
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and antioxidant activity of 1,3,4-oxadiazoles and their diacylhydrazine precursors derived from phenolic acids†
Nevena Mihailovića,
Violeta Markovića,
Ivana Z. Matićb,
Nemanja S. Stanisavljevićc,
Živko S. Jovanović c,
Snežana Trifunovićd and
Ljubinka Joksović*a
c,
Snežana Trifunovićd and
Ljubinka Joksović*a
aFaculty of Science, Department of Chemistry, University of Kragujevac, R. Domanovića 12, 34000 Kragujevac, Serbia. E-mail: ljubinka@kg.ac.rs
bInstitute of Oncology and Radiology of Serbia, Pasterova 14, 11000 Belgrade, Serbia
cInstitute of Molecular Genetics and Genetic Engineering, University of Belgrade, Vojvode Stepe 444a, 11000 Belgrade, Serbia
dFaculty of Chemistry, University of Belgrade, Studentski trg 12–16, 11000 Belgrade, Serbia
First published on 26th January 2017
Abstract
Eight 1,3,4-oxadiazole derivatives containing phenolic acid moieties (7a–h) and eight of their diacylhydrazine precursors (6a–h) were synthesized, characterized using spectroscopic methods and examined by scavenging of stable DPPH (2,2-diphenyl-1-picrylhydrazyl) radicals. The most potent phenolic 1,3,4-oxadiazoles showed better DPPH scavenging activity in comparison with their corresponding diacylhydrazine precursors as a result of participation of both aromatic rings and a 1,3,4-oxadiazole moiety in resonance stabilization of the formed phenoxyl radical. Four diacylhydrazines (6d, 6e, 6g, and 6h) and four 1,3,4-oxadiazoles (7d, 7e, 7g and 7h) with the best DPPH scavenging activity, were chosen for further evaluation of their antioxidant potential through various assays. The investigated compounds exerted pronounced ABTS radical scavenging capacity, moderate to good H2O2 scavenging properties and strong ferric ion reducing capacity. Further in vitro evaluation of the antioxidant properties of the most active compounds demonstrated their protective effects in normal lung fibroblasts MRC-5 against hydrogen peroxide induced oxidative stress. Diacylhydrazine 6h increased two times the activity of glutathione peroxidase in treated cells in comparison with a control sample and did not affect the superoxide dismutase activity.
1. Introduction
Reactive oxygen species (ROS) in the form of free radicals (superoxide, singlet oxygen, hydroxyl radical) and neutral molecules (hydrogen peroxide) induce damage of biological macromolecules under oxidative stress and contribute to the pathogenesis of many health problems such as cancer, inflammation, atherosclerosis, cardiovascular and neurodegenerative diseases.1–3 The antioxidants prevent the oxidation of biological substrates, lowering oxidative stress, DNA mutations, malignant changes and thus, block the loss of cell function.4Owing to the possibility of facile hydrogen atom transfer and resonance stabilization of the resulting phenoxyl radical, phenolic acids act as chain-breaking antioxidants able to scavenge free radical species.5 Natural and synthetic phenolic acids often contain both hydroxyl and methoxy substituents and the positioning of OH groups is crucial for their antioxidant properties.6–8 The combination of the known phenolic antioxidants with heterocyclic pharmacophores in the same unit can be an interesting approach to discover novel more potent radical scavengers as a result of their synergistic effects. Furthermore, one of such heterocycles, substituted 1,3,4-oxadiazole scaffold has been reported to exhibit a broad spectrum of biological activity.9–12 Literature searches reveal that 2,5-disubstituted 1,3,4-oxadiazole derivatives commonly obtained from diacylhydrazines show antioxidant potential. This class of compounds includes sulfonamidomethane linked 1,3,4-oxadiazoles,13 substituted bis(1,3,4-oxadiazoles),14 1,3,4-oxadiazoles possessing benzoxazole,15 1,3,4-oxadiazole tagged thieno[2,3-d]pyrimidines,16 2-benzoylamino-5-hetaryl-1,3,4-oxadiazoles,17 and 1,3,4-oxadiazoles containing 3-fluoro-4-methoxyphenyl moiety.18 To the best of our knowledge, there is no available data regarding antioxidant activity of hydroxy-substituted dibenzoylhydrazines in the literature. In this study, a series of 1,3,4-oxadiazoles and their diacylhydrazine precursors derived from phenolic acids was synthesized and screened for radical scavenging properties using several assays.
2. Results and discussion
2.1. Chemistry
The synthesis of 1,3,4-oxadiazoles from 1,2-diacylhydrazines requires various cyclodehydration agents such as SOCl2,19 POCl3,20,21 Burgess reagent,22 and 2-chloro-1,3-dimethylimidazolinium chloride.23 A series of 2,5-disubstituted 1,3,4-oxadiazole antioxidants incorporating phenol moiety was prepared by reacting aryl hydrazides with substituted 4-hydroxybenzoic acids in the presence of phosphorus oxychloride.20 Nieddu et al. performed a synthesis of cytotoxic unsymmetrical 1,3,4-oxadiazoles also using POCl3 as cyclodehydration agent.21 For cyclization of our 1,2-diacylhydrazines containing more phenolic hydroxyl groups, thionyl chloride was the best choice, and for this purpose, we modified literature procedure for the synthesis 1,3,4-oxadiazoles containing adamantane ring19 replacing solvent and excluding base during the preparation of 1,2-diacylhydrazines as well as excluding toluene in the next cyclization step.The synthetic pathway for the targeted 1,3,4-oxadiazole derivatives 7a–h is presented in Scheme 1. In the first step, commercially available aromatic acids 1a–h were converted into corresponding methyl esters 2a–h in the presence of H2SO4.24 The obtained esters were then reacted with excess of hydrazine hydrate yielding acylhydrazines (5a–h).25 In order to dissolve polar impurities after the evaporation of the solvent, small amount of water was added to the crude residue and the formed suspension was filtrated and dried. On the other side, commercially available aromatic acids 3a–h were transformed into acid chlorides 4a–h in the reaction with thionyl chloride in the presence of catalytic amounts of N,N-dimethylformamide, according to slightly modified procedure.26 The next step was performed immediately due to the low stability of the formed acid chlorides. The obtained acylhydrazine (5a–h) and corresponding acid chloride (4a–h) were stirred at room temperature in dry THF, giving the corresponding diacylhydrazines19 (6a–h) with satisfactory purity without a need for further purification. It was possible to combine here the condensation of acid chloride ArCOCl with acylhydrazine Ar′CONHNH2 or acid chloride Ar′COCl with acylhydrazine ArCONHNH2. The selection of precursors for this type of reaction only depends on higher purity and better yields of diacylhydrazines 6a–h. Finally, diacylhydrazines 6a–h were cyclised to 1,3,4-oxadiazole derivatives 7a–h by treatment with SOCl2 in the reflux conditions.19
 | ||
| Scheme 1 Reagents and conditions: (a) H2SO4, MeOH, 5 h, reflux; (b) N2H4 × H2O, EtOH, 12 h, reflux; (c) SOCl2, DMF, CH2Cl2, 2 h, r.t.; (d) THF, 6 h, r.t.; (e) SOCl2, 6 h, reflux. | ||
The exact structure of synthesized compounds (6a–h and 7a–h) was confirmed by means of 1H and 13C NMR spectroscopy (see ESI, Fig. S1–S32†). All aromatic protons for both classes of compounds displayed signals in the characteristic region ranging from 6.3 to 8.3 ppm. Signals present at the lower field in 1H NMR spectra of compounds 6a–h correspond to OH and NH protons. Their acidic nature was proven by the addition of two drops of D2O to NMR sample, which resulted in the disappearance of the corresponding signals as a consequence of fast deuterium exchange. In 1H NMR spectra of 7a–h, due to the cyclization to 1,3,4-oxadiazole derivatives, signals attributed to NH protons disappeared and remained only D2O exchangeable signals assigned to phenolic OH protons at the highest chemical shift values.
2.2. DPPH scavenging activity of diacylhydrazines and their corresponding 1,3,4-oxadiazoles
The results of DPPH radical scavenging activity of the synthesized diacylhydrazines and corresponding 1,3,4-oxadiazoles, expressed as concentration of tested compound that reduce 50% (IC50) of the DPPH radicals are presented in Table 1. From all tested compounds, four diacylhydrazines and four 1,3,4-oxadiazoles showed good antioxidant activity, with IC50 values in the range of 13.59 to 22.17 μM, while IC50 values of ascorbic acid and nordihydroguaiaretic acid (NDGA) were 38.78 and 20.83 μM, respectively.| Diacylhydrazines | 1,3,4-Oxadiazoles | ||
|---|---|---|---|
| Compound | IC50 (μM) ± SD | Compound | IC50 (μM) ± SD |
| a Results are mean values ± SD from three measurements. | |||
| 6a | >1000 | 7a | >1000 |
| 6b | >1000 | 7b | >1000 |
| 6c | >1000 | 7c | >1000 |
| 6d | 16.06 ± 0.40 | 7d | 14.63 ± 0.34 |
| 6e | 20.97 ± 0.47 | 7e | 13.59 ± 0.30 |
| 6f | >1000 | 7f | >1000 |
| 6g | 17.28 ± 0.41 | 7g | 15.81 ± 0.38 |
| 6h | 22.17 ± 0.53 | 7h | 14.93 ± 0.36 |
| Referent antioxidants | |
|---|---|
| Ascorbic acid | 38.78 ± 0.60 |
| NDGA | 20.83 ± 0.23 |
Compared to the corresponding 1,3,4-oxadiazoles (7a–h), their diacylhydrazine precursors derived from phenolic acids (6a–h) have shown up to 50% weaker antioxidant activity (Table 1). In these compounds the formation of radicals is possible through heterolytic OH and NH cleavage, as it was previously demonstrated by DFT calculations for triazole derivatives.6 The radical species formed from derivatives 6a–h after the cleavage of NH bond have no ability for resonance stabilization through the corresponding aromatic ring. Therefore, their role in the antioxidant activity of parent compounds is not significant. On the other hand, in the case of heterolytic OH-cleavage, the unpaired electron of the formed radical can be delocalized across the benzene ring (Scheme 2A), causing an easy formation of phenoxyl radical with lower reactivity and higher stability.
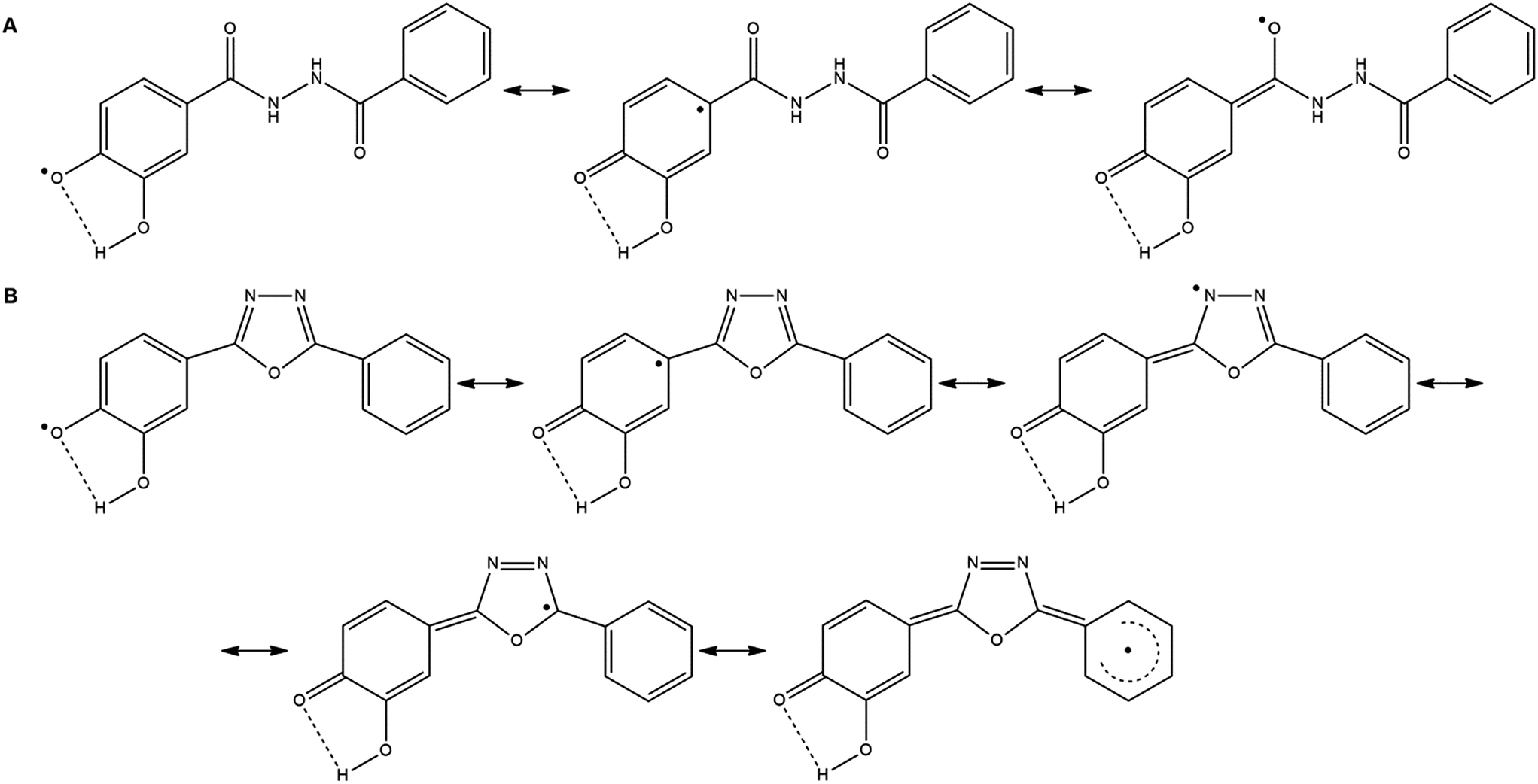 | ||
| Scheme 2 Resonance stabilization of radicals formed from compound 6e (A), and 7e (B), after heterolytic cleavage of OH bond, and loss of one electron. | ||
The phenoxyl radical formed from the corresponding 1,3,4-oxadiazole derivative (7a–h) can be highly stabilized through resonance since the unpaired electron may be delocalized across both aromatic rings and 1,3,4-oxadiazole part of the molecule (Scheme 2B). All of the compounds which show excellent antioxidant activity (7d, 7e, 7g and 7h) contain in their structure two neighboring hydroxyl groups having the possibility to form an intramolecular hydrogen bond (Scheme 2B). The most active compound 7e possesses two hydroxyl groups in m- and p-positions of the aromatic ring. Substitution of the m-hydroxyl group with methoxy one (7c) leads to the loss of the activity due to the less effective radical stabilization by intramolecular hydrogen bonding. Comparing the activity of compound 7e with the activities of 7g and 7h it can be concluded that the presence of additional hydroxyl groups induces a slight decrease of activity. The presence of one or two OH groups at the second benzene ring in 7g and 7h does not contribute to resonance stabilization of radical due to the negative inductive effect of the hydroxyl group. Compound 7f exerts no antioxidant activity, as it was expected for the molecule which contains two m-hydroxyl groups in its structure and it does not allow extended resonance stabilization.
2.3. ABTS, H2O2 scavenging activity and reducing capacity
The compounds which showed the best DPPH radical scavenger activities 6d, 6e, 6g, 6h, 7d, 7e, 7g and 7h have been chosen for further examination of their in vitro antioxidant properties through ABTS radical scavenging, peroxide scavenging assays and reducing capacity. The obtained results are summarized in Table 2.| Compounds | FC reducing capacity assay (gGAE per g) | Ferric ion reducinga capacity assay (A700 nm) | ABTS assay IC50 (μM) | H2O2 scavenging assay IC50 (μM) |
|---|---|---|---|---|
| a Concentrations of tested compounds and ascorbic acid are set at 500 μM. | ||||
| 6d | 1.141 ± 0.013 | 0.296 ± 0.009 | 120.1 ± 5.7 | 37.41 ± 0.89 |
| 6e | 1.256 ± 0.007 | 0.478 ± 0.011 | 104.4 ± 11.6 | 62.24 ± 3.97 |
| 6g | 1.107 ± 0.003 | 0.461 ± 0.008 | 133.3 ± 35.7 | 43.92 ± 0.38 |
| 6h | 1.470 ± 0.019 | 0.478 ± 0.009 | 102.6 ± 7.5 | 57.00 ± 1.43 |
| 7d | 0.918 ± 0.008 | 0.270 ± 0.005 | 118.4 ± 31.2 | 100.33 ± 5.89 |
| 7e | 1.153 ± 0.016 | 0.367 ± 0.015 | 79.3 ± 2.6 | 107.58 ± 9.02 |
| 7g | 1.450 ± 0.006 | 0.313 ± 0.005 | 137.7 ± 30.4 | 84.89 ± 0.70 |
| 7h | 1.209 ± 0.006 | 0.426 ± 0.004 | 120.6 ± 18.5 | 75.17 ± 5.23 |
| Ascorbic acid | — | 0.173 ± 0.007 | 242.6 ± 7.3 | 22.40 ± 0.60 |
Radical scavenging capacity of synthesized compounds was tested using ABTS radical cation scavenging assay, which is another widely used method for the estimation of in vitro antioxidant ability. The obtained results (Table 2) showed that all tested compounds possessed more pronounced scavenging capacities than ascorbic acid (IC50 = 242.6 μM). Similarly to previous DPPH assay compound 7e (IC50 = 79.3 μM) showed to be the most potent scavenger of ABTS radical cation which was followed by samples 6e and 6h. Meanwhile 6d and 7h exhibited the weakest scavenging capacities (IC50 = 120.1 μM and IC50 = 120.6 μM respectively).
Hydrogen peroxide is a biologically relevant, oxidizing species which is formed in numerous animal tissues through several oxidative processes and can diffuse across biological membranes, causing oxidative damage to DNA, proteins, lipids and other biomolecules. According to our results presented in Table 2 all tested compounds showed weaker H2O2 scavenging properties than the ascorbic acid (IC50 = 22.4 μM). Among tested samples 6d showed the most prominent scavenging capacity with IC50 value approximately two times higher than the ascorbic acid (IC50 = 37.41 μM). The obtained results are comparable to data collected by Kotaiah et al.16 who examined H2O2 scavenging capacities of 1,3,4-oxidazole tagged thieno[2,3-d]pyrimidine derivatives using the same assay. On contrary to our results all compounds examined in the mentioned study exhibited H2O2 scavenging capacities near to the value obtained for the ascorbic acid (IC50 = 16.35).
Folin–Ciocalteu reducing capacity assay (F–C) has been widespread assay for measurement of total phenolics in natural products, although the basic mechanism is an oxidation–reduction process, thus it can be considered as a method suitable for the estimation of antioxidant capacity.27 It could be particularly useful for comparison of reducing capacities of natural products and newly synthesized compounds since the results are often expressed in mg or g of gallic acid or catechin equivalents per gram of tested sample which allows direct comparison with available literature data. In the present study majority of investigated compounds exhibited a stronger reduction of F–C complex than gallic acid which was used as standard with the exception of compound 7d which was approximately 10% less efficient in the reduction of F–C reagent than the gallic acid. Sample 6h possessed the strongest reducing capacity among tested compounds (Table 2).
Considering the results of ferric ion reducing capacity assay (Table 2) it can be observed that all examined compounds showed higher capacities for Fe3+ reduction than ascorbic acid which was used as standard. In concordance with the previous assay compound 7d exhibited the lowest reduction capacity towards Fe3+ ion and two samples with the strongest reduction capacity were 6e and 6h.
From the mechanistic standpoint discrepancies in two previous reducing capacity tests and ABTS assay could be explained either by different redox potential of counterparts reacting with the tested antioxidant compounds or by different reaction times needed for reaching the steady-state time point. The difference in steric accessibility of antioxidant compounds to ABTS radical cation, F–C complex and Fe3+ ion could be also the possible reason for observed discrepancies between the assays.27
2.4. Cytotoxic activity of diacylhydrazines and 1,3,4-oxadiazoles on MRC-5 cells
All synthesized compounds which exerted the most intensive antioxidant effects as shown by DPPH assay, were chosen for further evaluation of their protective properties in normal cells. For that reason, we first examined the cytotoxic activity of selected diacylhydrazines 6d, 6e, 6g, 6h, and 1,3,4-oxadiazoles 7d, 7e, 7g and 7h against normal human lung fibroblasts MRC-5 (results are presented in Table 3). Among tested compounds, 1,3,4-oxadiazoles 7h and 7d exerted the highest intensity of the cytotoxic activity on MRC-5 cells. The compounds 7g and 7e also showed pronounced cytotoxicity with IC50 values approximately 30% higher than 7h and 7d. However, the examined diacylhydrazines exhibited significantly lower cytotoxic effects in comparison with 1,3,4-oxadiazoles. The cytotoxicity of compounds 6g and 6h was approximately three times lower than the most active compounds 7d and 7h. The compound 6d showed very low cytotoxic activity, while the activity of 6e was quite poor.| Compound | IC50 ± SD [μM] |
|---|---|
| 6d | 118.35 ± 9.41 |
| 6e | 194.27 ± 2.69 |
| 6g | 86.33 ± 2.41 |
| 6h | 91.83 ± 8.57 |
| 7d | 26.88 ± 1.66 |
| 7e | 37.49 ± 0.20 |
| 7g | 34.88 ± 1.93 |
| 7h | 25.59 ± 1.76 |
| Cisplatin | 9.24 ± 0.18 |
2.5. Effects of pretreatment of MRC-5 cells with diacylhydrazines and 1,3,4-oxadiazoles on ROS generation induced by H2O2
To fully evaluate the antioxidant potential of synthesized diacylhydrazines and 1,3,4-oxadiazoles, their possible cytoprotective effects against the generation of ROS induced by hydrogen peroxide were examined in normal human lung fibroblasts MRC-5. As it could be seen in Fig. 1, pretreatment for 24 h of MRC-5 cells with non-toxic concentrations of all tested compounds (50 μM for diacylhydrazines and 20 μM for 1,3,4-oxadiazoles, as determined for 24 h treatment) reduced the levels of ROS in MRC-5 cells treated for 30 min with H2O2 compared with cells that were grown in the nutrient medium during 24 h and then exposed to H2O2. The percentages labeled in each histogram represent the subpopulation of cells with increased fluorescence levels, i.e. with increased levels of ROS. The effectiveness of diacylhydrazines and 1,3,4-oxadiazoles in attenuating the oxidative stress in normal fibroblasts in addition to the results of the antioxidant assays suggest the significant antioxidant potential of the investigated compounds. Higher intracellular level of ROS is implicated in the early steps of malignant transformation, as well as in the further progression of malignant tumors.28 Due to their prominent antioxidant effects, and especially the ability to reduce intracellular ROS levels, the examined 1,3,4-oxadiazoles and their diacylhydrazine precursors might serve as an useful cancer chemopreventive agents. | ||
| Fig. 1 Effects of 24 h pretreatment of MRC-5 cells with diacylhydrazines and 1,3,4-oxadiazoles on ROS production induced by hydrogen peroxide. (A) Fluorescence histograms; (B) fluorescence intensity of generated dichlorofluorescein in MRC-5 cells. Applied non-toxic concentrations of compounds were: 50 μM for 6d, 6e, 6g and 6h, and 20 μM for 7d, 7e, 7g and 7h. | ||
2.6. Effects of diacylhydrazines and 1,3,4-oxadiazoles on the activity of antioxidant enzymes
Several enzymatic defense mechanisms attempt to minimize the production and the action of harmful oxidants, such as superoxide dismutase (SOD), glutathione reductase (GR) and glutathione peroxidase (GPX). SOD dismutates the free radical superoxide by converting it to hydrogen peroxide, which in turn is decomposed by catalase at high concentration, and by GPX at low concentration. Most hydrogen peroxide in the cell is generated through the dismutation of superoxide by SOD action, even though it can be produced by other superoxide-generating enzymes.29,30 In our experiments we also investigated the effects of 1,3,4-oxadiazoles and their diacylhydrazine precursors derived from phenolic acids on the activities of antioxidant enzymes, GR, GPX and SOD in MRC-5 cells (Table 4). It is evident that applied compounds drastically reduced the activity of enzymes involved in glutathione metabolism – GR and GPX. The only exception is diacylhydrazine 6h which increased two times the GPX activity in exposed cells in comparison with the control cell sample. However, no significant changes were noticed on SOD activity. The decrease in GPX activity could suggest inactivation by ROS.31 Superoxide anions have been shown to inhibit GPX.32 However, it is more likely that such decrease in the activity of GR and GPX could be attributed to other mechanisms, not to the production of ROS, having in mind that there were no changes in SOD activity during applied treatment. It is well known that some 1,3,4-oxadiazoles, but only small group of them, could inhibit the enzymes of glutathione metabolism, dominantly GR.33 That is very important for using of 1,3,4-oxadiazoles and their derivatives as anthelmintic agents.| Glutathione reductase activity (mU mg−1 protein) | Glutathione peroxidase activity (U mg−1 protein) | Superoxide dismutase activity (U mg−1 protein) | |
|---|---|---|---|
| Control | 10.09 ± 0.44 | 1.17 ± 0.16 | 2.70 ± 0.24 |
| 6d | 11.18 ± 0.76 | 0.86 ± 0.11 | 2.66 ± 0.21 |
| 6e | 7.58 ± 1.02 | 0.87 ± 0.09 | 2.53 ± 0.14 |
| 6g | 5.67 ± 0.33 | 0.05 ± 0.02 | 2.57 ± 0.23 |
| 6h | 3.48 ± 0.07 | 2.26 ± 0.21 | 2.66 ± 0.31 |
| 7d | 3.59 ± 0.01 | 0.66 ± 0.08 | 2.64 ± 0.16 |
| 7e | 2.19 ± 0.06 | 0.67 ± 0.12 | 2.76 ± 0.15 |
| 7g | 0.86 ± 0.17 | 0.37 ± 0.07 | 2.73 ± 0.22 |
| 7h | 1.18 ± 0.11 | 0.37 ± 0.10 | 2.79 ± 0.30 |
3. Conclusion
Four diacylhydrazines (6d, 6e, 6g, and 6h) and four 1,3,4-oxadiazoles (7d, 7e, 7g and 7h) showing the best DPPH scavenging activity, with IC50 values in the range of 13.59 to 22.17 μM were selected for further evaluation of their antioxidant potential through various assays. The excellent ABTS radical scavenging potential displayed compounds 6h, 6e and 7e. Considering the structures of the compounds which showed the strongest antioxidant activity (6d, 6e, 6g, 6h, 7d, 7e, 7g and 7h), it can be noticed that they all contain two neighboring hydroxyl groups having the possibility to form an intramolecular hydrogen bond. The most potent compound 7e possesses two hydroxyl groups in m- and p-positions of the aromatic ring, while the presence of additional hydroxyl groups induces a slight decrease of activity (7g and 7h). The compounds 6d, 6e, 6g and 6h possessed moderate to good H2O2 scavenging properties; the most potent was compound 6d. The diacylhydrazines 6e and 6h showed the strongest ferric ion reducing capacities. In addition, the majority of compounds exhibited a stronger reduction of F–C complex than gallic acid; among them, compound 6h had the strongest capacity. The tested compounds applied at non-toxic concentrations showed the ability to reduce intracellular levels of oxidative stress induced by hydrogen peroxide in normal human lung fibroblasts MRC-5. Non-toxic concentrations of the examined compounds reduced the activities of glutathione reductase and glutathione peroxidase in treated MRC-5 cells. The exception was diacylhydrazine 6h which increased two times the activity of glutathione peroxidase in treated cells in comparison with control sample. However, the compounds did not affect the superoxide dismutase activity. Results of our study point out the significant antioxidant potential of selected 1,3,4-oxadiazole derivatives and their diacylhydrazine precursors, especially of compounds 6h and 7e.4. Experimental
4.1. Physical measurements and methods
Melting points were determined on a Mel-Temp capillary melting points apparatus, model 1001 and are uncorrected. Elemental (C, H, N, S) analysis of the samples was carried out in the Center for Instrumental Analysis, Faculty of Chemistry, Belgrade. UV spectra were recorded using an Agilent Technologies, Cary 300 Series UV-Vis Spectrophotometer. IR spectra were obtained on a Perkin Elmer Spectrum One FT-IR spectrometer with a KBr disc. 1H and 13C-NMR spectra were taken on a Varian Gemini 200 MHz spectrometer.4.2. Procedure for the preparation of 7a–h
4.2.2.1. N′-Benzoyl-2-hydroxybenzohydrazide × 0.5H2O (6a). White powder; yield: 0.24 g (89%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.92–7.00, (m, 2H, Ar-H); 7.43–7.62, (m, 4H, Ar-H); 7.93, (dd, 3H, J = 7.9 and 1.2 Hz, Ar-H); 10.69, (s, 2H, NH); 11.95, (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 114.7, 117.5, 119.1, 127.6 (2C), 128.4, 128.6 (2C), 132.0, 132.8, 134.2, 159.4, 165.7, 167.8; IR (KBr, cm−1): 3304; 3058; 1657; 1635; 1548; 1480; 1283; 759; anal. calcd for C14H12N2O3 × 0.5H2O (265.27 g mol−1): C, 63.39; H, 4.94; N, 10.56; found: C, 63.53; H, 4.92; N, 10.60.
4.2.2.2. N′-Benzoyl-3-hydroxybenzohydrazide × 0.5H2O (6b). Beige powder; yield: 0.20 g (74%); mp: >250 °C; for 1H and 13C NMR see ref. 34 or our spectral data placed in ESI.†
4.2.2.3. N′-Benzoyl-4-hydroxy-3-methoxybenzohydrazide × 0.5H2O (6c). Beige powder; yield: 0.18 g (60%); mp: 219–220 °C; 1H NMR (200 MHz, DMSO-d6): 3.83, (s, 3H, CH3); 6.86, (d, 1H, J = 8.2 Hz, Ar-H); 7.42–7.60, (m, 5H, Ar-H); 7.92, (dd, 2H, J = 7.4 and 1.6 Hz, Ar-H); 9.73, (s, 1H, OH); 10.28, (s, 1H, NH); 10.42, (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 55.8, 111.6, 115.1, 121.3, 123.6, 127.5 (2C), 128.5 (2C), 131.8, 132.9, 147.3, 150.2, 165.6, 166.0; IR (KBr, cm−1): 3218; 3009; 1633; 1593; 1513; 1287; 690; anal. calcd for C15H14N2O4 × 0.5H2O (295.29 g mol−1): C, 61.01; H, 5.12; N, 9.49; found: C, 60.94; H, 5.14; N, 9.47.
4.2.2.4. N′-Benzoyl-2,3-dihydroxybenzohydrazide × 0.5H2O (6d). White powder; yield: 0.23 g (82%); mp: 226–227 °C (Dec.); 1H NMR (200 MHz, DMSO-d6): 6.76, (t, 1H, J = 7.8 Hz, Ar-H); 6.99, (dd, 1H, J = 7.8 and 1.2 Hz, Ar-H); 7.39, (dd, 1H, J = 8.0 and 1.2 Hz, Ar-H); 7.49–7.66, (m, 1H, Ar-H); 7.93, (dd, 2H, J = 7.4 and 1.6 Hz, Ar-H); 9.38, (s, 1H, OH); 10.64, (s, 1H, NH); 10.72, (s, 1H, NH); 11.96, (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 114.3, 117.7, 118.6, 119.5, 127.6 (2C), 128.6 (2C), 132.0, 132.5, 146.3, 149.2, 165.8, 168.9; IR (KBr, cm−1): 3434; 3224; 3057; 1636; 1602; 1579; 1535; 1310; 1265; 737; anal. calcd for C14H12N2O4 × 0.5H2O (281.27 g mol−1): C, 59.78; H, 4.66; N, 9.96; found: C, 59.73; H, 4.67; N, 9.93.
4.2.2.5. N′-Benzoyl-3,4-dihydroxybenzohydrazide × 1.5H2O (6e). Beige powder; yield: 0.17 g (57%); mp: 166–167 °C; 1H NMR (200 MHz, DMSO-d6): 6.80, (d, 1H, J = 8.2 Hz, Ar-H); 7.29, (d, 1H, J = 8.2 and 2.0 Hz, Ar-H); 7.34, (d, 1H, J = 2.0 Hz, Ar-H); 7.46–7.59, (m, 3H, Ar-H); 7.91, (dd, 2H, J = 7.2 and 1.6 Hz, Ar-H); 9.25, (s, 1H, OH); 9.59, (s, 1H, OH); 10.16, (s, 1H, NH); 10.36, (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 115.1, 115.5, 119.5, 123.9, 127.5 (2C), 128.5 (2C), 131.8, 132.9; 145.1, 149.0, 165.9, 166.0; IR (KBr, cm−1): 3215; 3055; 1618; 1600; 1576; 1516; 1489; 1296; 688; anal. calcd for C14H12N2O4 × 1.5H2O (299.28 g mol−1): C, 56.38; H, 4.73; N, 9.32; found: C, 56.40; H, 4.74; N, 9.34.
4.2.2.6. N′-Benzoyl-3,5-dihydroxybenzohydrazide (6f). Light beige powder; yield: 0.23 g (84%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.41, (t, 1H, J = 2.2 Hz, Ar-H); 6.75, (d, 2H, J = 2.2 Hz, Ar-H); 7.47–7.60, (m, 3H, Ar-H); 7.91, (dd, 2H, J = 8.0 and 1.6 Hz, Ar-H); 9.56, (s, 2H, OH); 10.26, (s, 1H, NH); 10.41, (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 105.8 (3C), 127.5 (2C), 128.5 (2C), 131.8, 132.8, 134.9; 158.5 (2C); 165.8; 166.2; IR (KBr, cm−1): 3340, 3198; 1676; 1649; 1599; 1541; 1501; 1160; 706; anal. calcd for C14H12N2O4 (272.26 g mol−1): C, 61.76; H, 4.44; N, 10.29; found: C, 61.73; H, 4.45; N, 10.31.
4.2.2.7. 3,4-Dihydroxy-N′-(2-hydroxybenzoyl)benzohydrazide × 1.5H2O (6g). Beige powder; yield: 0.28 g (88%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.81, (d, 1H, J = 8.2 Hz, Ar-H); 6.91–6.99, (m, 2H, Ar-H); 7.29, (dd, 1H, J = 8.2 and 2.0 Hz, Ar-H); 7.34, (d, 1H, J = 2.0 Hz, Ar-H); 7.46, (td, 1H, J = 8.0 and 1.6 Hz, Ar-H); 7.92, (dd, 1H, J = 8.0 and 1.6 Hz, Ar-H); 9.27, (s, 1H, OH); 9.63, (s, 1H, OH); 10.32, (s, 1H, NH); 10.58, (s, 1H, NH); 12.03, (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 114.7, 115.2, 115.4, 117.5, 119.1, 119.6, 123.5, 128.3, 134.2, 145.1, 149.2, 159.5, 165.6, 168.0; IR (KBr, cm−1): 3288, 3055; 2956; 1638; 1605; 1526; 1489; 1302; 1219; 754; anal. calcd for C14H12N2O5 × 1.5H2O (315.28 g mol−1): C, 53.34; H, 4.80; N, 8.88; found: C, 53.35; H, 4.79; N, 8.90.
4.2.2.8. N′-(3,5-Dihydroxybenzoyl)-3,4-dihydroxybenzohydrazide × 0.5H2O (6h). Beige powder; yield: 0.19 g (62%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.39, (t, 1H, J = 2.2 Hz, Ar-H); 6.73, (d, 1H, J = 2.2 Hz, Ar-H); 6.79, (d, 1H, J = 8.2 Hz, Ar-H); 7.27, (dd, 1H, J = 8.2 and 2.0 Hz, Ar-H); 7.33, (d, 1H, J = 2.0 Hz, Ar-H); 9.24, (s, 1H, OH); 9.54, (s, 2H, OH); 9.58, (s, 1H, OH); 10.06, (s, 1H, NH); 10.11, (s, 1H, NH); 13C NMR (50 MHz, DMSO-d6): 105.7, 105.8 (2C), 115.1, 115.5, 119.5, 124.0, 135.1, 145.1, 149.0, 158.4 (2C), 165.7, 166.3; IR (KBr, cm−1): 3385; 3279; 1598; 1508; 1299; 1168; 859; anal. calcd for C14H12N2O6 × 0.5H2O (313.27 g mol−1): C, 53.68; H, 4.18; N, 8.94; found: C, 53.66; H, 4.19; N, 8.92.
4.2.2.9. 2-(5-Phenyl-1,3,4-oxadiazol-2-yl)phenol (7a). White powder; yield: 0.16 g (66%); mp: 158–159 °C; for 1H and 13C NMR see ref. 35 or our spectral data placed in ESI.†
4.2.2.10. 3-(5-Phenyl-1,3,4-oxadiazol-2-yl)phenol × 0.5H2O (7b). Beige powder; yield: 0.14 g (56%); mp: 179–180 °C; for 1H and 13C NMR see ref. 34 or our spectral data placed in ESI.†
4.2.2.11. 2-Methoxy-4-(5-phenyl-1,3,4-oxadiazol-2-yl)phenol × 2.5H2O (7c). Grey powder; yield: 0.17 g (53%); mp: 162–163 °C; 1H NMR (200 MHz, DMSO-d6): 3.90, (s, 3H, CH3); 6.99, (d, 1H, J = 8.8 Hz, Ar-H); 7.58–7.65, (m, 5H, Ar-H); 8.11–8.15, (m, 2H, Ar-H); 9.99, (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 56.0, 110.4, 114.4, 116.2, 120.7, 123.6, 126.6 (2C), 129.5 (2C), 131.9, 148.3, 150.6, 163.5, 164.4; IR (KBr, cm−1): 3432; 3204; 1600; 1502; 1432; 1288; 726; anal. calcd for C15H12N2O3 × 2.5H2O (313.31 g mol−1): C, 57.50; H, 5.47; N, 8.94; found: C, 57.53; H, 5.46; N, 8.96.
4.2.2.12. 3-(5-Phenyl-1,3,4-oxadiazol-2-yl)benzene-1,2-diol (7d). Beige crystals; yield: 0.15 g (58%); mp: 172–173 °C; for 1H and 13C NMR see ref. 36 or our spectral data placed in ESI.†
4.2.2.13. 4-(5-Phenyl-1,3,4-oxadiazol-2-yl)benzene-1,2-diol × 2H2O (7e). Beige powder; yield: 0.16 g (56%); mp: 220–221 °C; 1H NMR (200 MHz, DMSO-d6): 6.93, (d, 1H, J = 8.0 Hz, Ar-H); 7.45, (dd, 1H, J = 8.0 and 2.0 Hz, Ar-H); 7.50, (d, 1H, J = 2.0 Hz, Ar-H); 7.61–7.64, (m, 3H, Ar-H); 8.05–8.10, (m, 2H, Ar-H); 9.58, (s, 1H, OH); 9.86, (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 113.8, 114.4, 116.4, 119.1, 123.7, 126.6 (2C), 129.5 (2C), 131.9; 146.0, 149.5, 163.4, 164.5; IR (KBr, cm−1): 3421; 3253; 1608; 1506; 1448; 1296; 698; anal. calcd for C14H10N2O3 × 2H2O (290.28 g mol−1): C, 57.92; H, 4.86; N, 9.65; found: C, 57.90; H, 4.87; N, 9.63.
4.2.2.14. 5-(5-Phenyl-1,3,4-oxadiazol-2-yl)benzene-1,3-diol (7f). Light yellow powder; yield: 0.19 g (71%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.46, (t, 1H, J = 2.2 Hz, Ar-H); 6.98, (d, 2H, J = 2.2 Hz, Ar-H); 7.62–7.66, (m, 3H, Ar-H); 8.06–8.11, (m, 2H, Ar-H); 9.84, (s, 2H, OH); 13C NMR (50 MHz, DMSO-d6): 104.8 (2C), 106.2, 123.5, 124.7, 126.7 (2C), 129.5 (2C); 132.1; 159.3 (2C); 163.9; 164.3; IR (KBr, cm−1): 3493, 3386; 3102; 1624; 1608; 1567; 1555; 1448; 1160; 853; anal. calcd for C14H10N2O3 × H2O (272.26 g mol−1): C, 61.76; H, 4.44; N, 10.29; found: C, 61.75; H, 4.43; N, 10.27.
4.2.2.15. 4-(5-(2-Hydroxyphenyl)-1,3,4-oxadiazol-2-yl)benzene-1,2-diol × 0.5H2O (7g). Light brown powder; yield: 0.22 g (78%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.94, (d, 1H, J = 8.2 Hz, Ar-H); 7.00–7.12, (m, 2H, Ar-H); 7.42, (dd, 1H, J = 8.2 and 1.6 Hz, Ar-H); 7.47–7.51, (m, 2H, Ar-H); 7.87, (dd, 1H, J = 8.0 and 1.0 Hz, Ar-H); 9.60, (s, 1H, OH); 9.86, (s, 1H, OH); 10.28, (s, 1H, OH); 13C NMR (50 MHz, DMSO-d6): 109.7, 113.8, 114.2, 116.4, 117.2, 119.1, 120.0, 128.4, 133.3, 146.0, 149.6, 156.4, 162.9, 163.7; IR (KBr, cm−1): 3435, 3204; 1615; 1595; 1517; 1491; 1451; 1301; 1253; 746; anal. calcd for C14H10N2O4 × 0.5H2O (279.25 g mol−1): C, 60.21; H, 3.97; N, 10.03; found: C, 60.23; H, 3.96; N, 10.05.
4.2.2.16. 4-(5-(3,5-Dihydroxyphenyl)-1,3,4-oxadiazol-2-yl)benzene-1,2-diol × 2H2O (7h). Brown powder; yield: 0.18 g (57%); mp: >250 °C; 1H NMR (200 MHz, DMSO-d6): 6.43, (t, 1H, J = 2.0 Hz, Ar-H); 6.91–6.95, (m, 3H, Ar-H); 7.38, (d, 1H, J = 8.2 and 2.0 Hz, Ar-H); 7.45, (d, 1H, J = 2.0 Hz, Ar-H); 9.58, (s, 1H, OH); 9.81, (s, 3H, OH); 13C NMR (50 MHz, DMSO-d6): 104.57 (2C), 106.0, 113.7, 114.4, 116.4, 119.0, 125.0, 146.0, 149.5, 159.2 (2C), 163.6, 164.2; IR (KBr, cm−1): 3237, 1605; 1573; 1505; 1453; 1285; 1162; 854; anal. calcd for C14H10N2O5 × 2H2O (322.27 g mol−1): C, 52.18; H, 4.38; N, 8.69; found: C, 52.16; H, 4.37; N, 8.70.
4.3. DPPH free radical scavenging method
The antioxidant activity of synthesized diacylhydrazines and 1,3,4-oxadiazoles was evaluated using DPPH free radical scavenging activity method by Kumarasamy et al.37 1 mL of methanol solution of DPPH (80 mg L−1) was mixed with the same volume of referent antioxidants and synthesized diacylhydrazines and 1,3,4-oxadiazoles dissolved in methanol. The absorbance was measured spectrophotometrically at 517 nm after incubation for 30 min in the dark. The DPPH radical scavenging activity of newly synthesized compounds was compared with well – known referent antioxidants, ascorbic acid and NDGA. DPPH radical scavenging activity was calculated as:where Ac is the absorbance of control (DPPH in methanol) and As is absorbance of the samples. Results were expressed as the concentration of the compounds providing 50% of scavenging of DPPH radicals (IC50), which were calculated through the sigmoidal dose response curve, as μM, using OriginPro8 statistical software. All radical scavenging assays of DPPH were analyzed in triplicates.
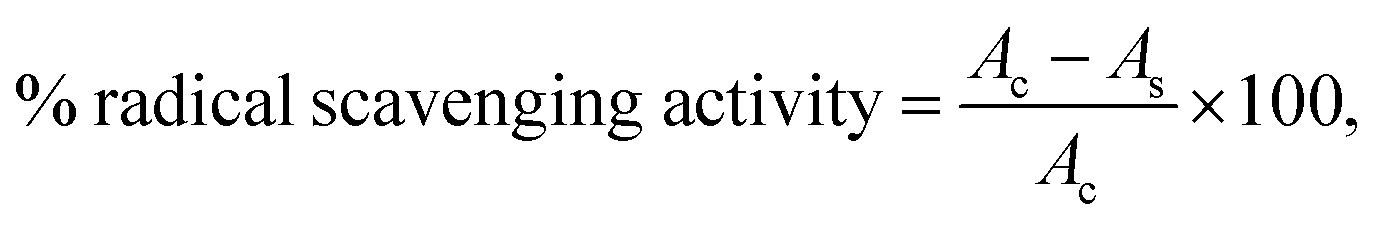
4.4. ABTS radical cation scavenging assay
For the measurement of total antioxidant capacity of synthesized compounds we have employed automated assay using stable ABTS radical cation described previously by Erel.38 Reaction mixture consisted of 200 μL of Reagent 1 (acetate buffer 0.4 M, pH 5.8) containing 5 μL of different sample dilutions prepared in DMSO was mixed with 20 μL of Reagent 2 (10 mM ABTS˙+, 2 mM H2O2 in 30 mM acetate buffer pH 3.6. Solution should be prepared 12–16 h prior to analysis). The first absorbance measurement at 660 nm was taken before mixing Reagent 1 and Reagent 2 and it served as sample blank, the last absorbance measurement was taken after the incubation period of 5 min after the mixing two reagents. The control sample consisted of the same reagents with 5 μL of DMSO instead of the sample in Reagent 1. Results were calculated as follows:| ABTS˙+ scavenging% = [A660 nm control − A660 nm sample]/A660 nm control × 100 |
The antioxidant capacity was expressed as IC50 (concentration of compounds in μM required for 50% reduction of ABTS radicals).
4.5. H2O2 scavenging assay
For the detection of hydrogen peroxide scavenging activity previously described method of Ruch et al. was employed.39 Reaction mixture contained 0.6 mL 40 mM H2O2 in 50 mM phosphate buffer pH 7.4 which was added to 3.4 mL of the same buffer containing different concentrations of test samples. Upon the addition of peroxide, mixture was shaken vigorously and left to incubate at room temperature for 10 min, than the absorbance was recorded at 230 nm. Control sample consisted of phosphate buffer and H2O2 without samples. Percent of H2O2 scavenging was calculated according to the following equation:| H2O2 scavenging% = [A230 nm control − A230 nm sample]/A230 nm control × 100 |
The antioxidant capacity was expressed as IC50 (concentration of compounds in μM required for 50% reduction of H2O2).
4.6. F–C reducing capacity assay
The ability of synthesized compounds to reduce Folin–Ciocalteu's reagent (FC) was measured according to the previously described method of Singleton and Rossi.40 The reaction mixture consisted of 1580 μL H2O, 20 μL sample in DMSO, 100 μL of FC reagent and 300 μL of 20% Na2CO3 solution. After 2 h of incubation at room temperature absorbance was measured at 765 nm and results were expressed as grams of gallic acid equivalents per gram of tested compound (gGAE per g) using gallic acid calibration curve 10–1000 μg mL−1 with linearity range r = 0.993.4.7. Ferric ion-reducing capacity assay
Reducing power of tested compounds was performed according to the method of Pownall et al.41 Sample dilutions were prepared in 50 mM phosphate buffer, pH 7.0 and 500 μL of dilutions were mixed with 250 μL of 1% potassium ferricyanide solution followed by incubation for 20 min at 50 °C. After the incubation 500 μL of 10% trichloroacetic acid was mixed with 500 μL of the incubated sample, 100 μL of 0.1% ferric chloride and 500 μL of distilled water. The mixture was left to incubate for 10 min at room temperature and the absorbance was immediately measured at 700 nm, against blank, which consisted of phosphate buffer and appropriate volume of solvent. The results were expressed as absorbance units at 700 nm which was considered as a measure of reducing power.4.8. Cell culture
Human normal lung fibroblasts MRC-5 were cultured as monolayers at 37 °C in an atmosphere of 5% CO2 and humidified air. The cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). The nutrient medium was RPMI-1640 supplemented with 2 mM L-glutamine, 100 μg mL−1 streptomycin, 100 IU mL−1 penicillin, 10% heat-inactivated (56 °C) fetal bovine serum and 25 mM HEPES, adjusted to pH 7.2 with a bicarbonate solution. RPMI-1640, fetal bovine serum and HEPES were products of Sigma Aldrich (Saint Louis, MO, USA).4.9. MTT cell survival assay
MRC-5 normal lung fibroblasts (5000 cells per well) were seeded in 96-well microtiter plates with flat bottom and 20 h later, after the cell adherence, five concentrations of the investigated compounds were added to the cells (range from 12.5 μM to 200 μM). Nutrient medium only was added to the cells in the control wells. Stock solutions of tested compounds were made in dimethyl sulfoxide (DMSO) at a concentration of 10 mM. After 72 h treatment, the survival of MRC-5 cells was assessed by MTT cell survival test according to the method of Mosmann42 and modified by Ohno and Abe,43 as described elsewhere by our research group.44All experiments were done in triplicate. A chemotherapy drug cisplatin was used as a positive control. The used chemicals were products of Sigma Aldrich.
4.10. Measurement of intracellular ROS levels
After 24 h exposure to non-toxic concentrations of investigated diacylhydrazines and 1,3,4-oxadiazoles (values were determined using MTT test for the 24 h treatment with compounds), the treated and control MRC-5 cells were loaded with a 30 μM 2′,7′-dichlorodihydrofluorescein diacetate (Sigma Aldrich, catalog number D6883) in a phosphate buffered saline (PBS) for 45 min at 37 °C. The cells were then washed with PBS and exposed to 2.5 mM hydrogen peroxide solution (H2O2) for 30 min at 37 °C to induce generation of ROS. Following incubation with H2O2, the cell samples were washed with PBS, collected and the intensity of green fluorescence emitted by the generated probe dichlorofluorescein was measured on a FACSCalibur flow cytometer (BD Biosciences Franklin Lakes, NJ, USA). The data (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events acquired for each cell sample) were analysed using CELLQuest software (BD Biosciences). Applied non-toxic concentrations of the investigated compounds were: 50 μM for the compounds 6d, 6e, 6g and 6h, and 20 μM for the compounds 7d, 7e, 7g and 7h.
000 events acquired for each cell sample) were analysed using CELLQuest software (BD Biosciences). Applied non-toxic concentrations of the investigated compounds were: 50 μM for the compounds 6d, 6e, 6g and 6h, and 20 μM for the compounds 7d, 7e, 7g and 7h.
4.11. Antioxidative enzymes assay
MRC-5 cells (control and cell samples exposed for 24 h to non-toxic concentrations of investigated compounds) were harvested from culture flasks, washed with PBS and centrifuged at 1500 × g for 10 min at 4 °C. Cell pellets were re-suspended in 0.5 mL of PBS and then sonicated on ice three times, for 30 s each. The total extract was centrifuged at 3000 × g for 15 min at 4 °C. Aliquots of the supernatant were used for enzyme assays. Protein concentration in the extracts was determined according to Bradford,45 using a BioRad assay kit with bovine serum albumin as standard.Glutathione reductase (EC 1.6.4.2) was measured according to Foyer and Halliwell.46 The assay medium contained 0.025 mM phosphate buffer pH 7.8, 0.5 mM GSSG, 0.12 mM NADPH-Na4 and 50 μL of protein extract. NADPH oxidation was determined by recording the decrease of absorbance at 340 nm. The activity of this enzyme was expressed as mU mg−1, using extinction co-efficient of NADPH (E) = 6.2 mM−1 cm−1. One unit of GR activity has been defined as one μM of NADPH per minute under standard conditions.
Glutathione peroxidase (EC 1.11.1.9) was assayed according to the procedure of Rotruck et al.47 with some modifications. The reaction mixture consisting of 0.4 mL of 0.4 M sodium phosphate buffer (pH 7.0), 0.1 mL of 10 mM sodium azide, 0.2 mL of 4 mM reduced glutathione, 0.1 mL of 2.5 mM H2O2, 0.2 mL of water and 0.5 mL of enzyme was incubated at 0, 30, 60, 90 seconds respectively. The reaction was terminated with 0.5 mL of 10% TCA and after centrifugation, 2 mL of the supernatant was added to 3 mL of phosphate buffer and 1 mL of DTNB reagent (0.04% DTNB in 1% sodium citrate). The color developed was read at 412 nm and the enzyme activity is expressed in terms of μg of glutathione utilized per min per mg protein.
Superoxide dismutase (EC 1.15.1.1) activity was measured according to Beuchamp and Fridovich.48 Protein extract (50 μL) was added to the reaction mixture (1.5 mL) containing 50 mM potassium phosphate buffer (pH 7.0), 0.1 mM EDTA, 13 mM methionine, 2 μM riboflavin and 75 μM nitro blue tetrazolium (NBT). Riboflavin was added last and the tubes were shaken. The reaction was started by exposing the mixture to a cool white fluorescent light. After 15 min the light was switched off, the tubes were mixed and the absorbance measured at 560 nm. One unit of enzyme activity was measured as the amount of SOD capable of inhibiting 50% of nitrite formation under the assay conditions.
4.12. Statistical evaluations
The results of DPPH scavenging activity are expressed as mean of three experiments ± standard deviation (SD). The IC50 was calculated using OriginPro8 statistical software. The results were statistically analyzed by one way analysis of variance (ANOVA) using the SPSS statistical software package (version13.0). Comparison between IC50 values was performed with T3 Dunnett test or with Bonferroni test. The results were considered to be statistically significant at p < 0.05.Acknowledgements
The authors are grateful to the Ministry of Education, Science and Technological Development of the Republic of Serbia for financial support (Projects 172016, 175011 and 173005).References
- A. M. Pisoschi and A. Pop, Eur. J. Med. Chem., 2015, 97, 55–74 CrossRef CAS PubMed.
- M. Eberlein, K. A. Scheibner, K. E. Black and W. Willet, Science, 1994, 264, 532–537 Search PubMed.
- R. Roberta, G. M. Luciana, C. C. Luciana and P. Glaucia, J. Food Sci., 2006, 71, C102–C107 CrossRef.
- B. Halliwell and J. M. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, Midsomer Norton, Avon, England, 3rd edn, 1999 Search PubMed.
- C. Siquet, F. Pavia-Martins, J. L. Lima, S. Reis and F. Borges, Free Radical Res., 2006, 40, 433–442 CrossRef CAS PubMed.
- N. Ivanović, L. Jovanović, Z. Marković, V. Marković, M. D. Joksović, D. Milenković, P. T. Djurdjević, A. Ćirić and L. Joksović, ChemistrySelect, 2016, 1, 3870–3878 CrossRef.
- X. Ma, H. Li, J. Dong and W. Qian, Food Chem., 2011, 126, 698–704 CrossRef CAS.
- P. C. Eklund, O. K. Långvik, J. P. Wärnå, T. O. Salmi, S. M. Willför and R. E. Sjöholm, Org. Biomol. Chem., 2005, 3, 3336–3347 CAS.
- C. Soares de Oliveira, B. Freitas Lira, J. M. Barbosa-Filho, J. G. Fernandez Lorenzo and P. Filgueiras de Athayde-Filho, Molecules, 2012, 17, 10192–10231 CrossRef CAS PubMed.
- H. Khalilullah, M. J. Ahsan, M. Hedaitullah, S. Khan and B. Ahmed, Mini-Rev. Med. Chem., 2012, 12, 789–801 CrossRef CAS PubMed.
- P. Neeraja, S. Srinivas, K. Mukkanti, P. K. Dubey and S. Pal, Bioorg. Med. Chem. Lett., 2016, 26, 5212–5217 CrossRef CAS PubMed.
- A. Lukin, R. Karapetian, Y. Ivanenkov and M. Krasavin, Lett. Drug Des. Discovery, 2016, 13, 198–204 CrossRef CAS.
- M. Swapna, C. Premakumari, S. N. Reddy, A. Padmaja and V. Padmavathi, Chem. Pharm. Bull., 2013, 61, 611–617 CrossRef CAS PubMed.
- E. A. Musad, R. Mohamed, B. A. Saeed, B. S. Vishwanath and K. M. L. Rai, Bioorg. Med. Chem. Lett., 2011, 21, 3536–3540 CrossRef CAS PubMed.
- V. B. Iyer, B. M. Gurupadayya, B. Inturi, K. Venkata Sairam and G. V. Pujar, RSC Adv., 2016, 6, 24797–24807 RSC.
- Y. Kotaiah, N. Harikrishna, K. Nagaraju and C. V. Rao, Eur. J. Med. Chem., 2012, 58, 340–345 CrossRef CAS PubMed.
- S. Bondock, S. Adel and H. A. Etman, Res. Chem. Intermed., 2016, 42, 1845–1861 CrossRef CAS.
- Dinesha, S. Viveka, S. Chandra and G. K. Nagaraja, Monatsh. Chem., 2015, 146, 207–214 CrossRef CAS.
- S. Olson, S. D. Aster, K. Brown, L. Carbin, D. W. Graham, A. Hermanowski-Vosatka, C. B. LeGrand, S. S. Mundt, M. A. Robbins, J. M. Schaeffer, L. H. Slossberg, M. J. Szymonifka, R. Thieringer, S. D. Wright and J. M. Balkovec, Bioorg. Med. Chem. Lett., 2005, 15, 4359–4362 CrossRef CAS PubMed.
- R. M. Shakir, A. Ariffin and M. A. Abdulla, Molecules, 2014, 19, 3436–3449 CrossRef PubMed.
- V. Nieddu, G. Pinna, I. Marchesi, L. Sanna, B. Asproni, G. A. Pinna, L. Bagella and G. Murineddu, J. Med. Chem., 2016, 59, 10451–10469 CrossRef CAS PubMed.
- C. T. Brain, J. M. Paul, Y. Loong and P. J. Oakley, Tetrahedron Lett., 1999, 40, 3275–3278 CrossRef CAS.
- T. Isobe and T. Ishikawa, J. Org. Chem., 1999, 64, 6989–6992 CrossRef CAS.
- A. I. Vogel, in Textbook of Practical Organic Chemistry, Longman Group Ltd., London, 3rd edn, 1956, p. 782 Search PubMed.
- L. Wang, D.-G. Guo, Y.-Y. Wang and C.-Z. Zheng, RSC Adv., 2014, 4, 58895–58901 RSC.
- G. Singh, S. Rani, A. Arora, D. Aulakh and M. Wriedt, New J. Chem., 2016, 40, 6200–6213 RSC.
- R. L. Prior, X. Wu and K. Schaich, J. Agric. Food Chem., 2005, 53, 4290–4302 CrossRef CAS PubMed.
- G. Y. Liou and P. Storz, Free Radic. Res., 2010, 44, 479–496 CrossRef CAS PubMed.
- N. Hauptmann, J. Grimsby, J. C. Shih and E. Cadenas, Arch. Biochem. Biophys., 1996, 335, 295–304 CrossRef CAS PubMed.
- T. Arimoto, M. B. Kadiiska, K. Sato, J. Corbett and R. P. Mason, Am. J. Respir. Crit. Care Med., 2005, 171, 379–387 CrossRef PubMed.
- M. Radu, M. Munteanu, S. Petrache, A. I. Serban, D. Dinu, A. Hermenean, C. Sima and A. Dinischiotu, Acta Biochim. Pol., 2010, 57, 355–360 CAS.
- J. Blum and I. Fridovich, Arch. Biochem. Biophys., 1985, 240, 500–508 CrossRef CAS PubMed.
- R. S. Treger, A. G. Cook, G. Rai, D. J. Maloney, A. Simeonov, A. Jadhav, C. J. Thomas, D. L. Williams, M. Cappello and J. J. Vermeire, International Journal for Parasitology: Drugs and Drug Resistance, 2012, 2, 171–177 CrossRef CAS PubMed.
- H. C. Hansen, F. S. Chiacchia, R. Patel, N. C. W. Wong, V. Khlebnikov, R. Jankowska, K. Patel and M. M. Reddy, J. Med. Chem., 2010, 45, 2018–2023 CrossRef CAS PubMed.
- M. Kidway, D. Bhatnar and N. K. Mishra, Green Chem. Lett. Rev., 2010, 3, 55–59 CrossRef.
- D. Suresh, K. Kanagaraj and K. Pitchumani, Tetrahedron Lett., 2014, 55, 3678–3682 CrossRef CAS.
- Y. Kumarasamy, M. Byres, P. J. Cox, M. Jaspars, L. Nahar and S. D. Sarker, Phytother. Res., 2007, 21, 615–621 CrossRef PubMed.
- O. Erel, Clin. Biochem., 2004, 37, 277–285 CrossRef CAS PubMed.
- R. J. Ruch, S. J. Cheng and J. E. Klaunig, Carcinogenesis, 1989, 10, 1003–1008 CrossRef CAS PubMed.
- V. L. Singleton and J. A. Rossi, Am. J. Enol. Vitic., 1965, 16, 144–158 CAS.
- T. L. Pownall, C. C. Udenigwe and R. E. Aluko, J. Agric. Food Chem., 2010, 58, 4712–4718 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. Ohno and T. Abe, J. Immunol. Methods, 1991, 145, 199–203 CrossRef CAS PubMed.
- M. V. Rodić, V. M. Leovac, L. S. Jovanović, V. Spasojević, M. D. Joksović, T. Stanojković, I. Z. Matić, L. S. Vojinović-Ješić and V. Marković, Eur. J. Med. Chem., 2016, 115, 75–81 CrossRef PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- C. H. Foyer and B. Halliwell, Planta, 1976, 133, 21–25 CrossRef CAS PubMed.
- J. T. Rotruck, A. L. Pope, H. E. Ganther, A. B. Swanson, D. G. Hafeman and W. G. Hoekstra, Science, 1973, 179, 588–590 CAS.
- C. O. Beuchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28787e |
| This journal is © The Royal Society of Chemistry 2017 |