DOI:
10.1039/C6RA27709H
(Paper)
RSC Adv., 2017,
7, 8542-8549
The construction of electrochemical chiral interfaces using hydroxypropyl chitosan
Received
3rd December 2016
, Accepted 20th December 2016
First published on 26th January 2017
Abstract
A simple chiral electrochemical sensor based on hydroxypropyl chitosan (HPCS) covalently bound to multi-walled carbon nanotubes (MWCNTs) was developed for the recognition of mandelic acid (MA) enantiomers. HPCS preferably combines with (S)-MA over (R)-MA, which was attributed to the favorable host–guest interactions between HPCS and the MA enantiomers. In addition, the recognition efficiency was significantly increased after hydroxypropyl groups were grafted onto chitosan (CS), which may be due to a stereo-hindrance effect. Moreover, the electrochemical recognition of amino acid enantiomers was investigated; the functional groups of the enantiomers play an important role in the enantioselective discrimination.
Introduction
The chiral recognition of active synthetic and natural compounds is a fundamental field, which is attracting increasing attention, particularly in the enantioselective recognition of chiral drugs.1 A critical step in the fabrication of chiral electrochemical sensors is to build a chiral surface with recognition sites for discriminating enantiomers; the modified chiral selector plays a key role in the fabrication of the chiral surface.2 Thus far, various chiral selectors have been applied in enantioselective recognition such as bovine serum albumin,3,4 human serum albumin,5,6 deoxyribonucleic acid (DNA),7 sodium alginate,8 β-cyclodextrin9 and amino acids.10
Chitosan (CS) is similar to DNA with a double helix structure; its wide substrate scope and significant selectivity are attributed to the existence of a large amount of chiral sites.11 CS was prepared by alkaline deacetylation of chitin with a high degree of deacetylation, it has poor solubility in water but is soluble in aqueous dilute acids.12 The limited solubility of CS restricts its use in chiral electrochemical sensors;13 however, there is an enormous potential for structural modification to impart special chemical functions and properties. Apart from the hydroxyl groups, there are several ways to chemically modify the amino groups in CS.14 X. Q. Chen et al. (2015) constructed a chiral electrochemical sensor based on CS functionalized multi-walled carbon nanotubes (MWCNTs) used for the discrimination of tryptophan (Trp) enantiomers.15 Y. Kong et al. (2015) developed a novel enantioselective system to enantioselectively recognize Trp based on sulfonated chitosan and the recognition efficiency was improved remarkably because of the introduction of the alkyl sulfonate, which generated significant steric hindrance in the formation of intermolecular hydrogen bonds in the enantioselective recognition system.16 Hydroxypropyl chitosan (HPCS) is regarded as an important functional derivative of chitosan. The water solubility is enhanced when the hydroxypropyl group is introduced into CS.17 Moreover, there is a possible role in the discrimination of chiral enantiomers due to the introduced hydroxypropyl groups leading to the possibility of different affinities between CS and enantiomers.18
MA enantiomers are important structural units of several natural products and drug molecules, particularly, (R)-MA serves as chiral synthon for the synthesis of drugs, such as semi-synthetic penicillin and cephalosporin.19 Y. Z. Fu et al. developed a strategy for the recognition of mandelic acid (MA) enantiomers in the presence of Zn(II) ions based on chiral ligand exchange.20 E. Zor et al. reported an electrochemical biosensor for the discrimination of MA enantiomers based on a three-points interaction model, which has a shorter response time and higher recognition efficiency than the chiral ligand exchange model.21 However, a fast electrochemical chiral sensor used for the discrimination of MA enantiomers based on MWCNTs–HPCS has not been reported to date.
In this study, the water-soluble chitosan derivative, HPCS was prepared through amino protection using benzaldehyde in order to selectively introduce the hydroxypropyl group. HPCS was linked to the carboxylic multi-walled carbon nanotubes (MWCNTs–COOH) through covalent bonding after deprotection of the amino groups. The MWCNTs–HPCS was used to construct an electrochemical sensor used for the stereoselective recognition of MA enantiomers via cyclic voltammetry (CV) and the results were satisfactory.
Experimental
Reagents and materials
Chitosan (with a degree of deacetylation of 95%). N-Hydroxy succinimide (NHS, 98%) and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDC, 99%) were purchased from Aladdin Industrial Corporation. 1,2-Epoxypropane was supplied by Sinopharm Chemical Reagent Co., Ltd. Tetramethylammonium hydroxide and benzaldehyde were supplied by Tianjing Kaixing Chemical Reagent Co., Ltd (Tianjing, China). The multi-walled carbon nanotubes (MWCNTs) were obtained from Shenzhen Bier technology Co., Ltd (Shenzhen, China). The MA enantiomers were purchased from Energy Chemical Corporation. All chemicals were of analytical grade and used without further purification. Ultrapure water was used throughout the experiments.
Apparatus
All electrochemical measurements were carried out on a CHI60E (Shanghai Chenhua Instruments Co., China). A three-electrode system included a glassy carbon electrode (GCE, Φ = 3 mm) as the working electrode, a platinum wire as the auxiliary electrode, and a saturated calomel electrode as the reference electrode. The morphology and functional groups of the MWCNTs–COOH, HPCS and MWCNTs–HPCS hybrids were recorded using a JSM-6701F cold field emission scanning electron microscope (Japan). Fourier-transform infrared (FT-IR) spectra were obtained using a QUINOX55 spectrometer. X-ray photoelectron spectroscopy (XPS) analysis was characterized using an Axis Ultra spectrometer.
Preparation of hydroxypropyl chitosan
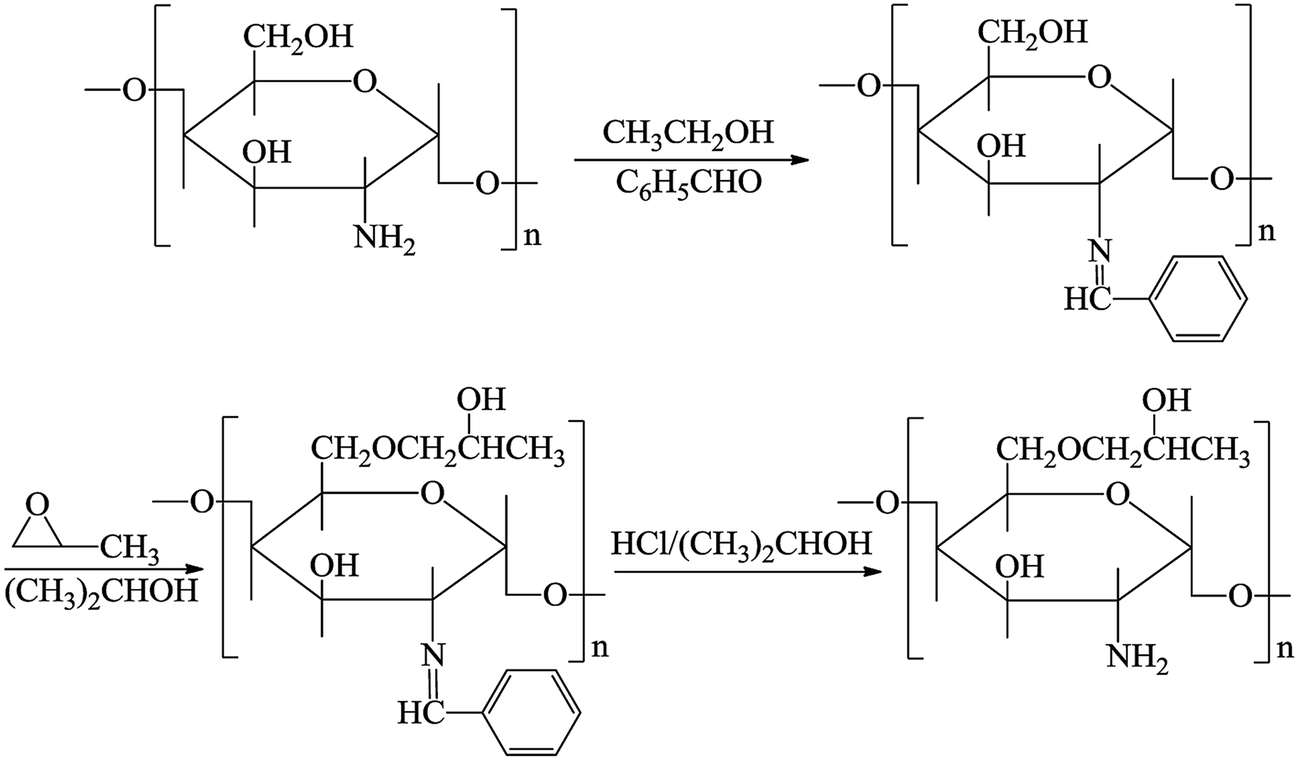
0.5 g of CS was added into 100 mL of an acetic acid solution (1 wt%) in a round-bottom flask. A mixture of 2.7 mL of benzaldehyde and 15 mL of ethanol was added and the CS solution was stirred at room temperature for 3 h. The mixture was adjusted to pH = 7 using sodium hydroxide solution, filtered and washed with acetone and absolute ethanol to remove the unreacted benzaldehyde, and the N-benzylidene chitosan (B-CS) was obtained after being dried at 40 °C.
B-CS (0.3 g) was basified using 2.0 g of a sodium hydroxide aqueous solution (35 wt%) in an isopropanol solution (4 mL) for 12 h. Alkali CS was incorporated into 4 mL of isopropanol, 5 drops of tetramethylammonium hydroxide (25 wt%) and 3 mL of propylene oxide were added, and the mixture was heated at 45 °C for 12 h. The reaction mixture was adjusted to pH = 7 using hydrochloric acid (20 wt%), cooled and washed several times with acetone. Afterwards the products were soaked in 0.25 N hydrochloric acid/ethanol solution overnight as the deprotection step and white HPCS powder was obtained after filtration and vacuum drying at 50 °C for 12 h.22
Preparation of hydroxypropyl chitosan functionalized multi-walled carbon nanotubes
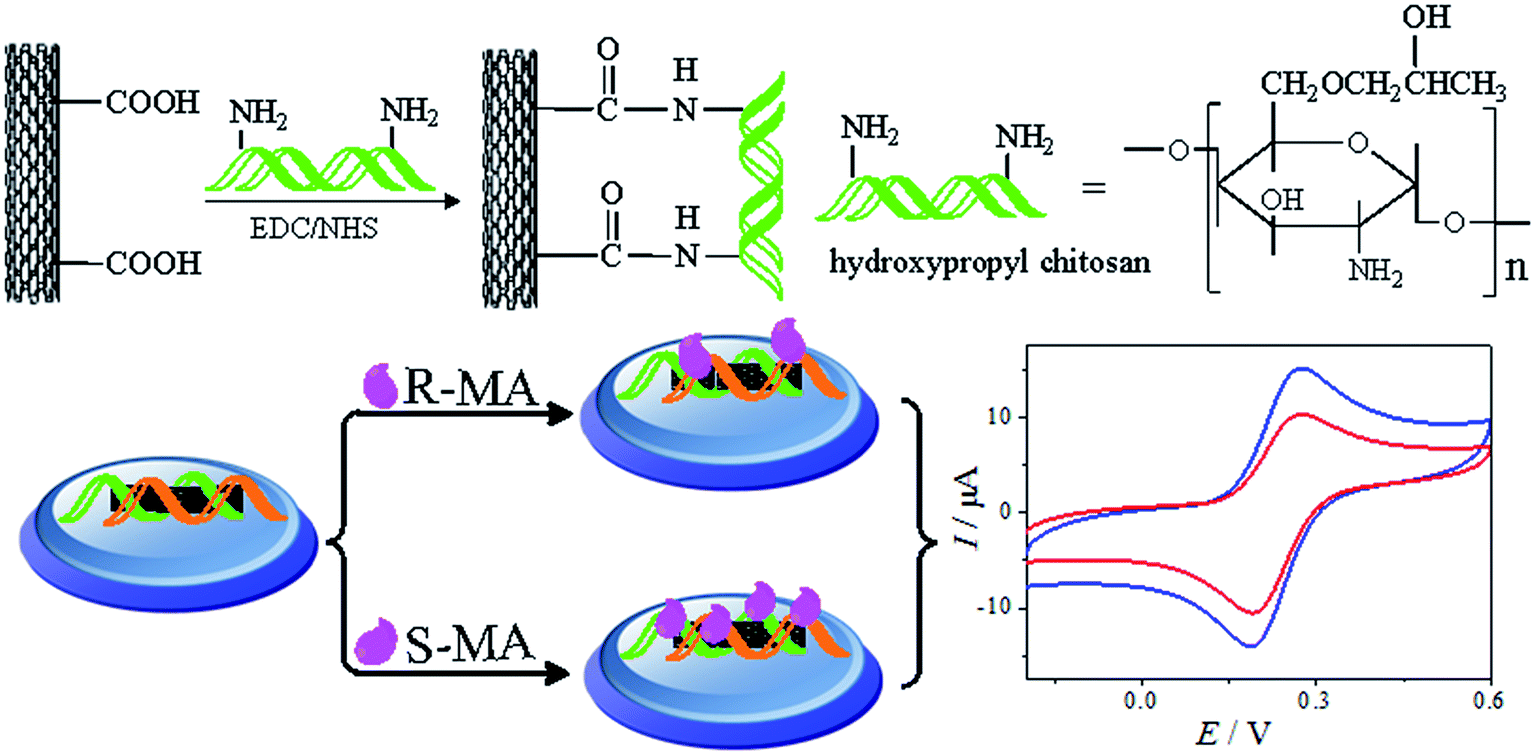
The MWCNTs–COOH was synthesized as described previously.23 First, 0.03 g of HPCS was dissolved in 5 mL of an acetic acid solution (1 wt%). Next, 0.006 g of MWCNTs–COOH was dispersed in 2 mL of ultrapure water via ultrasonication. 0.08 g of EDC and 0.2 g of NHS were added to the abovementioned solution. Subsequently, the HPCS solution was added under continuous stirring for 24 h. The product was isolated by filtration and dried under vacuum at 60 °C.24 The synthesis process used to prepare hydroxypropyl chitosan is shown in Fig. 1.
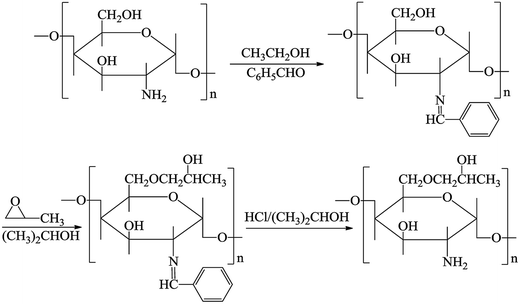 |
| | Fig. 1 The procedures used for the preparation of hydroxypropyl chitosan. | |
Fabrication of the MWCNTs–HPCS chiral sensor
Prior to the experiment, the glassy carbon electrode (GCE, Φ = 3 mm) was polished with 1.0, 0.3 and 0.05 μm alumina slurry. Afterwards, the GCE was sonicated in ethanol and ultrapure water and dried in air. Then, 6 mL of a homogeneous MWCNTs–HPCS dispersed solution was dropped on the surface of the freshly polished GCE and then dried using an infrared lamp. As a comparison, a MWCNTs–CS/GCE was prepared using the same procedure. The fabrication process is illustrated in Scheme 1.
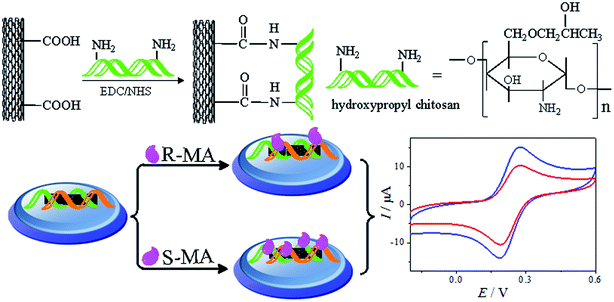 |
| | Scheme 1 A schematic of the MWCNTs–HPCS chiral sensor fabrication and the electrochemical responses to MA enantiomers. | |
Results and discussion
Characterization of the prepared MWCNTs–HPCS
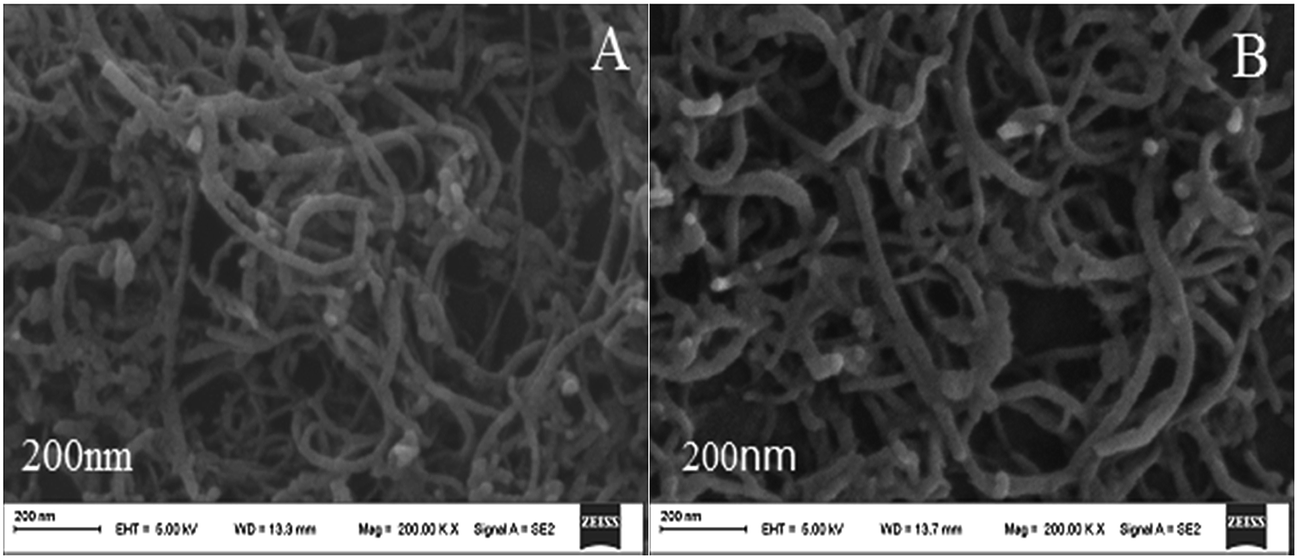
The surface morphologies of the MWCNTs–HPCS films at different stages were studied using scanning electron microscopy (SEM). As shown in Fig. 2A, the MWCNTs–COOH have a kinkled tubular structure and are intertwined with each other. Compared with MWCNTs–COOH, the diameter of MWCNTs–HPCS was changed, which indicated that the HPCS was grafted onto the sides of the MWCNTs (Fig. 2B).
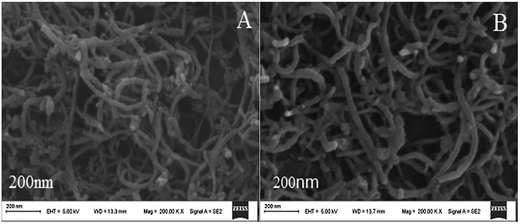 |
| | Fig. 2 SEM images of (A) MWCNTs–COOH and (B) MWCNTs–HPCS. | |
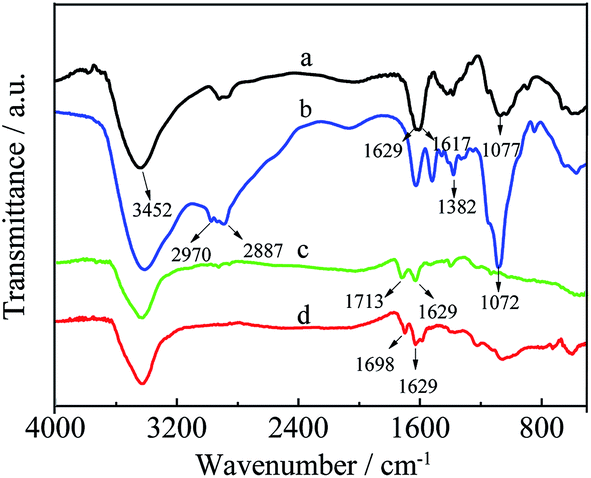
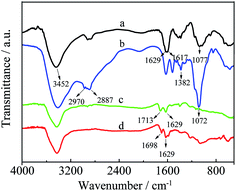
As shown in Fig. 3, the products of HPCS and MWCNTs–HPCS were confirmed via the FT-IR spectra of CS, HPCS, MWCNTs–COOH and MWCNTs–HPCS. For CS (curve a), the stretching vibrations of O–H are detected at approximately 3452 cm−1, and the two characteristic peaks located at 1629 cm−1 and 1617 cm−1 are also observed, which can be assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the bending vibrations of N–H, respectively.25 HPCS exhibits similar absorption peaks to those of CS (curve b), the broad peak at around 3410 cm−1 was ascribed to the stretching vibrations of O–H due to intermolecular or intramolecular hydrogen bonding. The stretching vibration enhancement of –CH2– was located at 2887 cm−1. Moreover, the characteristic peaks of –CH3 appeared at 2970 cm−1 and 1382 cm−1. In addition, the stronger peak of C–O moved to 1072 cm−1. This evidence suggests that the hydroxylpropyl group had been linked to CS.26 For MWCNTs–COOH (curve c), the characteristic peaks of MWCNTs–COOH were centered at 1713 m−1 (CO), 1370 cm−1 (C–O) and 3439 cm−1 (–OH). The peak of CO stretching for MWCNTs–COOH (1713 cm−1) shifts to 1698 cm−1 due to the amidization reaction (curve d). In addition, a new strong band at 1629 cm−1 appeared in curve d of the MWCNTs–HPCS due to the formation of –NHCO–, which indicated that the hydroxypropyl group was grafted onto the MWCNTs–COOH.27,28
O and the bending vibrations of N–H, respectively.25 HPCS exhibits similar absorption peaks to those of CS (curve b), the broad peak at around 3410 cm−1 was ascribed to the stretching vibrations of O–H due to intermolecular or intramolecular hydrogen bonding. The stretching vibration enhancement of –CH2– was located at 2887 cm−1. Moreover, the characteristic peaks of –CH3 appeared at 2970 cm−1 and 1382 cm−1. In addition, the stronger peak of C–O moved to 1072 cm−1. This evidence suggests that the hydroxylpropyl group had been linked to CS.26 For MWCNTs–COOH (curve c), the characteristic peaks of MWCNTs–COOH were centered at 1713 m−1 (CO), 1370 cm−1 (C–O) and 3439 cm−1 (–OH). The peak of CO stretching for MWCNTs–COOH (1713 cm−1) shifts to 1698 cm−1 due to the amidization reaction (curve d). In addition, a new strong band at 1629 cm−1 appeared in curve d of the MWCNTs–HPCS due to the formation of –NHCO–, which indicated that the hydroxypropyl group was grafted onto the MWCNTs–COOH.27,28
 |
| | Fig. 3 The FT-IR spectra of CS (a), HPCS (b), MWCNTs–COOH (c) and MWCNTs–HPCS (d). | |
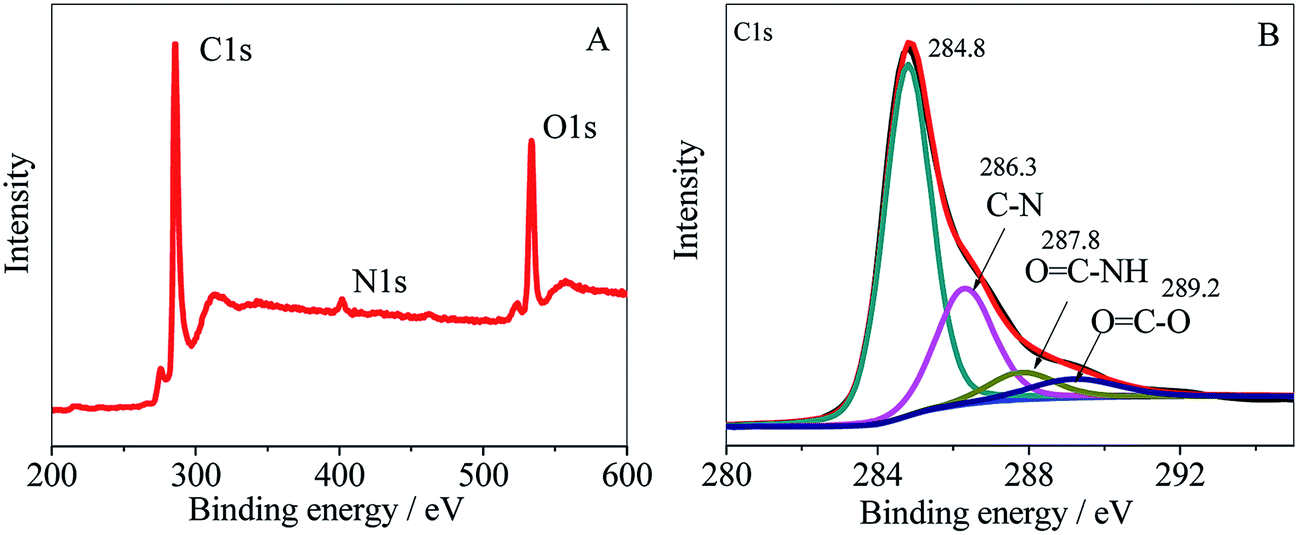
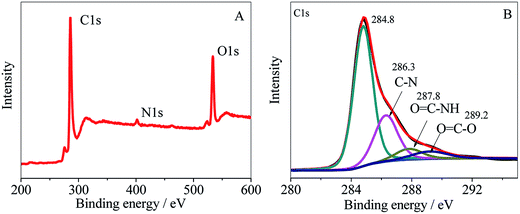
HPCS was grafted onto MWCNTs–COOH covalently, which can be confirmed from the FT-IR spectra. The full XPS spectrum of MWCNTs–HPCS is shown in Fig. 4A. The XPS survey scan of the MWCNTs–HPCS indicates the presence of C 1s (286.3 eV), O 1s (533 eV) and N 1s (401 eV) peaks. The high resolution XPS C 1s is shown in Fig. 4B; the peak located at 286.3 eV was due to the C–N bond of HPCS. Moreover, the peaks at 287.8 eV (OC–NH) and 289.2 eV (OC–O) were observed. More importantly, the peak observed at 287.8 eV, which originated from the amidation reaction between MWCNTs–COOH and HPCS, suggests that HPCS was successfully grafted onto the MWCNTs–COOH.29,30
 |
| | Fig. 4 The XPS spectrum of the (A) survey scan and (B) C 1s region of the MWCNTs–HPCS composite. | |
Electrochemical recognition of MA enantiomers with MWCNTs–HPCS
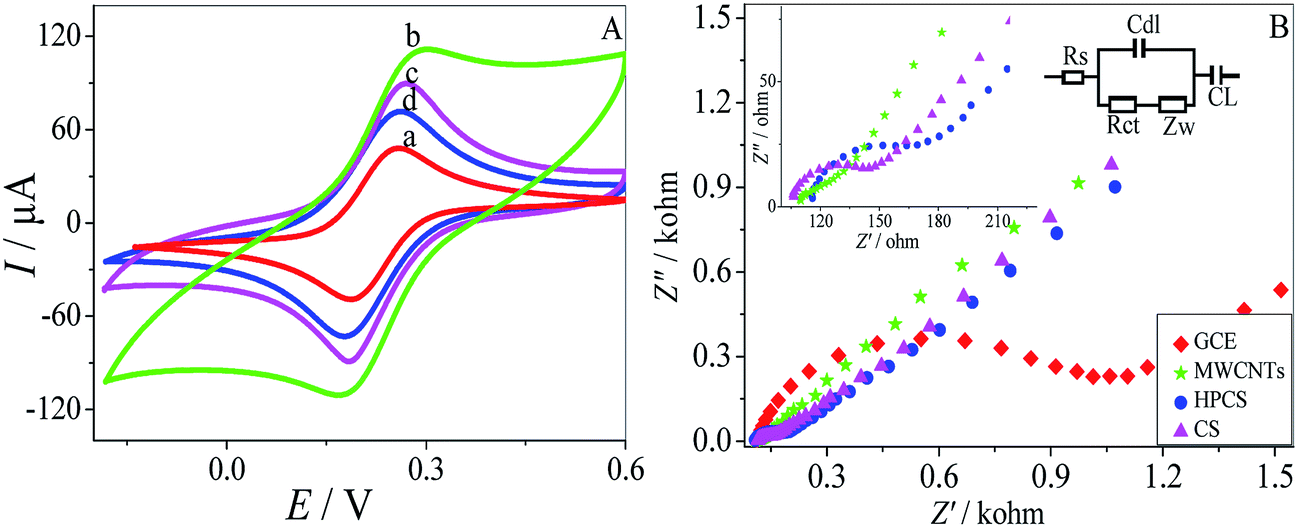
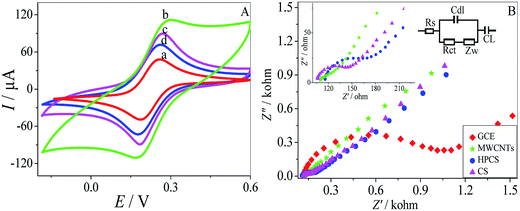
CV was performed to investigate the electrochemical properties of the MWCNTs–HPCS modified GCE in 5.0 mM [Fe(CN)6]4−/3− containing 0.1 M KCl as the electroactive probe couple31,32 (Fig. 5A). The CV of the bare GCE in [Fe(CN)6]4−/3− was also carried out for comparison (a), a couple of well-defined redox peaks appeared at the MWCNTs–COOH/GCE, which were caused by the redox transition between the electroactive probe couple. The peak currents at the MWCNTs–COOH/GCE increased significantly, and the ΔIP was increased due to the high conductivity of the introduced MWCNTs (b). Moreover, the peak currents of MWCNTs–HPCS/GCE (d) were lower than those of MWCNTs–CS (c) due to the introduction of the hydroxylpropyl group. The result suggests that the HPCS provides more hindrance to the electronic transition of the electrode.33
 |
| | Fig. 5 (A) CV of 5.0 mM [Fe(CN)6]4−/3− containing 0.1 M KCl at the bare GCE (a), MWCNTs–COOH/GCE (b), MWCNTs–CS/GCE (c) and MWCNTs–HPCS (d). (B) Nyquist plots at the bare GCE, MWCNTs–COOH, MWCNTs–HPCS and MWCNTs–CS. Inset: magnified Nyquist plots at the MWCNTs, HPCS and CS (left); equivalent circuit (right). | |
Electrochemical impedance spectroscopy (EIS) was applied to investigate the interfacial properties of the modified electrodes. Moreover, the equivalent circuit was provided in Fig. 5B. The bare GCE has the biggest semicircle, and the Ret value of the bare GCE was about 1073 Ω. However, the Ret value of the MWCNTs–COOH was 114 Ω, which acts as a conducting bridge, and it was found that the value of the charge transfer resistance was significantly lower than the other electrode. Moreover, as for the HPCS (167 Ω), the value of resistance was larger than CS (143 Ω) due to the introduction of the hydroxypropyl groups, which was attributed to the obstruction of electron transfer from HPCS. The results from EIS and CV were confirmed with each other.
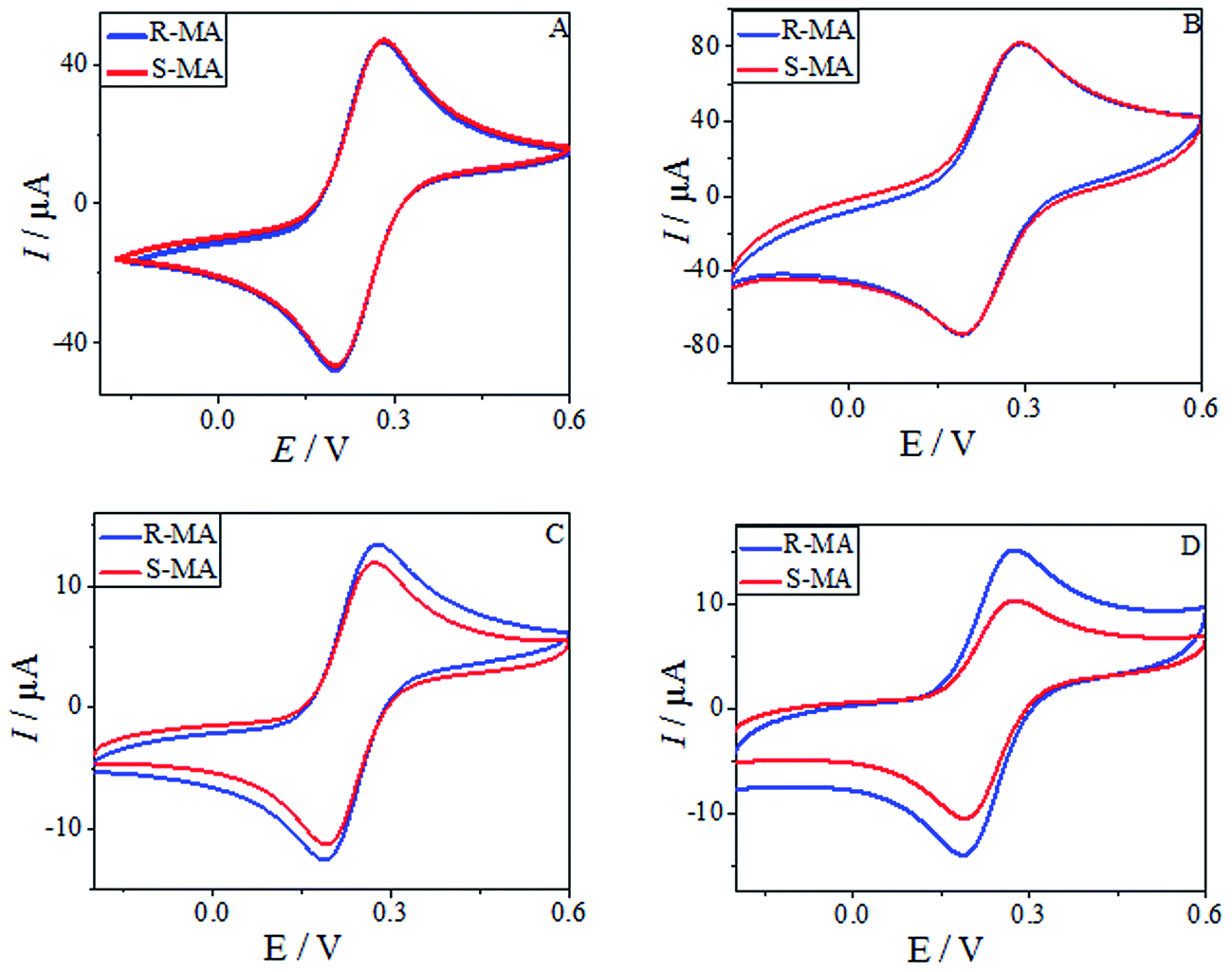
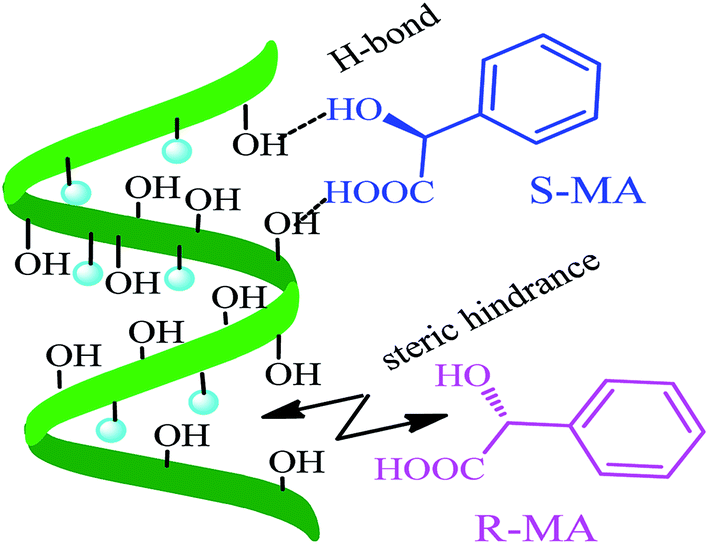
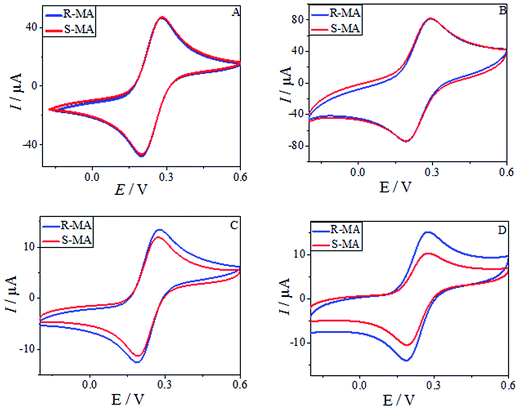
CV was used to study the electrochemical properties of the modified electrodes, and [Fe(CN)6]4−/3− was applied as the electroactive probe to record the peak currents of the different modified electrodes. The current difference (ΔIp) of the [Fe(CN)6]4−/3− redox couple indicates the changes in the progress of GCE modification. Fig. 6 shows the CV of (R)- and (S)-MA at the different electrochemical interfaces. The MA enantiomers exhibited completely similar responses at the bare GCE (Fig. 6A), suggesting that the bare GCE was incapable of recognizing the MA enantiomers due to the absence of a chiral selector. Similarly, there was no significant difference in the peak currents (Ip) observed in the CV of the MWCNTs (Fig. 6B). When compared with CS as a chiral selector (Fig. 6C), discernable differences in the Ip appeared in the CV of the HPCS modified GCE (Fig. 6D); HPCS exhibited a higher affinity for (S)-MA than (R)-MA. It was noteworthy that a remarkable difference can be observed from the Ip based HPCS, which may be attributed to the stereo-hindrance of the hydroxypropyl groups. In addition, after the hydroxypropyl groups were grafted onto the CS, the hydrophility of HPCS was greatly improved, which lead to the formation of hydrogen bonds between the MA enantiomers and HPCS. Furthermore, the hydrogen bonds were more easily formed between (S)-MA and HPCS compared with (R)-MA, which was attributed to the different stereoisomeric configurations of the MA enantiomers. The results indicate that the peak current ratio (IR-MA/IS-MA) with the HPCS chiral interfaces (1.48) was significantly higher than CS (1.13). The possible recognition mechanism is shown in Fig. 7.
 |
| | Fig. 6 CV for the redox reactions of 10 mM MA on the different electrodes in 0.2 M PBS buffer solution (pH 7.0) containing 5.0 mM [Fe(CN)6]4−/3−. (A) Bare GCE, (B) MWCNTs, (C) MWCNTs–CS and (D) MWCNTs–HPCS. | |
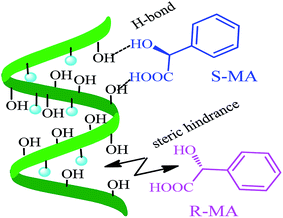 |
| | Fig. 7 The chiral recognition mechanism between MWCNTs–HPCS and the MA enantiomers. | |
Analysis of single MA enantiomers
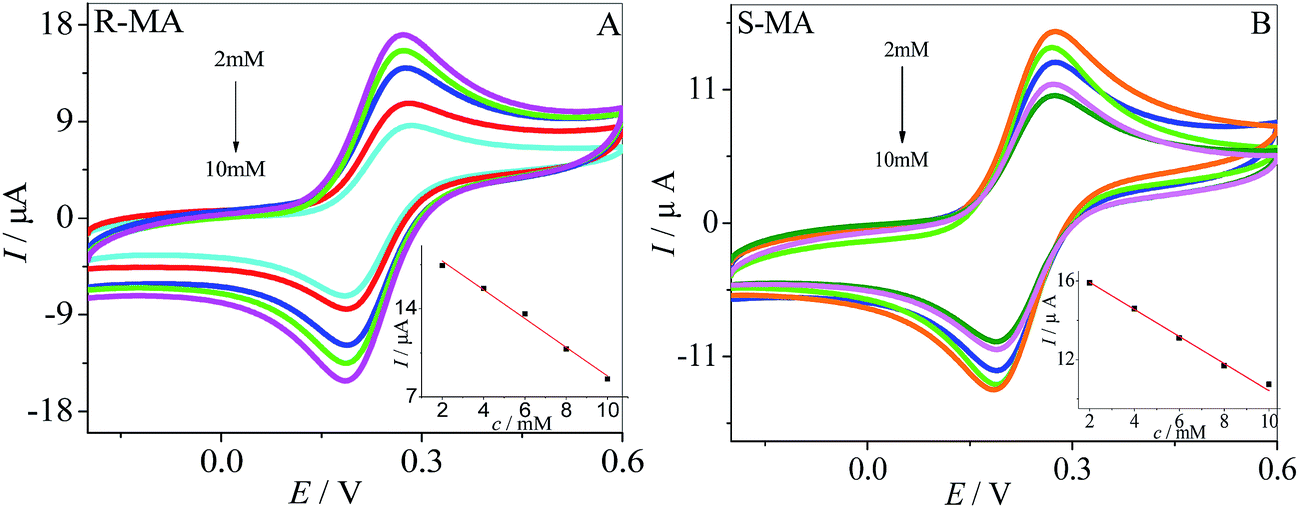
The current response of the MWCNTs–HPCS modified GCE to (R)-MA and (S)-MA at various concentrations is shown in Fig. 8A. Along with the (R)-MA concentration increasing from 2 to 10 mM, more and more of the (R)-MA interact with the surface of the MWCNTs–HPCS/GCE, and the peak current gradually decreases. The linear relationship between peak current and the concentration of (R)-MA is shown in the inset of Fig. 8A. The peak current decreased upon increasing the (S)-MA concentration from 2 to 10 mM and the relationship of (S)-MA concentration and peak current is illustrated (Fig. 8B). The linear regression equations for (R)-MA and (S)-MA can be presented: Ip (μA) = −1.14 c + 20.06 (R2 = 0.9963) (curve A) and Ip (μA) = −0.66 c + 17.18 (R2 = 0.9930) (curve B), respectively. In addition, the (R)-MA and (S)-MA enantiomers can be discriminated by the MWCNTs–HPCS/GCE in this concentration range.
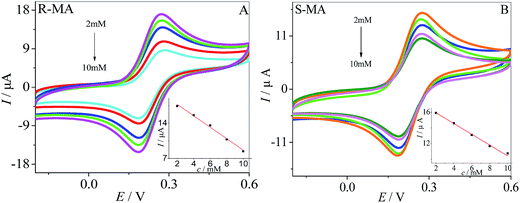 |
| | Fig. 8 CV images of (R)-MA (A) and (S)-MA (B) at MWCNTs–HPCS modified GCE in 0.2 M PBS buffer solution (pH 7.0) containing 5.0 mM [Fe(CN)6]4−/3−. Inset: linear relationship of the concentration of MA enantiomers and Ip of CV corresponding. | |
Optimization of the experimental conditions
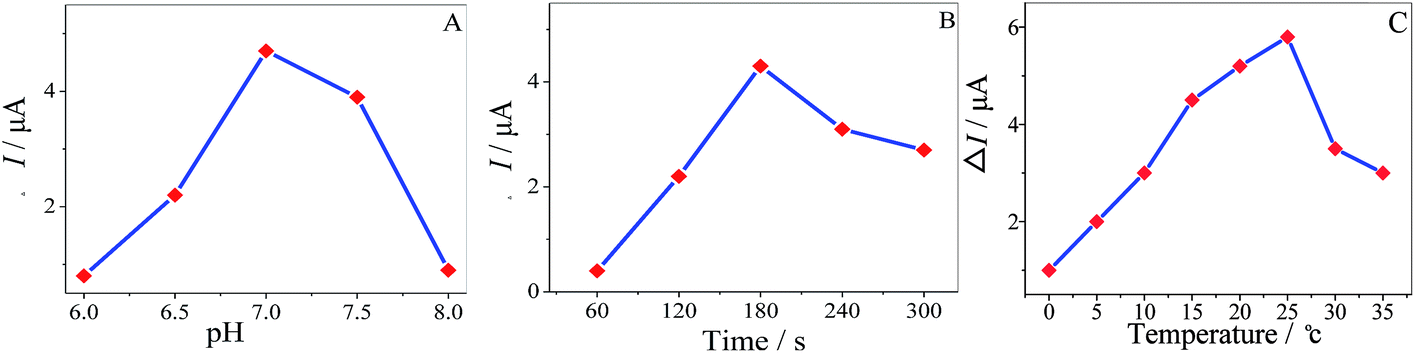
The pH of the solution has an influence in enantiospecificity of the MA enantiomers. The effect of pH on the enantioselective recognition was studied from 6.0 to 8.0 in 10 mM MA, as shown in (Fig. 9A) and the maximum ΔI value appeared at pH 7.0 (ΔI = IR-MA − IS-MA). Consequently, pH 7.0 was the optimized value for chiral recognition. It was important to investigate the incubation time of the MWCNTs–HPCS/GCE in the MA enantiomers; the peak currents increased with the incubation time, as shown in (Fig. 9B), and the maximum ΔI value appeared at 180 s. The greater water solubility of HPCS may play an important role in enantioselective recognition. Similar chiral sensing platforms are reported in Table 1. The influence of temperature on the ΔI value at the MWCNTs–HPCS based chiral interfaces is shown in Fig. 9C. Clearly, the highest ΔI value was achieved at 25 °C. When the temperature was below 10 °C, H-bond formation was favorable, which has a close relationship with angles of the relative orientation. Then, the kinetic energy increased with temperature, and thus the increasing orientational disorder broke the H-bonds,34–36 which resulted in a low recognition efficiency.
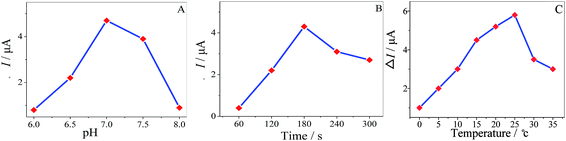 |
| | Fig. 9 The influence of pH (A), incubation time (B) and temperature (C) on the current response of the MWCNTs–HPCS in 5.0 mM [Fe(CN)6]4−/3− containing 10 mM MA enantiomers. | |
Table 1 The electrochemical methods used for recognition of MA enantiomers reported to datea
| Modified electrode |
Chiral recognition principle |
pH |
Interaction time |
Ref. |
| L-Cys, L-cysteine; GLOB, γ-globulin; rGO, reduced graphene oxide. |
| L-Cys–Au |
Ligand exchange model |
5.5 |
10 min |
22 |
| GLOB/rGO/GCE |
Three-point interaction model |
7.2 |
1440 min |
23 |
| MWCNTs–HPCS/GCE |
Three-point interaction model |
7.0 |
3 min |
Our study |
Electrochemical discrimination of the amino acid enantiomers
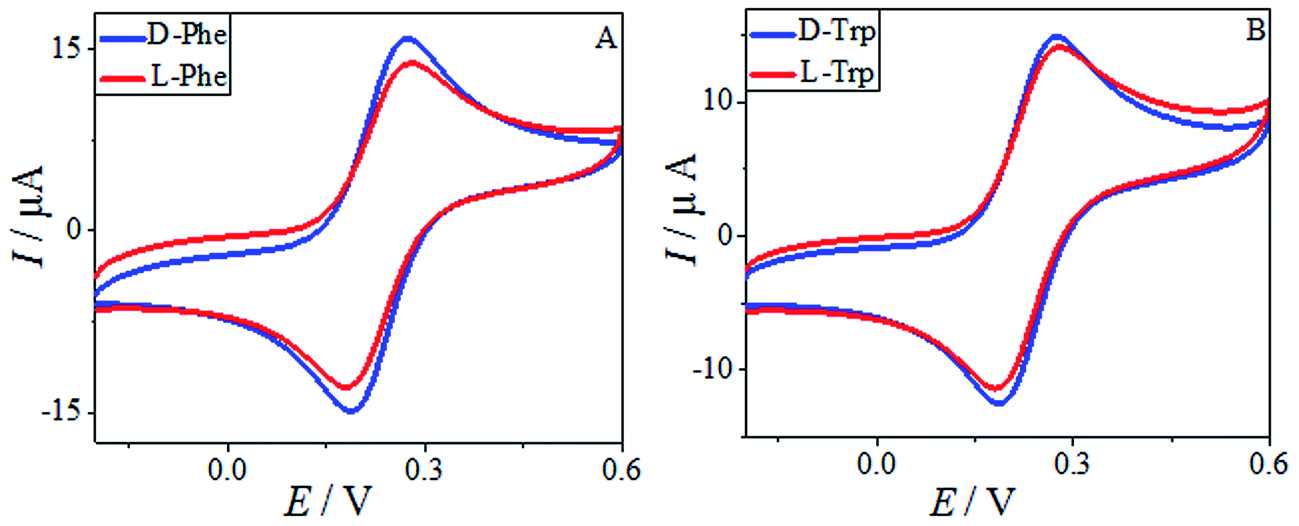
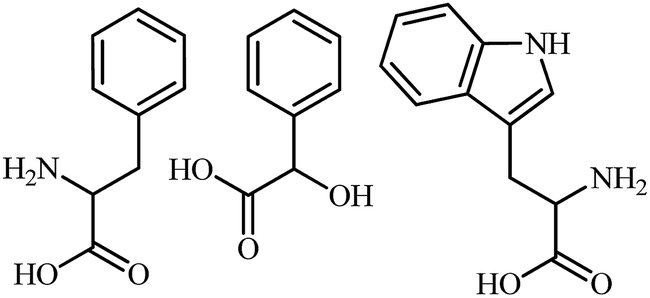
The HPCS, as a chiral selector, was further applied to the chiral discrimination of the enantiomers of phenylalanine (Phe) and Trp under the same conditions, and the results are shown in Fig. 10. The Phe and Trp enantiomers show differences in Ip based on the HPCS chiral interfaces; the ID-Trp/IL-Trp value for the Trp enantiomers (1.14) was much higher than those of Phe (1.05), which suggests that HPCS was capable of recognizing the Trp and Phe enantiomers as well as MA. According to the three-point interaction model, the chemical structures of MA, Trp and Phe play an important role in the enantioselective discrimination process. As shown in Fig. 11, Trp enantiomers provide more steric hindrance due to the benzopyrole group during the formation of hydrogen bonds. Compared with the Phe enantiomers, the recognition efficiency for the Trp isomers decreased significantly. The chiral sites of MA are different to the Phe and Trp enantiomers; the hydroxy and carboxyl groups are linked to the chiral carbon of MA. In addition, the functional groups of MA more easily form hydrogen bonds.37 Furthermore, there were many kinds of electron-donating groups in the enantiomers, such as benzopyrole and benzene; the distance between the functional group and chiral carbon is an important factor during the enantioselective discrimination process.38
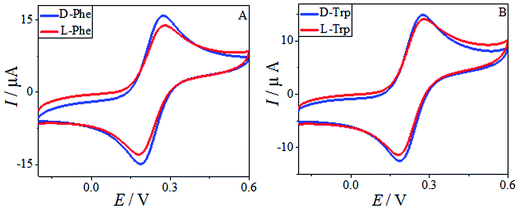 |
| | Fig. 10 The CV of 10 mM Phe enantiomers (A) and Trp enantiomers (B) in 0.2 M PBS buffer solution (pH 7.0) containing 5.0 mM [Fe(CN)6]4−/3−. | |
 |
| | Fig. 11 The chemical structures of phenylalanine (Phe), mandelic acid (MA) and tryptophan (Trp). | |
Chiral recognition of MA enantiomers in a racemic solution
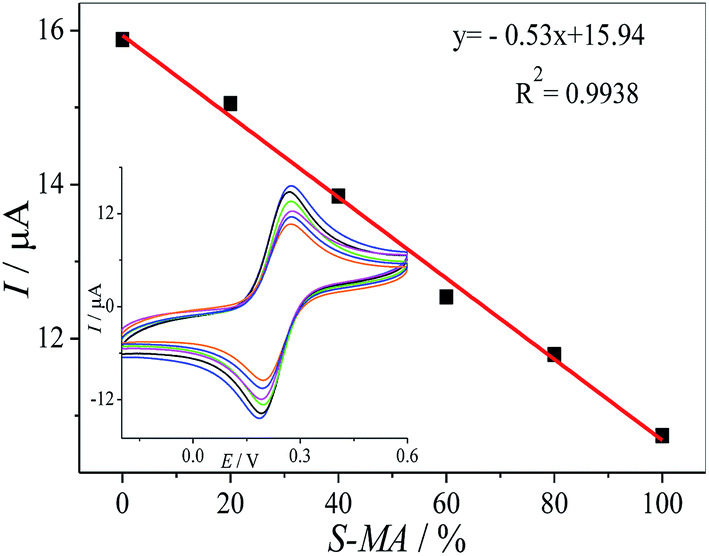
We tried to develop a simple method for the discrimination of one enantiomer in the racemic solution, in this manner, the MWCNTs–HPCS/GCE was used for determination of the mixture of (S)- and (R)-MA. The peak currents of the MA enantiomers were recorded as a ratio of (S)- and (R)-MA (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 20:80, 40:60, 60:40, 80:20 and 100:0, v/v) via CV (Fig. 12). It is important for the chiral discrimination in racemic mixtures of chiral compounds, especially chiral drugs, and it is highly desirable for the determination of the chiral enantiomers in pharmaceutical factories and institutions engaged in drug study and development.
:100, 20:80, 40:60, 60:40, 80:20 and 100:0, v/v) via CV (Fig. 12). It is important for the chiral discrimination in racemic mixtures of chiral compounds, especially chiral drugs, and it is highly desirable for the determination of the chiral enantiomers in pharmaceutical factories and institutions engaged in drug study and development.
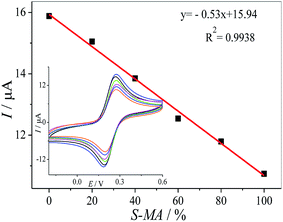 |
| | Fig. 12 The linear relationship between Ip and the ratio of the concentrations of the MA enantiomers. Inset: CV of the different ratios of the concentrations of (S)-MA and (R)-MA (0:100, 20:80, 40:60, 60:40, 80:20 and 100:0) on the HPCS modified GCE. | |
Reproducibility and stability
A pair of modified electrodes was fabricated for discriminating MA enantiomers in repeated tests; the peak current differences were very close to each other and the relative standard deviation (RSD) was 5.19%. The stability of the prepared chiral sensors was also investigated via CV measurements over 50 successive scanning cycles in [Fe(CN)6]4−/3− at 0.5 V s−1; the peak current decreased by 0.43%, and it was confirmed that the stability of the MWCNTs–HPCS chiral sensors was fairly satisfactory.
Conclusion
In summary, MWCNTs–HPCS was prepared via covalent bonds for an effective chiral interface, and the water solubility of HPCS was improved as a result of the introduction of the hydroxypropyl groups, which was applied to the enantioselective recognition of MA enantiomers. The electrochemical recognition of MA enantiomers was obtained at the MWCNTs–HPCS based chiral interface with a peak current ratio of 1.48. The steric hindrance of HPCS plays an important role during the formation of hydrogen bonds between the MA enantiomers and HPCS. Moreover, the molecular structure of the enantiomers was a key factor in the enantioselective recognition process; for example, the distance between the functional group and chiral carbon, steric hindrance and the type of functional group, and thus, functionalized CS may open up a new way for the fabrication of chiral interfaces used for electrochemical recognition.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (51262027), the Natural Science Foundation of Gansu Province (0803RJZA009), Science and Technology Tackle Key Problem Item of Gansu Province (2GS064-A52-036-08), Gansu Key Laboratory of Polymer Materials (ZD-04-14) and the support from the fund of the State Key Laboratory of Solidification Processing in NWPU (SKLSP201011).
Notes and references
- R. I. Stefan van Staden, R. G. Bokretsion, K. I. Ozoemena, J. F. van Staden and H. Y. Aboul-Enein, Electroanalysis, 2006, 18, 1718 CrossRef CAS.
- A. Zehnacker and M. A. Suhm, Angew. Chem., Int. Ed., 2008, 47, 6970 CrossRef CAS PubMed.
- C. Z. Xuan, Q. Xia, J. J. Xu, Q. H. Wang, X. Lin and Y. Z. Fu, Anal. Methods, 2016, 8, 3564 RSC.
- B. D. Liu, X. Zhang, Y. P. Ding, L. Q. Luo and F. F. Zhang, Anal. Methods, 2015, 7, 3022 RSC.
- Q. Zhang, L. J. Guo, Y. H. Huang, Y. H. Wang, Q. Han and Y. Z. Fu, Anal. Methods, 2013, 5, 4397 RSC.
- E. Zor, I. H. Patir, H. Bingol and M. Ersoz, Biosens. Bioelectron., 2013, 42, 321 CrossRef CAS PubMed.
- Q. Zhang, Y. H. Huang, L. J. Guo, C. Chen, D. M. Guo, Y. Chen and Y. Z. Fu, New J. Chem., 2014, 38, 4600 RSC.
- L. P. Bao, J. Y. Dai, L. Yang, J. F. Ma, Y. X. Tao, L. H. Deng and Y. Kong, J. Electrochem. Soc., 2015, 162, H486 CrossRef CAS.
- W. L. Feng, C. Liu, S. Y. Lu, C. Y. Zhang, X. H. Zhu, Y. Liang and J. M. Nan, Microchim. Acta, 2014, 181, 501 CrossRef CAS.
- D. M. Guo, P. Y. Ran, C. Chen, Y. Chen, C. Z. Xuan and Y. Z. Fu, J. Electrochem. Soc., 2015, 162, B354 CrossRef CAS.
- J. Ou, Y. X. Tao, J. J. Xue, Y. Kong, J. Y. Dai and L. H. Deng, Electrochem. Commun., 2015, 57, 5 CrossRef CAS.
- N. M. Alves and J. F. Mano, Int. J. Biol. Macromol., 2008, 43, 401 CrossRef CAS PubMed.
- X. K. Lu, S. J. Tang, B. L. Huang, X. Y. Shen and F. Hong, Fibers Polym., 2013, 14, 935 CrossRef CAS.
- C. K. S. Pillai, W. Paul and C. P. Sharma, Prog. Polym. Sci., 2009, 34, 641 CrossRef CAS.
- L. Y. Yu, Q. Liu, X. W. Wu, X. Y. Jiang, J. G. Yu and X. Q. Chen, RSC Adv., 2015, 5, 98020 RSC.
- X. G. Gu, Y. X. Tao, Y. Pan, L. H. Deng, L. P. Bao and Y. Kong, Anal. Chem., 2015, 87, 9481 CrossRef CAS PubMed.
- Y. G. Wang, L. Shi, L. Gao, Q. Wei, L. M. Cui, L. H. Hu, L. G. Yan and B. Du, J. Colloid Interface Sci., 2015, 451, 7 CrossRef CAS PubMed.
- L. P. Bao, Y. X. Tao, X. G. Gu, B. Z. Yang, L. H. Deng and Y. Kong, Electrochem. Commun., 2016, 64, 21 CrossRef CAS.
- K. W. Tang, J. M. Yi, K. L. Huang and G. L. Zhang, Chirality, 2009, 21, 390 CrossRef CAS PubMed.
- Y. Z. Fu, L. L. Wang, Q. Chen and J. Zhou, Sens. Actuators, B, 2011, 155, 140 CrossRef CAS.
- E. Zora, A. O. Safc and H. Bingolc, Anal. Biochem., 2014, 449, 83 CrossRef PubMed.
- M. Z. Sai, S. L. Zhong, Y. Tang, W. T. Ma, Y. G. Sun and D. R. Ding, J. Appl. Polym. Sci., 2014, 131, 40535 CrossRef.
- Y. H. Huang, D. M. Guo, Q. Zhang, L. J. Guo, Y. Chen and Y. Z. Fu, RSC Adv., 2014, 4, 33457 RSC.
- D. M. Guo, P. Y. Ran, C. Chen, Y. Chen, C. Z. Xuan and Y. Z. Fu, J. Electrochem. Soc., 2015, 162, B354 CrossRef CAS.
- C. S. Wu and H. T. Liao, Polymer, 2007, 48, 4449 CrossRef CAS.
- Y. Z. Zhang, H. Y. Ma, K. Y. Zhang, S. J. Zhang and J. Wang, Electrochim. Acta, 2009, 54, 2385 CrossRef CAS.
- C. L. Chen, A. Ogino, X. K. Wang and M. Nagatsu, Appl. Phys. Lett., 2010, 96, 131504 CrossRef.
- S. Y. Yang, C. C. M. Ma, C. C. Teng, Y. W. Huang, S. H. Liao, Y. L. Huang, H. W. Tien, T. M. Lee and K. C. Chiou, Carbon, 2010, 48, 592 CrossRef CAS.
- Q. Bi, S. Q. Dong, Y. M. Sun, X. Q. Lu and L. Zhao, Anal. Biochem., 2016, 508, 50 CrossRef CAS PubMed.
- H. Gou, J. X. He, Z. L. Mo, X. J. Wei, R. R. Hu, Y. W. Wang and R. B. Guo, J. Electrochem. Soc., 2016, 163, B272 CrossRef CAS.
- D. M. Guo, Y. H. Huang, C. Chen, Y. Chen and Y. Z. Fu, New J. Chem., 2014, 38, 5880 RSC.
- J. J. Xu, Q. H. Ying, Q. Xia, N. Wang, X. Lin and Y. Z. Fu, New J. Chem., 2016, 40, 6955 RSC.
- L. P. Bao, X. H. Chen, B. Z. Yang, Y. X. Tao and Y. Kong, ACS Appl. Mater. Interfaces, 2016, 8, 21710 CAS.
- Y. X. Tao, J. Y. Dai, Y. Kong and Y. Sha, Anal. Chem., 2014, 86, 2633 CrossRef CAS PubMed.
- F. H. Stillinger and A. Rahman, J. Chem. Phys., 1972, 57, 1281 CrossRef CAS.
- T. I. Mizan, P. E. Savage and R. M. Ziff, J. Phys. Chem., 1996, 100, 403 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS PubMed.
- L. L. Wang, W. C. Gong, F. Wang, Z. Y. Yu and Z. L. Chen, Anal. Methods, 2016, 8, 3481 RSC.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Zunli Mo
,
Zunli Mo