H & H3, d, Jm = 2.74, 1H), 8.82 (NH, s, 1H), 7.88 (H5, dd, Jo = 8.96, Jm = 2.74, 1H), 6.98 (H6, d, Jo = 8.96, 1H), 2.52 (H8, q, J = 7.14, 1H), 1.50 (H9, m, 4H), 1.38 (H10, m, 4H), 0.92 (H11, t, J = 7.14, 6H). NMR 13C (δ = ppm, CDCl3): 176.3 (C7, 1C), 153.4 (C1, 1C), 140.2 (C4, 1C), 126.7 (C2, 1C), 120.8 (C5, 1C), 116.8 (C3, 1C), 116.0 (C6, 1C), 47.8 (C8, 1C), 35.1 (C9, 2C), 20.6 (C10, C2), 14.0 (C11, 2C).O)–N–), 1592, 1506 (–C(O)N–), 1465, 1424, 1400, 1346 (–N(O)–O), 1327, 1268, 1247, 1194, 896, 875, 825, 880, 743. NMR 1H (δ = ppm, CDCl3, J = Hz): 9.07, 8.35 (OH, sb, 1H & NH, s, 1H), 8.20 (H3, d, Jo = 8.88, 1H), 7.76 (H4, d, Jo = 8.88, 1H), 7.63 (H6, s, 1H), 2.43 (H8, q, J = 7.14, 1H), 1.48 (H9, m, 4H), 1.36 (H2, m, 4H), 0.92 (H11, t, J = 7.14, 6H). NMR 13C (δ = ppm, CDCl3): 175.0 (C7, 1C), 146.1 (C1, 1C), 142.5 (C5, 1C), 132.6 (C2, 1C), 118.7 (C3, 1C), 114.7 (C4, 1C), 109.5 (C6, 1C), 47.1 (C8, 1C), 34.5 (C9, 2C), 19.9 (C10, C2), 13.4 (C11, 2C).O)–N–), 1541, 1524 (–C(O)N–), 1291, 867, 821 (C–Cl). NMR 1H (δ = ppm, CDCl3): 8.17, 7.86 (OH, s, 1H & NH, s, 1H), 7.40 (H3, s, 1H), 6.95 (H5, d, 1H), 6.95 (H6, d, 1H), 2.35 (H8, q, J = 6.60, 1H), 1.45 (H9, m, 4H), 1.20 (H10, m, 4H), 0.92 (H11, t, J = 6.60, 6H). NMR 13C (δ = ppm, CDCl3): 177.3 (C7, 1C), 147.0 (C1, 1C), 127.0 (C5, 1C), 126.3 (C2, 1C), 125.0 (C4, 1C), 121.9 (C3, 1C), 119.5 (C6, 1C), 48.3 (C8, 1C), 35.5 (C9, 2C), 21.0 (C10, C2), 14.2 (C11, 2C). Suitable monocrystals for the X-ray diffraction analysis were obtained in a 4:1 hexane/ethyl acetate solvent mixture.O)–N–), 1531, 1495 (–C(O)N–) 919, 896, 807 (C–Cl). NMR 1H (δ = ppm, CDCl3, J = Hz): 8.50, 7.95 (OH, s, 1H & NH, s, 1H), 7.18 (H3, d, Jo = 8.40, 1H), 7.00 (H6, d, Jm = 2.04, 1H), 6.80 (H4, dd, Jo = 8.40, Jm = 2.04, 1H), 2.36 (H8, q, J = 6.60, 1H), 1.47 (H9, m, 4H), 1.37 (H10, m, 4H), 0.92 (H11, t, J = 6.60, 6H). NMR 13C (δ = ppm, CDCl3): 177.4 (C7, 1C), 149.1 (C1, 1C), 131.9 (C5, 1C), 124.8 (C2, 1C), 123.1 (C3, 1C), 120.6 (C4, 1C), 119.3 (C6, 1C), 48.4 (C8, 1C), 35.5 (C9, 2C), 21.0 (C10, C2), 14.3 (C11, 2C).Semiempirical determination of partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
A partition coefficient (logP) calculation was developed in Hyperchem v.8.0 computational chemistry software. This was performed for VPA and VA1–7, starting from the X-ray structures obtained herein and through the replacement of particular functional groups and substituents for the whole series. Minimization was carried out with the PM3 semiempirical approach for these means and logP was computed for each compound at this level of theory.
Cell culture and drug treatment
Human U373 and rat C6 cells were cultured in Dulbecco's Modified Eagle's F12 Medium (DMEM-F12, Gibco, Thermo Fisher Scientific) with 10% fetal calf serum and 1% penicillin/streptomycin in a humidified atmosphere containing 5% CO2 at 37 °C. Due to drug hydrophobicity, the drug stocks were dissolved in Tween 80 (1:80 weight ratio) and then in DMSO (1:11 v/v). C6 and U373 cells were generously donated by Drs José Segovia and José Antonio Arias-Montaño, both from CINVESTAV México.
Cell viability
The cells were seeded at a density of 15625 cells per cm2 in a 96 well plate. The C6 cells were treated for 24 h with the drugs at different concentrations, while the U373 cells were treated for 48 h. Viability was determined using WST-1 reagent (Roche) and the absorbance was measured at 440 nm in a plate reader (Infinite M1000, Tecan). The cell viability was used to obtain the inhibitory concentration at 50% (IC50) of the tested compounds in the C6 and U373 cells. At least three independent assays were performed, and in each assay triplicates were included. The IC50 determination was done by non-linear logarithmic curve fitting using GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla California USA, http://www.graphpad.com).
Statistical analysis
One-way ANOVA, followed by Dunnett's multiple comparisons test were performed also using GraphPad Prism v. 6.00.Results and discussion
Synthetic proposal for valproic amides (VA)
In this research, the molecular modifications performed on the VPA parent were aimed on the acidic side, maintaining the valproyl moiety in an attempt to preserve the VPA biological activity. This is because acidic side modifications have been shown to cause a variety of effects that enhance the therapeutic capacity of VPA, e.g., the simplest VPA amide, valproyl amide (VA), which despite having antiepileptic activity has been demonstrated to lack iHDAC activity.20 Consequently, the exchange between acidic and amidic functionalities is not enough to produce the desired effect or even maintain it, because the iHDAC activity is suppressed for VA. Anyway, the preparation of other valproic amides is forthcoming, with the aim of preserving iHDAC activity as well as other particular biological properties like cell viability.A common strategy in pharmaceutical design has been the inclusion of aminophenols to generate an amidic functionality, where the nitrogen atom forms a peptidic bond or derived groups, in order to form new chemical chimeras through molecular design.21 For example, in 2011, Yamazaki et al.22 patented a benzoxazole generated through aminophenols and intermediary amides. Their invention refers to a pharmaceutical composition containing this active ingredient to treat illnesses such as hyperlipidemia, atherosclerosis, diabetes and complications, inflammation, and heart disease. Another example, patented in 2013 by Casado-Centellas et al.,23 is a catechol-O-methyltransferase inhibitor (iCOMT) that again is a benzoxazole derived from aminophenols and intermediary amides, employed in the prevention and treatment of amyloidosis. Moreover, one of the most commonly employed drugs is paracetamol or acetaminophen (APAP), which has analgesic properties and a chemical structure that contains a para-aminophenol moiety and an acetyl group attached as an amide to the aromatic ring.24 Considering the latter, it is evident that the incorporation of fragments such as aminophenols in diverse structures provides special characteristics in drugs and prototypes, thus signifying a promising way to design new VPA derivatives.
Molecular design of VPA analogues of valproic amide (VA) containing aminophenols
If one considers the use of aminophenols to generate an amidic functionality, the nitrogen atom is employed for the peptidic bond, leaving the OH untouched and thus recovering this group that is present in the VPA. The incorporation of OH should be as close as possible to the amide functional group, and the ortho-aminophenol (oAMPOH) would provide this requisite, thus generating the desired parent compound that connects VPA with oAMPOH. With the inclusion of an aromatic ring in oAMPOH, it is also possible to include structural modifications with substituents that are electro-donating or electro-withdrawing in nature, in order to modulate a plausible biological response and determine what substitution enhanced the activity.25 These variations include fragments with a slight effect on electronic density (–H), electro-donating methyl (–Me), electro-withdrawing nitro (–NO2) and chlorine (–Cl), which has an electro-withdrawing nature but also has the capacity for pi retrodonation to the aromatic ring.Synthesis of VA1–7
To obtain the designed compounds, VPA was reacted with SOCl2 in a 1:1 ratio in acetonitrile to yield valproyl chloride (VPCl), which was reacted again to achieve amide formation.18 Once the VPCl was formed, the corresponding oAMPOH1–7 was added to form a 1:4 mixture with triethylamine to neutralize the hydrochloric acid obtained during both synthetic steps. In this way, it was possible to generate the amidic bond that joined VPA with oAMPOH1–7 to obtain seven N-(2-hydroxyphenyl)-valproamides (VA1–7) (Scheme 1) in yields of 24.6–95.7%. In all cases, the completion of the reaction was followed by FTIR analysis, due to the change of the carbonyl band from acid to valproyl chloride and finally to valproyl amide. Thin layer chromatography of the crude products was also performed. All compounds were characterized using FTIR, 1H, 13C and HETCOR NMR, and melting point analysis. Also, VA2, VA3 and VA6 were characterized by their single crystal X-ray diffraction structures.
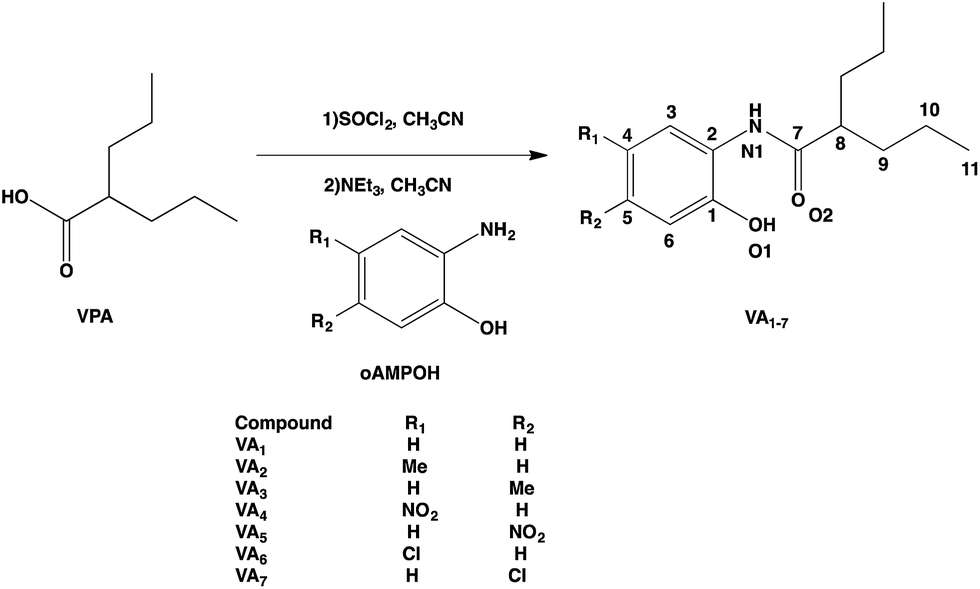 | ||
| Scheme 1 Synthetic procedure for obtaining VA1–7. The numbering scheme employed was followed for the NMR and X-ray characterization. | ||
Spectroscopic and structural analysis of VA1–7
In this section, the experimental and spectroscopic evidence for the obtained VA type compounds through Fourier transform infrared (FTIR) spectroscopy is presented. For the case of VA1, a single band at 3256 cm−1 is characteristic of the N–H bond present in a secondary amide (R–NH–CO). A very broad band is observed around 3600–2600 cm−1, which is characteristic of a phenolic O–H bond. A band belonging to Csp2–H from an aromatic ring at 3077 cm−1 and bands due to Csp3–H at 2957, 2929 and 2873 cm−1 belonging to the valproyl moiety were identified. The bands of the amidic group appear doubled, and could be due to two possibilities of conformers. These bands appear since the amide and phenolic OH interact, (a) 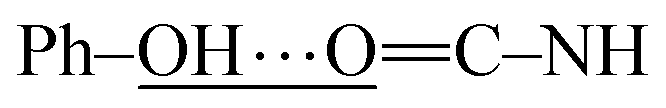 and (b)
and (b) 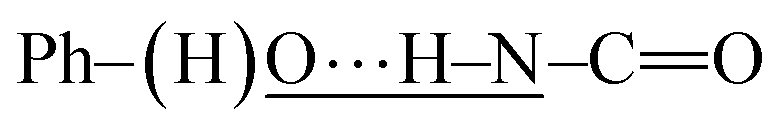 , gives four amidic bands in 1626, 1603, 1545 and 1497 cm−1. Analogously to VA1, the FTIR band assignment was done for the remaining N-(2-hydroxyphenyl)-valproamides (VA2–7). For VA4 and VA5, there were also NO2 bands at 1400 and 1341–1346 cm−1. Finally, the C–Cl bands appear at around 867 and 821 cm−1 for VA6, and 896 and 807 cm−1 for VA7 (see Experimental section for further details).
, gives four amidic bands in 1626, 1603, 1545 and 1497 cm−1. Analogously to VA1, the FTIR band assignment was done for the remaining N-(2-hydroxyphenyl)-valproamides (VA2–7). For VA4 and VA5, there were also NO2 bands at 1400 and 1341–1346 cm−1. Finally, the C–Cl bands appear at around 867 and 821 cm−1 for VA6, and 896 and 807 cm−1 for VA7 (see Experimental section for further details).
Compounds VA1–7 were also characterized using 1H (Table 1), 13C (Table 2) and HETCOR (13C–1H) NMR experiments. Multiplicities resulted according to their substitution patterns. The HETCOR spectra results were very useful to ascertain the unequivocal assignment of resonances in VA1–7, both for the 1H and 13C spectra. Quaternary carbons, secondary amide and phenol signals were confirmed as not having correlations in the HETCOR experiments. Protons H8, H9, H10 and H11, which belong to the valproyl moiety, appear in the ranges of 2.52–2.35 (H8), 1.55–1.45 (H9), 1.38–1.20 (H10) and 0.92 ppm (H11), these resonances are slightly influenced by the function of distance among aromatic and valproyl fragments as well as by the particular substituents at the aromatic ring. In the case of 13C NMR, the carbonyl signal C7 appears at 177.4–175.0 ppm, characteristic of a secondary amide with a double hydrocarbon chain, and is slightly influenced by aromatic substitution. In the case of C1, ipso to OH, the peak appears in the range of 153.4–145.5 ppm, clearly evidencing the para electro-withdrawing effect of the –NO2 substituent in VA4, as well as the –Cl substituent for VA7, which is instead in the meta position. The C2 position, which is ipso to the amide, appears at 132.6–123.4 ppm, and is mostly affected by the para –NO2 in VA5. The remnant C3, C4, C5 and C6 positions appear at 123.1–116.8, 140.2–114.4, 142.5–120.8, and 119.5–109.5 ppm, respectively.
| Cmpd | OH | NH | H3 | H4 | H5 | H6 | H8 | H9 | H10 | H11 | H12 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a The numbering employed is shown in Scheme 1. Samples were dissolved in DMSO-d6. δ = ppm. | |||||||||||
| VA1 | 8.95 | 8.73 | 7.44 | 6.95 | 7.02 | 6.83 | 2.46 | 1.49 | 1.23 | 0.92 | — |
| VA2 | 9.20 | 8.94 | 7.37 | — | 6.83 | 6.81 | 2.48 | 1.55 | 1.36 | 0.92 | 2.24 |
| VA3 | 8.90 | 8.45 | 7.16 | 6.72 | — | 6.72 | 2.49 | 1.48 | 1.29 | 0.92 | 2.27 |
| VA4 | 9.22 | 8.82 | 8.86 | — | 7.88 | 6.98 | 2.52 | 1.50 | 1.38 | 0.92 | — |
| VA5 | 9.07 | 8.35 | 8.20 | 7.76 | — | 7.63 | 2.43 | 1.48 | 1.36 | 0.92 | — |
| VA6 | 8.17 | 7.86 | 7.40 | — | 6.95 | 6.95 | 2.35 | 1.45 | 1.20 | 0.92 | — |
| VA7 | 8.50 | 7.95 | 7.18 | 6.80 | — | 7.00 | 2.36 | 1.47 | 1.37 | 0.92 | — |
| Cmpd | C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | C11 | C12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The numbering employed is shown in Scheme 1. Samples were dissolved in DMSO-d6. δ = ppm. | ||||||||||||
| VA1 | 148.2 | 126.5 | 120.4 | 122.4 | 126.0 | 118.4 | 177.3 | 48.0 | 35.4 | 20.8 | 14.1 | — |
| VA2 | 145.5 | 125.8 | 122.0 | 128.8 | 125.8 | 117.2 | 175.9 | 47.3 | 35.2 | 20.5 | 13.9 | 20.4 |
| VA3 | 148.4 | 123.4 | 122.3 | 121.1 | 136.9 | 119.4 | 177.0 | 47.9 | 35.4 | 20.0 | 14.2 | 20.8 |
| VA4 | 153.4 | 126.7 | 116.8 | 140.2 | 120.8 | 116.0 | 176.3 | 47.8 | 35.1 | 20.6 | 14.0 | — |
| VA5 | 146.1 | 132.6 | 118.7 | 114.7 | 142.5 | 109.5 | 175.0 | 47.1 | 34.5 | 19.9 | 13.4 | — |
| VA6 | 147.0 | 126.3 | 121.4 | 125.0 | 127.0 | 119.5 | 177.3 | 48.3 | 35.5 | 21.0 | 14.2 | — |
| VA7 | 149.6 | 124.8 | 123.1 | 120.6 | 131.9 | 119.3 | 177.4 | 48.4 | 35.5 | 21.0 | 14.3 | — |
Moreover, out of the seven synthesized VA compounds, suitable monocrystals for three of them were obtained from ethyl acetate/hexane solvent mixtures. These cases were (crystalline systems and space groups) VA3 (orthorhombic: P212121), VA2 and VA6 (both as monoclinic: P21/c), for which crystallographic data were determined through single crystal X-ray diffraction and analysis. Refined data and structures are presented in Tables 3 and 4. Fig. 1 shows the molecular perspectives following the same numbering as that used in the spectroscopic assignment. Selected structural parameters are shown in Table 4. These are aimed to analyse the amidic junction between VPA and oAMPOH, including bond distances (Å), bond angles (°) and dihedral angles (°) for these three analogous structures VA2, VA3 and VA6 (for which data will appear in this same order in this section). A very important observation is that aside from the attached substituent, these three amides are very similar in conformation. This behaviour clearly indicates a structural consensus depending on the characteristics of those attached moieties, and will be discussed as follows. For these three structures, there are clear hydrogen bond interactions between O1–H1a⋯O2–C7, the former belonging to the phenol and the latter being the carbonyl fragment, with bond distances of 1.75, 1.68 and 2.01 Å, where these values are smaller than the sum of the van der Waals radii (1.2 Å for hydrogen and 1.52 Å for oxygen).26 The bonding angles (O1–H1a⋯O2) for this same interaction are 160, 158 and 160°, tending towards 180° due to the strong directionality between these two fragments. Meanwhile, the torsion angles (O1–H1a⋯O2–C7) present for this hydrogen bond are 65, 41 and 58°, evidencing a needed angularity to avoid steric hindrance between the aminophenol and valproyl moieties. This finding of hydrogen bonding correlates with the observation in the FTIR spectra, where four amidic bands instead of only two were identified, as is common in simpler amides. One pair may be due to the non-hydrogen bonded structure and the other may be due to the hydrogen-bonded counterpart.
| a Numbers in parentheses are std. deviations. | |||
|---|---|---|---|
| Compound | VA2 | VA3 | VA6 |
| Molecular formula | C15H23NO2 | C15H23NO2 | C14H20ClNO2 |
| Molecular weight [g mol−1] | 249.34 | 249.34 | 269.76 |
| Crystalline system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/c | P212121 | P21/c |
| Cell dimensions | |||
| a [Å] | 8.9236(5) | 8.5924(17) | 9.0054(4) |
| b [Å] | 8.7861(4) | 12.938(3) | 8.7908(4) |
| c [Å] | 19.7125(10) | 14.250(3) | 19.4618(9) |
| β [°] | 102.535(3) | 90 | 102.377(3) |
| Volume [Å3] | 1508.69(13) | 1584.1(6) | 1504.88(12) |
| Z | 4 | 4 | 4 |
| ρ (calc.) [g cm−3] | 1.0977 | 1.0455 | 1.191 |
| μ [mm−1] | 0.072 | 0.069 | 0.249 |
| 2θ range [°] | 8.42 to 56.80 | 9.74 to 55.10 | 6.31 to 55.16 |
| Parameters/restraints | 207/30 | 218/30 | 222/18 |
| Collected reflections | 22042 |
22986 |
10105 |
| Independent reflections | 3457 | 3584 | 3348 |
| R (int.) | 0.1127 | 0.0517 | 0.0294 |
| GOOF | 1.032 | 1.091 | 1.029 |
| R | 0.0681 | 0.0558 | 0.0551 |
| Rw | 0.1490 | 0.1151 | 0.1469 |
| a Numbers in parentheses are std. deviations. | |||
|---|---|---|---|
| Compound | VA2 | VA3 | VA6 |
| Bonding distances (Å) | |||
| O1–H1a | 0.91 (3) | 0.99 (4) | 0.67 (3) |
| O2⋯H1a | 1.75 (3) | 1.68 (4) | 2.01 (3) |
| N1–H1 | 0.83 (2) | 0.83 (3) | 0.82 (2) |
| C7O2 |
1.247 (2) | 1.246 (3) | 1.247 (2) |
| C7–N1 | 1.338 (3) | 1.331 (3) | 1.335 (2) |
| N1–C2 | 1.429 (3) | 1.424 (3) | 1.426 (2) |
| C2–C1 | 1.392 (3) | 1.388 (4) | 1.392 (2) |
| C1–O1 | 1.362 (3) | 1.364 (3) | 1.356 (2) |
|
|||
| Bonding angles (°) | |||
| O2–C7–N1 | 122.0 (2) | 121.9 (2) | 121.76 (15) |
| C7–N1–C2 | 127.56 (19) | 127.6 (2) | 127.13 (15) |
| N1–C2–C1 | 123.4 (2) | 123.3 (2) | 123.45 (16) |
| C2–C1–O1 | 123.4 (2) | 122.9 (2) | 123.60 (16) |
| H1a–O1–C1 | 105.7 (18) | 105.0 (2) | 107 (2) |
| H1–N1–C2 | 115.7 (13) | 112.8 (18) | 116.2 (14) |
| O1–H1a⋯O2 | 160 (3) | 158 (4) | 160 (3) |
|
|||
| Dihedral angles (°) | |||
| H1a–O1–C1–C2 | −33.2 (18) | −39 (3) | −36 (3) |
| O1–H1a⋯O2–C7 | 65 (7) | 41 (11) | 58 (9) |
| H1–N1–C2–C1 | −148.6 (15) | −144.9 (19) | −148.8 (15) |
| H1–N1–C7–O2 | −176.2 (15) | −173 (2) | −175.8 (15) |
| N1–C7–O2–C8 | 178.5 (4) | −179.0 (4) | 178.4 (4) |
| C1–C2–N1–C7 | 45.4 (3) | 44.3 (4) | 46.8 (3) |
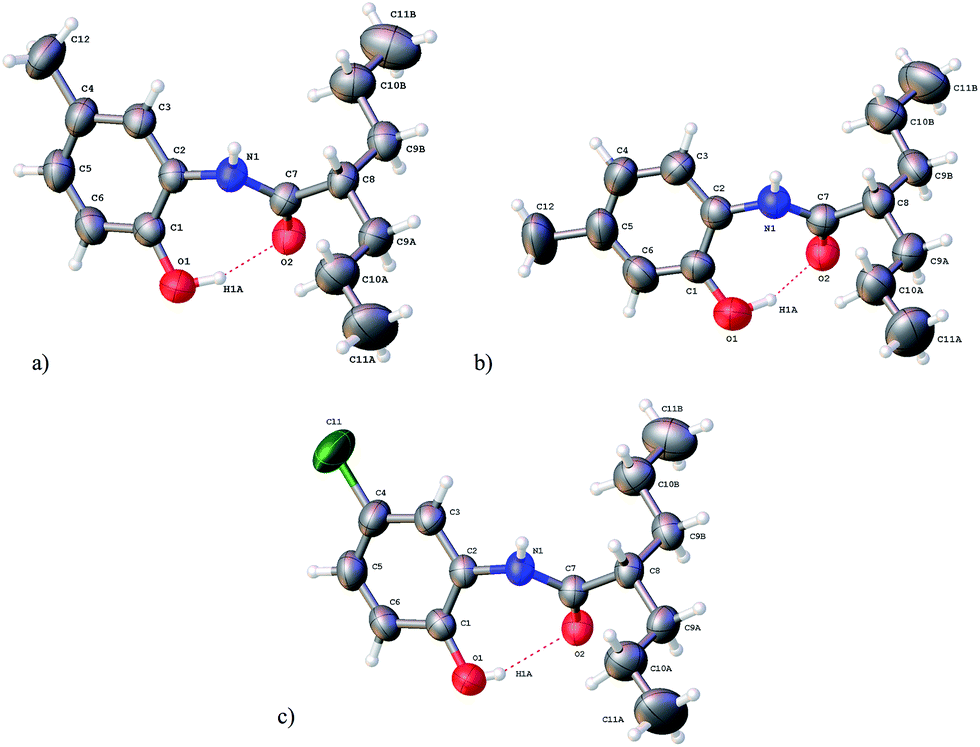 | ||
| Fig. 1 X-ray diffraction structures for (a) VA2, (b) VA3 and (c) VA6 (ellipsoids at 50% of probability). | ||
For the covalent behaviour of the structures, the bond distances of O1–H1a and N1–H1 were 0.91, 0.99 and 0.67 Å for the former, and 0.83, 0.83 and 0.82 Å for the latter, showing bond contractions only for VA6. In all cases, the double bonds between C7 and O2 are practically the same at 1.247, 1.246 and 1.247 Å, evidencing a negligible substituent effect through the structure. This last observation is reinforced, as C7–N1 also remains almost constant at 1.338, 1.331 and 1.35 Å for these compounds. As examples of the general structural behaviour, the remaining bond distances, bond angles and torsion angles almost behave the same for the amidic junction between VPA and oAMPOH. In particular, the amidic fragment is practically planar with dihedral angles for N1–C7–O2–C8 of 178.5, 179.0 and 178.4°. Meanwhile, the dihedral angles for the C1–C2–N1–C7 fragment are 45.4, 44.3 and 46.8°, clearly tending to minimize the steric hindrance among the VPA and oAMPOH moieties.
Cell viability of VA1–7
For the cell viability tests, all the VA1–7 compounds were predissolved in TWEEN 80 in 1:9 (w/w) ratios. This mixture was reformulated using 1:11 (v/v) TWEEN 80/DMSO ratios due to the hydrophobicity of these derivatives, and the final concentrations were related to the respective VA compound. According to the employed ratios of TWEEN 80 and DMSO, the tested concentrations were below the toxicity levels stated for these substances in cell viability assays, and blank tests were also carried out.27,28 At the tested concentrations there were no toxic effects. For VPA and all VA1–7 compounds, their cell viabilities in C6 (rat glioma) and U373 (human glioblastoma) lines were measured. The results show that all VA compounds presented more activity than VPA (see Table 5), hence indicating that the molecular design of these derivatives was clever in generating new molecules that fulfilled the desired goal. The cell viability of VPA was measured as the control, and the inhibitory concentration at 50% (IC50) obtained for C6 cells (1179 ± 130.8 μM) and for U373 cells (936.5 ± 174.4 μM) was similar to that of previous reports,29 requiring a millimolar concentration for 48 h. Meanwhile, for the VA1–7 compounds, the required concentrations to achieve IC50 ranged from 19.7 to 74.8 μM for C6, and 1.5 to 212.8 μM for U373 cells, which is comparable with other designed compounds aimed for cancer treatment.30
| Compound | Substituent* | C6 (μM) | U373 (μM) |
|---|---|---|---|
| a Data represents median ± standard error of at least three independent experiments. Glioma cells were in contact with each compound (VA1–7) in complete medium for 48 h. *Position relative to the amidic group. | |||
| VA1 | –H | 74.8 ± 8.4 | 212.8 ± 12.3 |
| VA2 | m-Me | 41.8 ± 8.4 | 151.8 ± 14.4 |
| VA3 | p-Me | 55.8 ± 11.0 | 161.8 ± 11.2 |
| VA4 | m-NO2 | 67.9 ± 9.7 | 64.9 ± 9.0 |
| VA5 | p-NO2 | 45.3 ± 12.3 | 39.7 ± 4.4 |
| VA6 | m-Cl | 19.7 ± 4.7 | 24.6 ± 2.2 |
| VA7 | p-Cl | 21.8 ± 2.7 | 15.5 ± 2.2 |
| VPA | — | 1179 ± 130.8 | 936.5 ± 174.4 |
Structure activity analysis of VA1–7
VA1 is the base compound and it was found to be 15.8 times more active than VPA in C6 cells (74.8 ± 8.4 μM) and 4.4 times more active than VPA in U373 cells (212.8 ± 12.3 μM), in the same range as other VPA amidic derivatives.31 In this case the molecular design gained credibility, due to the fact that this simple derivative augmented the biological response due to the recovery of the hydrogen-bonding group nearby, where it was originally placed in VPA. Furthermore, this modification not only recovers this interaction due to the presence of the aromatic ring, the biological effect is also enhanced by at least 4 times in U373 cells and almost 16 times in C6 cells. These results clearly show that the inclusion of an aromatic ring in this side of the parent molecule is a key method for developing other more potent derivatives. Therefore, the inclusion of steric and electronic modifiers of this aromatic ring, as is the case for VA2–7, should provide insight into the types of preferred interactions in a given molecular receptor in the cell. In this trend, the modification of the base compound VA1 with an electro-donating –Me substituent in the meta or para position relative to the amide group indeed increased the cell viability of the base compound to 41.8 ± 8.4 μM for VA2 (28.2 times more activity than VPA) and 55.8 ± 11.0 μM (21.1 times more activity than VPA) for VA3 in C6 cells. In U373 cells, VA2 and VA3 also showed an increased effect on cell viability (151.8 ± 14.4 and 161.8 ± 11.2 μM). This result clearly indicates that a bigger structure causes an important enhancement in the cell viability inhibition. Then, the compounds with an electro-withdrawing group, such as the –NO2 substituent at the meta or para position relative to the amide, presented a similar effect on the cell viability of C6 cells (67.9 ± 9.7 μM for VA4 and 45.3 ± 12.3 μM for VA5) as VA2 and VA3. The effect of these amides was more pronounced in U373 cells for the compounds with –NO2 (64.9 ± 9.0 μM for VA4 and 39.7 ± 4.4 μM for VA5). Finally, for both cell lines, VA6 and VA7 were the most active compounds. They contain –Cl as the substituent, behaving in a pi electro-donating/sigma electro-withdrawing manner, placed in the para or meta position relative to the amide. For C6 cells, the IC50 for VA6 was 19.7 ± 4.7 μM and 21.8 ± 2.7 μM for VA7; in U373 cells, the doses to obtain the IC50 were 24.6 ± 2.2 μM for VA6 and 15.5 ± 2.2 μM for VA7. This is mainly due to the fact that a chlorine atom attached at an aromatic ring prevents a wide variety of chemical reactions, enhancing the possible interactions of the compound with biological receptors.32 Aromatic moieties possessing chlorine (and halogen substituents in general) are a key pharmacological feature15 justified because these fragments are present in an important amount of already tested and used pharmaceutical drugs and pesticides, and because of the plausible interactions that they present among them, such as R–Cl⋯CO, R–Cl⋯H–R, R–Cl⋯Ar, R–Cl⋯Cl–R, etc.
Moreover, in order to track important physicochemical properties responsible for the biological findings, a semiempirical (Hyperchem v.8.0) partition coefficient (logP) calculation was developed. In this line, logPVPA was 2.61, logP for VA1 was 3.39, for VA2–3 was 3.85, for VA4–5 was 3.34 and for VA6–7 was 3.90. This calculation was carried out for each compound to track hydrophobic/hydrophilic ratios and correlate them with biologic trends. The results clearly evidence that the most hydrophobic ratio, resulting from the amide in VA6–7, led to the most potent inhibition, and this was attained due to the –Cl substituent.
In general, VA1, VA2 and VA3 presented a greater effect on C6 cells than U373 cells, but VA4 VA5, VA6 and VPA required similar doses for the IC50. From the analysis of this data we can conclude that the new compounds not only maintain the effect of VPA on the cell viability, but can also achieve the same viability with lower doses. Related references clearly state that amides of VPA do not undergo hydrolysis33,34 or further biotransformations, due to the steric hindrance of the valproyl moiety not only under chemical but also biological conditions. Therefore, this kind of compound should have a major effect on cell viability due to the newly obtained chemical structure and not due to the VPA metabolite. This demonstrates that the new base structure (with the neutral substituent) can be modified to increase the activity, and the best activity was found with –Cl, followed by –NO2 and finally with –Me in U373 cells. But in C6 cells, there was not a significant difference between –NO2 and –Me groups. This may reflect different histone deacetylase expression or regulation in these cell lines and further work is under development on this issue.
Conclusions
Finally, seven designed valproic acid derivatives, N-(ortho-hydroxyphenyl)valproamides (VA1–7), were obtained in moderate yields by coupling valproyl chloride with seven ortho-aminophenols. This led to a family of amides with similar structural characteristics but tuneable electronic and steric contributions, through the inclusion of electro-donating (–Me), electro-withdrawing (–NO2) or pi electro-donating/sigma electro-withdrawing (–Cl) substituents at the aromatic ring. The identity of such derivatives was evidenced through spectroscopic characterization using FTIR, 1H, 13C and HETCOR NMR, as well as by melting point analysis. In particular, for three of the derivatives (VA2, VA3 and VA6) it was feasible to determine their chemical crystalline structures, in all cases behaving in a similar fashion and with very similar conformations independent of the attached substituents. In general, VA1, VA2 and VA3 affected C6 cells more than U373 cells, but VA4, VA5, VA6 and VPA required similar doses for the IC50. From the analysis of this data we can conclude that the new compounds not only maintain the effect of VPA on cell viability, but also require lower doses. This demonstrates that the new base structure (with a neutral substituent) can be modified to increase the activity, and the best activity was found with –Cl, which was as expected due to the inclusion of this substituent in a huge amount of tested drugs. The hydrophobic effect was also an important variable in enhancing the potency of these derivatives, and the chlorine-bearing molecules produced the best results. In the biological trend –Cl is then followed by –NO2 and finally by –Me in U373 cells. However, in C6 cells, there was not a significant difference between the –NO2 and –Me groups, since they have almost the same hydrophobic/hydrophilic ratio according to the logP results. This may reflect different histone deacetylase expression or regulation in these cell lines, and work is under development on this issue. An important perspective is that VA6 and VA7 may become lead structures in the search for more potent pharmaceutical prototypes.
Acknowledgements
We acknowledge CONACyT projects 47310337 (L. Arregui) and 222872 (H. I. Beltrán), UAM and SEP-PROMEP for financial support. A. Alpuche-García and X. Dávila-González acknowledge university scholarships from CONACyT and UAM.References
- C. Wang, Z. Luan, Y. Yang, Z. Wang, Y. Cui and G. Gu, Neurosci. Lett., 2011, 497, 122–127 CrossRef CAS PubMed.
- S. Chateauvieux, F. Morceau, M. Dicato and M. Diederich, J. Biomed. Biotechnol., 2010, 2010, 479364 Search PubMed.
- R. W. Johnstone, Nat. Rev. Drug Discovery, 2002, 1, 287–299 CrossRef CAS PubMed.
- M. Dokmanovic, C. Clarke and P. A. Marks, Mol. Cancer Res., 2007, 5, 981–989 CrossRef CAS PubMed.
- D. Marchion and P. Münster, Expert Rev. Anticancer Ther., 2007, 7, 583–598 CrossRef CAS PubMed.
- M. Mottamal, S. Zheng, T. L. Huang and G. Wang, Molecules, 2015, 20, 3898–3941 CrossRef CAS PubMed.
- A. V. Krauze, S. D. Myrehaug, M. G. Chang, D. J. Holdford, S. Smith, J. Shih, P. J. Tofilon, H. A. Fine and K. Camphausen, Int. J. Radiat. Oncol., Biol., Phys., 2015, 92, 986–992 CrossRef CAS PubMed.
- D. Thotala, R. M. Karvas, J. A. Engelbach, J. R. Garbow, A. N. Hallahan, T. A. DeWees, A. Laszlo and D. E. Hallahan, Oncotarget, 2015, vol. 6, pp. 35004–35022 Search PubMed.
- S. Eyal, B. Yagen, J. Shimshoni and M. Bialer, Biochem. Pharmacol., 2005, 69, 1501–1508 CrossRef CAS PubMed.
- H. Deubzer, B. Busche, G. Rönndahl, D. Eikel, M. Michaelis, J. Cinatl, S. Schulze, H. Nau and O. Witt, Leuk. Res., 2006, 30, 1167–1175 CrossRef CAS PubMed.
- G. G. Mackenzie, L. Huang, N. Alston, N. Ouyang, K. Vrankova, G. Mattheolabakis, P. P. Constantinides and B. Rigas, PLoS One, 2013, 8, e61532 CAS.
- C. Harmon and D. Myles, Valproic acid salts, US Pat., WO2009142968 A2, 2009.
- N. Tarasenko, S. M. Cutts, D. R. Phillips, G. Berkovitch-Luria, E. Bardugo-Nissim, M. Weitman, A. Nudelman and A. Rephaeli, Biochem. Pharmacol., 2014, 88, 158–168 CrossRef CAS PubMed.
- E. Perrino, G. Cappelletti, V. Tazzari, E. Giavini, P. D. Soldato and A. Sparatore, Bioorg. Med. Chem. Lett., 2008, 18, 1893–1897 CrossRef CAS PubMed.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- E. Bechar and H. Astroug, Arch. Pharm., 1997, 330, 273–276 CrossRef CAS PubMed.
- N. Pariente-Cohen, M. Weitman, N. Tania, D. T. Major, H. E. Gottlieb, S. Hoz and A. Nudelman, RSC Adv., 2015, 5, 24038–24043 RSC.
- C. U. Johannessen and S. I. Johannessen, CNS Drug Rev., 2003, 9, 199–216 CrossRef CAS PubMed.
- H. Andleeb, Y. Tehseen, S. J. Ali Shah, I. Khan, J. Iqbal and S. Hameed, RSC Adv., 2016, 6, 77688–77700 RSC.
- Y. Yamazaki, T. Toma, M. Nishikawa, H. Ozawa, A. Okuda, K. Abe and S. Oda, Benzoxazole compound and pharmaceutical composition containing the compound, Germany Pat., EP1433786 B1, 2011.
- M. Casado Centellas, B. R. Insa, B. N. Reig and B. N. Gavaldà, New therapy for transthyretin-associated amyloidosis, Spain Pat., WO2013060668 A1, 2013.
- J. A. Clements, R. C. Heading, W. S. Nimmo and L. F. Prescott, Clin. Pharmacol. Ther. Pediatr. Perspect., 1978, 24, 420–431 CrossRef CAS.
- J. Fu, K. Cheng, Z. M. Zhang, R. Q. Fang and H. L. Zhu, Eur. J. Med. Chem., 2010, 45, 2638–2643 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- B. Arechabala, C. Coiffard, P. Rivalland, L. J. M. Coiffard and Y. D. Roeck-Holtzhauer, J. Appl. Toxicol., 1999, 19, 163–165 CrossRef CAS PubMed.
- Y. He, H. Liu, Z. Xie, Q. Liao, X. Lai and Z. Du, Drug Dev. Ind. Pharm., 2014, 40, 237–243 CrossRef CAS PubMed.
- J. A. Benitez, L. Arregui, G. Cabrera and J. Segovia, Neuroscience, 2008, 156, 911–920 CrossRef CAS PubMed.
- V. Di Bussolo, E. C. Calvaresi, C. Granchi, L. Del Bino, I. Frau, M. C. Dasso Lang, T. Tuccinardi, M. Macchia, A. Martinelli, P. J. Hergenrother and F. Minutolo, RSC Adv., 2015, 5, 19944–19954 RSC.
- M. Farooq, A. El-Faham, S. N. Khattab, A. M. Elkayal, M. F. Ibrahim, N. A. Taha, A. Baabbad, M. A. M. Wadaan and E. A. Hamed, Asian Pac. J. Cancer Prev., 2014, 15, 7785–7792 CrossRef PubMed.
- H. Marquardt, Angew. Chem., 1995, 107, 1017 CrossRef.
- M. Bialer, A. Haj-Yehia, K. Badir and S. Hadad, Pharm. World Sci., 1994, 16, 2–6 CrossRef CAS PubMed.
- S. Blotnik, F. Bergman and M. Bialer, Drug Metab. Dispos., 1996, 24, 560–564 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC no. 1525302 for VA2, 1525303 for VA3, and 1525301 for VA6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00048k |
| This journal is © The Royal Society of Chemistry 2017 |