
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient metal organic framework (MOF)-based copper catalysts for the base-free aerobic oxidation of various alcohols†
Abu Taher,
Dong Wook Kim and
Ik-Mo Lee *
*
Department of Chemistry, Inha University, Incheon 402-751, South Korea. E-mail: imlee@inha.ac.kr; Fax: +82-32-867-5604; Tel: +82-32-860-7682
First published on 23rd March 2017
Abstract
Copper (Cu) containing metal organic frameworks (MOFs) are found to be highly efficient heterogeneous catalysts for oxidation of various alcohols. A wide range of alcohols, including alcohols containing inactive hetero-aryl and long-chain alkyl units, are selectively converted into their corresponding products. The catalytic efficiency of the Cu containing MOFs, along with the co-catalyst 2,2,6,6-tetramethyl-piperidyl-1-oxy (TEMPO), was demonstrated by the high conversion of the reactants with 100% selectivity under base-free conditions. The use of an inexpensive copper containing catalyst, the broad substrate scope, and the open air conditions and absence of base additive make this protocol very practical. The catalyst maintained a unique structural framework and it could be reused at least five times without significant loss of activity.
Introduction
In recent years, metal organic frameworks (MOFs) have been actively investigated and successfully used for different purposes in various branches of the pure and applied sciences.1 MOFs are three dimensional porous structures made of metal ions or clusters linked by organic molecules. The most attractive features of MOFs are their extremely high surface areas, as well as their tunable pore size and topology.1c These key features, along with other characteristics, make them excellent candidates for a wide range of applications in heterogeneous catalysis.2 These catalysts can be readily recovered and reused,3 which is highly desirable for cost effectiveness and sustainability considerations in organic synthesis. In addition, copper (Cu),4 as a classical transition metal, is considered to be one of the most attractive catalysts for use in the oxidation of alcohols due to its abundance of resources, low cost, non-toxic properties and high catalytic efficiency. In particular, Cu(II) complexes combined with TEMPO have been proven to have good catalytic performance for alcohol oxidation.5 Up to now, even though many efficient Cu(II)/TEMPO catalytic systems have been reported,6 there are only a limited number of studies on MOF-based Cu(II)/TEMPO systems for alcohol oxidation.7 However, Cu/TEMPO catalysts often require a large excess of base additives, which always results in a carboxylate product and basic waste disposal problems,8 as well as separation difficulties for catalytic recycling.9 Importantly, the presence of a base is not desirable from an industrial point of view due to corrosion, and it can adversely affect the stereoselectivity and cannot be used for a base-sensitive alcohol,10 a fact that is very important in the pharmaceutical industry. Interestingly, some Cu(II) complexes can catalyze alcohol oxidation by using TEMPO as a co-catalyst without base additives.11 Unfortunately, these copper catalysts are still homogeneous and are difficult to separate after the reaction.On the other hand, the oxidation of alcohols to their corresponding carbonyl compounds is one of the most important reactions in organic synthesis, due to the wide-ranging utility of these carbonyl products as essential precursors and intermediates for the development of many drugs, fragrances, and fine chemicals.12 It traditionally requires numerous stoichiometric oxidants, and most of these alcohols are oxidized by toxic, corrosive and expensive oxidants, such as DMSO-coupled reagents,13 hypervalent iodines,14 and heavy-metal reagents.12a,b Unfortunately, these reactions result in large quantities of toxic materials and call for the use of stoichiometric quantities of moisture-sensitive, unrecoverable and expensive chemical reagents. Importantly, satisfactory results were obtained in only limited cases, in which a large excess of additives was required. Therefore, the development of potential catalysts for oxidation under mild conditions is highly desirable, but challenging, and a hot topic in both green chemistry and current organic synthesis. Herein, we report a highly efficient and reusable Cu(II)/MOF (3) composite assisted by TEMPO, as the co-catalyst, for the selective oxidation of various alcohols to their corresponding products under mild conditions, without the assistance of any base additives (Scheme 1).
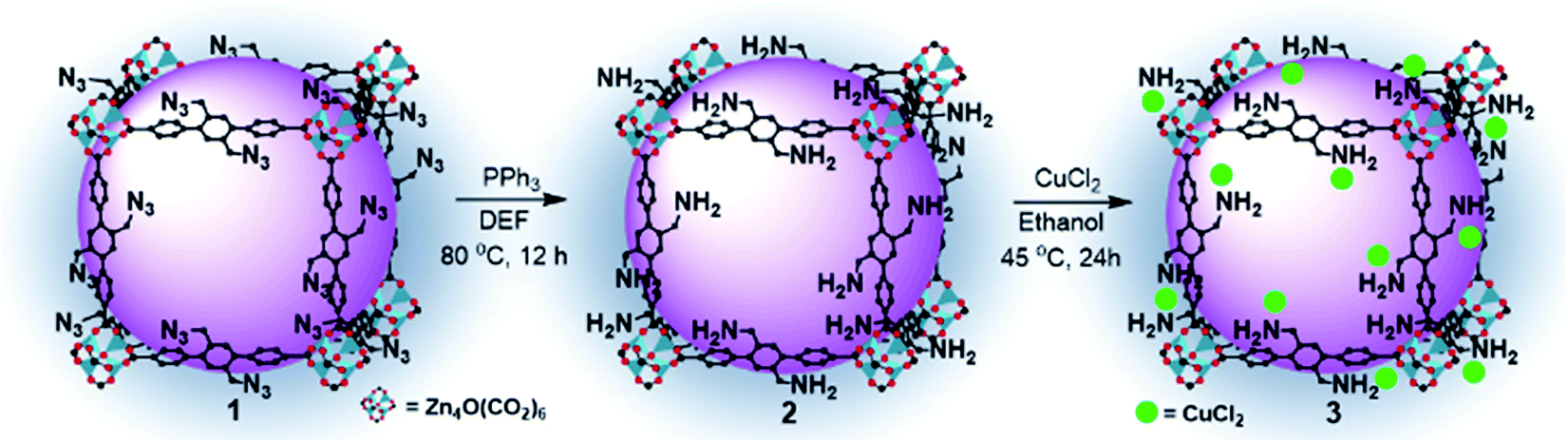 | ||
| Scheme 1 Post-functionalization route of the Cu(II)/MOF-NH2 (3) composite. | ||
Results and discussion
The azide-functionalized MOF-N3 (1) was prepared from the reaction of an organic linker, containing the azide functional group, with zinc nitrate hexahydrate in DEF, as described earlier.15 Subsequently, we tried to reduce the azide groups using triphenylphosphine (PPh3) in DEF, as described in our earlier reports, and the detailed synthesis and characterization procedure of MOF-NH2 (2) is presented elsewhere.16 The FTIR and XRD spectra of the as-synthesized compounds 1 and 2 are similar to those of the defined compound, as shown in Fig. S2a and b, respectively (ESI†). Moreover, an elemental analysis of 1 and 2 was in good agreement with their formula unit (Table S1, ESI†). The catalyst 3 was synthesized by loading the Cu precursor, copper(II) chloride, into 2 in ethanol. The colour of the solution changed from green to colourless (Fig. 1f). After separating the MOF crystal by filtration, we dried the crystal under vacuum at 80 °C to afford 3 as a green solid (Fig. S3, ESI†). The copper complex was anchored on the amine-functionalized MOF layer through coordination,17 and the presence of the Cu complexes was examined by FTIR, HRSEM, TEM, XPS, EDS, TGA and ICP spectroscopic analyses. The loading of Cu in MOF-NH2 was 5.02 wt%, which was measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES) and exactly matched the calculated value. The morphology of 3 was observed by SEM and TEM analyses. The HRSEM images at different magnifications are shown in Fig. 1a–c, and the TEM image of the catalyst 3 is shown in Fig. S4, (ESI†). The SEM, and corresponding elemental mapping by energy-dispersive X-ray spectroscopic analysis (EDS) of 3, demonstrated the homogeneous distribution of copper and chloride ions in the amine-functional MOF (Fig. 1d and e). The successful loading of Cu in 2 was confirmed by EDS spectroscopy. As shown in Fig. S5 (ESI†), a clear Cu peak was resolved when we probed the selected area 1 on the Cu loaded MOF. Furthermore, the XPS survey spectra of 3 confirmed the existence of Cu and Cl2 in 3 (Fig. S6b†). It can be remarked that the FTIR stretching band of the catalyst 3 at 1656 cm−1 is decreased, which may be attributed to the coordination interaction with CuCl2 (Fig. 2a). In particular, the amino group related peaks at 3373 cm−1 and 3290 cm−1 shifted to lower wave numbers at 3255 cm−1 and 3158 cm−1, respectively, in 3 because of the electron donation from the amino group to the Cu(II) center, which resulted in the formation of the Cu-amine functionalized-MOF complex.18 Furthermore, the characteristic XRD peak of Cu (112) at 2θ = 12.6° is assigned due to the insertion of Cu2+ in 2 (Fig. 2b), which means that the structure of the amine-functionalized MOF has coordinated with Cu2+, and a new complex has been formed that is in accordance with earlier reports.19 In addition, it is found that after the yellow amine-functionalized MOF powders were added into the green copper(II) chloride containing ethanoic solution, the solution turned colourless (Fig. 1f), which further confirmed the likely formation of the MOF copper complex under the defined conditions. The XRD pattern of the reacted 3 crystals exhibited no apparent changes of the pattern from the starting 2, as shown in Fig. 2b, and the C–O band was still maintained along with the Zn metal (Fig. S6a and b, ESI†), which indicated that the modified MOF framework was constructed by zinc ions and carboxylate anions with a similar structural framework. Thermogravimetric analysis (TGA) indicates that 3 is thermally stable, and it shows thermal stability that is identical to the parent compound (Fig. 2c). Fig. S7 (ESI†) shows the N2 adsorption–desorption isotherm profiles of 2 and 3. All of the adsorption–desorption isotherms show a type I shape, which is a characteristic of microporous materials. The BET surface area and pore volume of 2 were calculated to be 582.20 m2 g−1 and 0.31 cm3 g−1, respectively. Compared to 2, the BET surface area and pore volume of 3 decreased to 156.04 m2 g−1 and 0.07 cm3 g−1, respectively, mainly because of the added mass of copper(II) chloride to 2. | ||
| Fig. 1 SEM images of the surface of 3 at different magnifications (scale bars are shown at bottom left or right) for 5.00 μm, 1.00 μm and 300 μm respectively (a, b & c); the corresponding quantitative EDS mapping of Cu (d) and Cl (e); the color of the solution of catalyst 3 before and after reaction (f). | ||
 | ||
| Fig. 2 (a) FTIR and (b) XRD spectra of compounds 2 and 3; TGA of compounds 2 and 3 (c); XPS spectra of the nitrogen N 1s peaks for compound 2 (d) and for compound 3 (e); Cl S1 peaks for 3 (e, inset); XPS spectra of Cu 2p peaks of compound 3 (f). | ||
To gain further insights, we have also performed X-ray photoelectron spectroscopy (XPS) analysis. The N 1s spectrum is very useful for analyzing the nature of the N functionalities. Careful XPS analysis showed the presence of a split peak with maxima at 398.1 and 399.2 eV for the functional groups of the primary amine20 (Fig. 2d), whereas N 1s has a single peak at 400 eV for the catalyst (Fig. 2e) which suggested strong coordination with copper(II) chloride.21 We also tried to determine the oxidation state of Cu in 3 using XPS. The CuCl2 related peak at 934.8 eV was evident in the Cu 2p spectra of the catalyst, 3 (Fig. 2f).18 The spectra of CuCl2 in 3 was more clearly revealed in the Cl 1s spectra (284.1 eV), as shown in Fig. 2 (e, inset). Furthermore, the Cu 2p XPS spectrum of CuCl2/MOF showed shakeup satellite peaks of the Cu 2p3/2 at 942 and Cu 2p1/2 at 962 eV, which confirmed the presence of Cu(II) species (CuCl2) (Fig. 2f).22 Generally, the characteristic shakeup satellite is peculiar for the Cu(II) species, which relates to the d9 configuration of Cu. Whereas, in the case of Cu(I), no satellite peaks can be observed because the screening via a charge transfer into the d states is not possible due to the presence of a completely filled d shell.22
The catalytic activity of the prepared catalyst 3 was tested with a representative set of primary and secondary benzylic and aliphatic alcohols. Initially, to investigate the catalytically active components of the CuCl2/MOF-NH2 composite, a series of controlled experiments were carried out under exactly the same reaction conditions. The reactions were performed at 70 °C, and under atmospheric pressure, using air as the oxidant under base-free conditions, and the results are summarized in Table 1. As shown in Table 1 (entry 1), 3 was an active catalyst for this reaction and 100% conversion (GC yield) of benzyl alcohol to benzaldehyde was achieved. On the contrary, 1 and 2 gave no conversion of benzyl alcohol (entries 2–4) in the absence of any catalyst, which confirmed the oxidation of benzyl alcohol to benzaldehyde is catalysed by Cu.7 In addition, when only CuCl2 or a mixture of 2 and CuCl2 was used, relatively poor yields were obtained (entries 5 and 6). According to the literature, this may be due to the absence of coordinating ligands.23 Importantly, the results demonstrate that the connected ligand between the amine functionalized-MOF and the Cu moieties promoted the catalytic reaction of aerobic oxidation of benzyl alcohols under the defined conditions.
|
|||
|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) |
| a Reaction conditions: benzyl alcohol (1.0 mmol), TEMPO (1.0 mmol), CH3CN (6 mL) and air as an oxygen source.b GC yield.c Calculated to have the same amount of Zn2+ (when comparing 3 with 1 and 2). | |||
| 1 | CuCl2@MOF-NH2 (3) | 6 | 100 |
| 2c | MOF-N3 (1) | 6 | 0 |
| 3c | MOF-NH2 (2) | 6 | 0 |
| 4 | None | 6 | 0 |
| 5 | MOF-NH2 + CuCl2 | 6 | 48 |
| 6 | CuCl2 | 6 | 35 |

In order to obtain optimized catalytic performance, we investigated the effect of the solvent and found that CH3CN was the best solvent for this reaction (Fig. 3a). It is significant to note that 3 showed poor catalytic activity and the crystal structure deformed when water was used as the solvent (Fig. S9, ESI†). Then, in a quick survey of the bases, the conversion of alcohol without using any base was similar to that when using K2CO3 as a base (Fig. 3b). In this catalytic system, the reason for the high efficiency without base is supposedly that 3 acts not only as a catalyst but also as a base, which has a much higher tendency to coordinate to copper to form the active species. So it might be thought that the MOF containing an amino group had adequate basicity for deprotonation, and the oxidation could occur without any external base.24 Subsequently, we optimized the effect of the catalyst loading and found that 1 mol% of Cu had the best efficiency for alcohol oxidation (Fig. S1†). These obtained results suggested that 3 (1 mol%) is required to promote base-free air oxidation in CH3CN. With these optimized results on hand, the reactions of various aryl, alkyl and hetero-aryl substrates were investigated, with the assistance of TEMPO as the co-catalyst, and the results are summarized in Table 2.
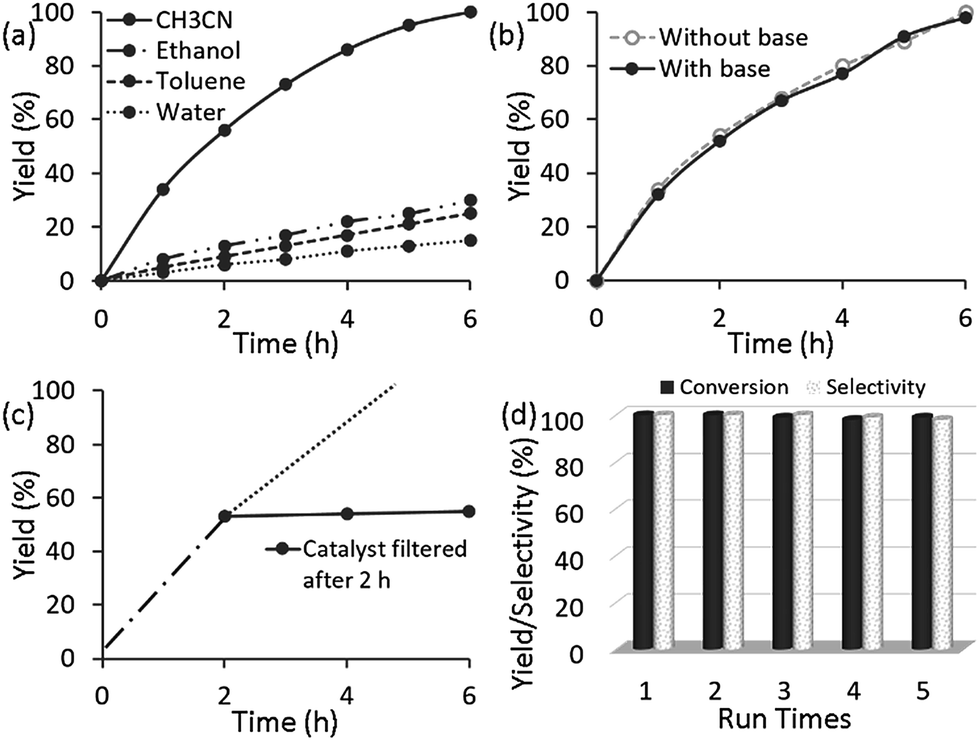 | ||
| Fig. 3 Aerobic oxidation of benzyl alcohol catalysed by 3 (a) in different solvents, (b) with base and without base, and the (c) hot filtration test and (d) recyclability test. In all cases, the reaction conditions are the same as in Table 2 (entry 1). | ||
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
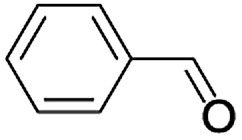
| a Reaction conditions: alcohol (1.0 mmol), TEMPO (1.0 mmol), CH3CN (6 mL) and air as an oxygen source.b Isolated yield. Selectivity 100%. | ||||
| 1 | 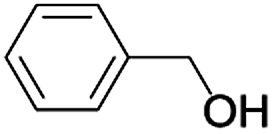 |
 |
6 | 98 |
| 2 |  |
 |
5.5 | 98 |
| 3 |  |
 |
5.5 | 96 |
| 4 |  |
 |
6 | 90 |
| 5 |  |
 |
7 | 96 |
| 6 | 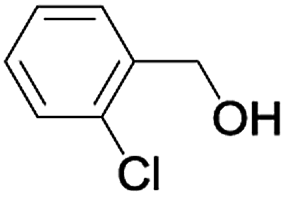 |
 |
7.5 | 82 |
| 7 |  |
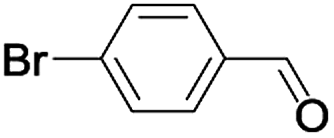 |
7 | 98 |
| 8 | 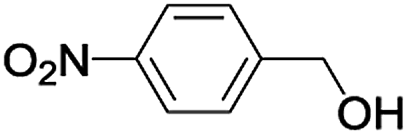 |
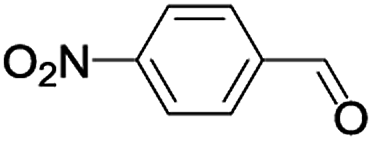 |
8 | 96 |
| 9 | 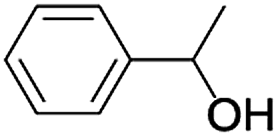 |
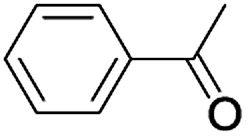 |
7 | 98 |
| 10 | 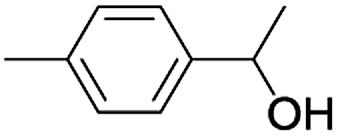 |
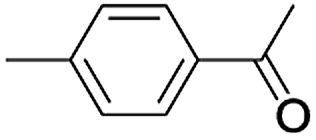 |
7 | 93 |
| 11 | 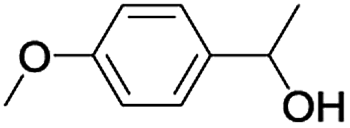 |
 |
7 | 94 |
| 12 | 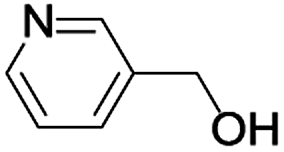 |
 |
11.5 | 61 |
| 13 |  |
 |
11 | 58 |
| 14 | 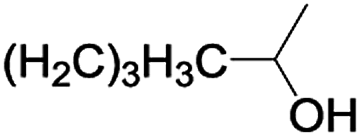 |
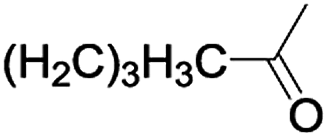 |
8 | 65 |
| 15 | 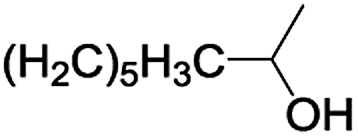 |
 |
8 | 48 |
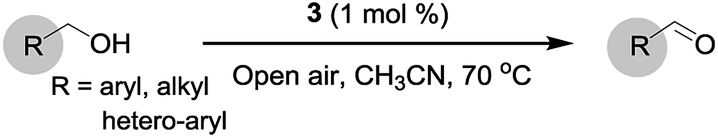
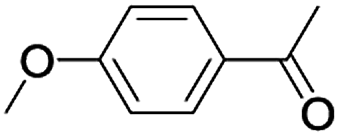
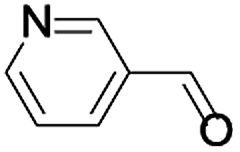
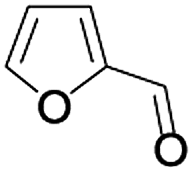
In order to extend the scope of the aerobic oxidation by 3, we performed the aerobic oxidation of various aromatic and aliphatic alcohols (Table 2). All of the reactions, such as for benzyl alcohol (entry 1), proceeded smoothly to give the corresponding products in high yield with 100% selectivity under base-free conditions. Where possible, the product peaks were compared to those reported in the literature.25,26 The electronic nature of the substituents on the benzylic alcohol did have an effect on the reaction. Electron-donating group (EDG) containing substrates, such as p-tolylmethanol, (4-methoxyphenyl)methanol and (2-methoxyphenyl)methanol, reacted to give the products (entries 2–4) with yields (90–98%) that are obviously higher than the yields for the electron-withdrawing group (EWG) containing substrates, such as (4-chlorophenyl)methanol, (2-chlorophenyl)methanol, (4-bromophenyl)methanol and (4-nitrophenyl)methanol (entries 5–8). Nevertheless, these results demonstrate that 3 successfully promoted the aerobic oxidation of various alcohols with either an EDG or EWG. It was noteworthy that the aerobic oxidation of secondary alcohols to their corresponding ketones is difficult, which is probably due to steric reasons.27 However, secondary alcohols such as 1-phenylethan-1-ol, 1-(p-tolyl)ethan-1-ol and 1-(4-methoxyphenyl)ethan-1-ol also gave the expected products (entries 9–11) in good yield (93–98%). The oxidation of heterocyclic alcohols, such as pyridin-3-yl-methanol and furan-2-yl-methanol, proceeded smoothly to provide the desired products (entries 12 and 13) in moderate yield (58–61%). In the case of the aliphatic alcohols, the product (entries 14–15) yields (48–65%) were lower than those of the benzylic alcohols. In all cases, the catalyst exhibited good activity in the oxidation of various alcohols. Importantly, the results demonstrated that the selectivity of the primary alcohols along with the secondary alcohols was excellent (100% selectivity), and the aliphatic alcohols showed similar trends.
In order to gain insight into the heterogeneous nature of 3, a leaching test was carried out, in which the solid catalyst was centrifuged and removed after 2 h, and the reaction continued. As indicated in Fig. 3c, no further reaction took place without 3 after initiation of the reaction at 4 h, and the possible leaching of Cu after completion of the reaction was monitored by ICP analysis. This finding indicates that no leaching of the catalytically active sites occurs, and that 3 exhibits a typical heterogeneous catalyst nature, demonstrating that there was excellent coordination interaction between Cu2+ and the amine functionalized MOF. As a heterogeneous catalyst, 3 can be reused. After each catalytic cycle, 3 could be easily recovered by centrifugation or filtration, and directly reused in the next run under the same reaction conditions. It was observed that similar yields (98–100%) were obtained by carrying out five runs without any significant loss of activity (Fig. 3d). More importantly, the catalyst maintained 100% selectivity during five consecutive runs. Evidently, the catalyst 3 is highly stable against the migration and sintering of Cu and exhibits excellent activity, selectivity, and stability for the oxidation of alcohols under mild reaction conditions without the assistance of any base additives. The FTIR, XRD and XPS spectra of the catalyst, 3, were similar to those of the fresh catalyst after recycling five times (Fig. S10 and S6c, ESI†), which suggests that the structure and composition of 3 was maintained.
The well-defined pores of MOFs may offer unique advantages for catalytic applications, compared with other catalysts in which the pores may favor size and shape selective catalysis.3b,28 The pore size of the as-synthesized catalyst, 3, was bigger than all of the substrates, as shown in Table 2. For example, the size of benzyl alcohol is 6.9 by 4.9 Å, and the size of (4-methoxyphenyl)methanol is 9.0 by 4.9 Å (Fig. 4), which is smaller than the pore size of 3 (18.3 Å) as shown in Fig. S7 and S8† respectively. Therefore, this enhancement and the high selectivity would be due to the structure of the pores and surface functionality of the MOF frameworks.
 | ||
| Fig. 4 Size and shape selectivity in the aerobic oxidation of benzyl alcohol and (4-methoxyphenyl)methanol using 3. | ||
Experimental
Catalyst preparation
MOF-N3 (1) was prepared according to the literature procedure,15 and the amine-functionalized MOFs (2) were synthesized by our earlier reported procedure.16 Typically, 0.31 g of MOF-N3 crystals was placed in a 30 mL screw vial, and a solution of 0.80 g of triphenylphosphine (3.0 mmol) in DEF (30 mL) was added. The vial was kept at 80 °C in an oil bath for 10 h without stirring. After all the starting materials had disappeared (monitored by FTIR spectroscopy), MOF-NH2 was given as a slightly yellow solid (Fig. S3c, ESI†).16 The crystals were filtered off and washed with fresh DEF (20 mL × 4) and ethanol, and then immersed in ethanol for 12 h. The material was dried at 80 °C under vacuum for 24 h. Finally, 0.04 g (0.30 mmol) of copper(II) chloride was dissolved in ethanol (12 mL), and 0.30 g of compound 2 was added to the solution in a 20 mL vial. The reaction mixture was kept in an oil bath at 45 °C for 24 h, and the color of the solution changed from blue to colorless (Fig. 1f, ESI†). The crystals were filtered off and washed with fresh ethanol (20 mL × 4), and Cu(II)/MOF-NH2 (3) was given as a blue solid (Fig. S3d, ESI†). The material was dried at 80 °C under vacuum for 24 h. Inductively coupled plasma (ICP) analysis showed 5.02% (wt) Cu in the resulting compound, 3.General procedure for the oxidation reaction
The oxidation of alcohols was carried out using air oxygen (open air) at atmospheric pressure in a reaction vial. In a typical run, a solution of substrate (1.0 mmol) and TEMPO (1.0 mmol) in CH3CN (6 mL) was added to 3 (1 mol%) without base, and the mixture was heated at 70 °C under atmospheric air. After completion of the reaction, the catalyst was filtered off and the conversion was measured using a GC (Agilent Technologies 7890A GC System). The crude product was obtained by concentrating the resultant solution under reduced pressure, and it was purified by column chromatography (on silica gel) using a mixture of EtOAc–hexane to give the pure corresponding product (Table 2). All the desired products are known compounds, which were characterized by comparison of their spectra with those reported in the previous literature.25,26 The separated catalyst was successively reused for the next reaction without any retreatment. A hot filtering experiment was carried out by separating the catalyst quickly from the reaction mixture after 2 h reaction time, and the filtrate was then maintained at 70 °C for an additional 4 h.Conclusions
In summary, we have developed a novel, highly efficient Cu(II) containing MOF catalytic system, which can be used with the co-catalyst TEMPO for alcohol oxidation. Numerous practical features associated with this method should facilitate its use in synthetic organic chemistry, including use without base additives, fast reaction rates, mild reaction conditions, and compatibility with 100% selectivity. More importantly, it is a highly active recyclable solid catalyst for the oxidation of various organic substrates under mild conditions. These results indicate that copper plays a central role in catalytic oxidation. Together, these catalyst systems provide compelling aerobic alternatives to traditional alcohol oxidation methods without the assistance of any base additives. Moreover, the exceptional reactivity of the novel catalyst system prompted our concurrent inquiry into the applications of these catalytic matrices in a multitude of catalytic reactions and engineering processes.Acknowledgements
This work is supported by the Inha University Research Grant (Inha 2016).Notes and references
- (a) A. U. Czaja, N. Trukhanb and U. Muller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC; (b) S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799–1812 CrossRef CAS; (c) H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (d) R. Ricco, C. Pfeiffer, K. Sumida, C. J. Sumby, P. Falcaro, S. Furukawa, N. R. Champness and C. J. Doonan, CrystEngComm, 2016, 18, 6532–6542 RSC.
- (a) D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed; (b) A. Corma, H. Garcia and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (c) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (d) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (e) J. W. Liu, L. F. Chen, H. Cui, J. Y. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- (a) X. Feng, C. Xu, Z.-Q. Wang, S.-F. Tang, W.-J. Fu, B.-M. Ji and L.-Y. Wang, Inorg. Chem., 2015, 54, 2088–2090 CrossRef CAS PubMed; (b) Y. Qi, Y. Luan, J. Yu, X. Peng and G. Wang, Chem.–Eur. J., 2015, 21, 1589–1597 CrossRef CAS PubMed; (c) J. Wang, F.-W. Ding, J.-P. Ma, Q.-K. Liu, J.-Y. Cheng and Y.-B. Dong, Inorg. Chem., 2015, 54, 10865–10872 CrossRef CAS PubMed.
- (a) S. Pande, A. Saha, S. Jana, S. Sarkar, M. Basu, M. Pradhan, A. K. Sinha, S. Saha, A. Pal and T. Pal, Org. Lett., 2008, 10, 5179–5181 CrossRef CAS PubMed; (b) S. E. Allen, R. R. Walvoord, R. P. Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6237 CrossRef CAS PubMed; (c) B. R. Kim, J. S. Oh, J. Kim and C. Y. Lee, Bull. Korean Chem. Soc., 2015, 36, 2799–2800 CrossRef CAS; (d) Z. Miao, Y. Luan, C. Qib and D. Ramellac, Dalton Trans., 2016, 45, 13917–13924 RSC; (e) B. R. Kim, J. S. Oh, J. Kim and C. Y. Lee, Catal. Lett., 2016, 146, 734–743 CrossRef CAS.
- (a) E. T. T. Kumpulainen and A. M. P. Koskinen, Chem.–Eur. J., 2009, 15, 10901–10911 CrossRef CAS PubMed; (b) K. T. Mahmudov, M. N. Kopylovich, M. Silva, P. J. Figiel, Y. Y. Karabach and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2010, 318, 44–50 CrossRef CAS; (c) X. Zhang, W. Dong, Y. Luan, M. Yang, L. Tan, Y. Guo, H. Gao, Y. Tang, R. Dang, J. Li and G. Wang, J. Mater. Chem. A, 2015, 3, 4266–4273 RSC.
- (a) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414–2415 RSC; (b) P. J. Figiel, A. Sibaouih, J. U. Ahmad, M. Nieger, M. T. Räisänen, M. Leskelä and T. Repo, Adv. Synth. Catal., 2009, 351, 2625–2632 CrossRef CAS; (c) Q. F. Wang, Y. Zhang, G. X. Zheng, Z. Z. Tian and G. Y. Yang, Catal. Commun., 2011, 14, 92–95 CrossRef CAS.
- (a) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 CrossRef CAS; (b) M. Paul, N. N. Adarsh and P. Dastidar, Cryst. Growth Des., 2014, 14, 1331–1337 CrossRef CAS; (c) L. C. Li, R. Matsuda, I. Tanaka, H. Sato, P. Kanoo, H. J. Jeon, M. L. Foo, A. Wakamiya, Y. Murata and S. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7543–7546 CrossRef CAS PubMed.
- (a) H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151–4154 CrossRef CAS PubMed; (b) H. Chen, Q. H. Tang, Y. T. Chen, Y. B. Yan, C. M. Zhou, Z. Guo, X. L. Jia and Y. H. Yang, Catal. Sci. Technol., 2013, 3, 328–338 RSC.
- N. F. Zheng and G. D. Stucky, Chem. Commun., 2007, 3862–3864 RSC.
- B. A. D’Sa, D. McLeod and J. G. Verkade, J. Org. Chem., 1997, 62, 5057–5061 CrossRef.
- (a) M. F. Semmelhack, C. R. Schmid, D. A. Cortés and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374–3376 CrossRef CAS; (b) A. Cecchetto, F. Fontana, F. Minisci and F. Recupero, Tetrahedron Lett., 2001, 42, 6651–6653 CrossRef CAS.
- (a) R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidation of Organic Compounds, Academic Press, New York, 1981 Search PubMed; (b) M. Hudlicky, Oxidations in Organic Chemistry, American Chemical Society, Washington, D.C., 1990 Search PubMed; (c) R. A. Sheldon, I. W. C. E. Arends, G. J. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed; (d) J. Muzart, Tetrahedron, 2003, 59, 5789–5816 CrossRef CAS; (e) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed.
- B. A. Steinhoff, S. R. Fix and S. S. Stahl, J. Am. Chem. Soc., 2002, 124, 766–767 CrossRef CAS PubMed.
- (a) R. D. Ri-chardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402–4404 CrossRef CAS PubMed; (b) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC; (c) M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251–262 CrossRef CAS PubMed.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef CAS PubMed.
- A. Taher, D.-J. Lee, B.-K. Lee and I.-M. Lee, Synlett, 2016, 27, 1433–1437 CrossRef CAS.
- (a) R. K. Sharma, S. Dutta and S. Sharma, Dalton Trans., 2015, 44, 1303–1316 RSC; (b) A. Ghorbani-Choghamarani, Z. Darvishnejad and M. Norouzi, Appl. Organomet. Chem., 2015, 29, 170–175 CrossRef CAS; (c) Q.-L. Yan, A. Cohen, N. Petrutik, A. Shlomovich, J.-G. Zhang and M. Gozin, ACS Appl. Mater. Interfaces, 2016, 8, 21674–21682 CrossRef CAS PubMed.
- (a) Y.-H. Choi and S.-H. Hong, Langmuir, 2015, 31, 8101–8110 CrossRef CAS PubMed; (b) K. J. Lee, Y.-I. Lee, I.-K. Shim, J. Joung and Y. S. Oh, J. Colloid Interface Sci., 2006, 304, 92–97 CrossRef CAS PubMed.
- Y. Zhang, Z. Xie, Z. Wang, X. Feng, Y. Wang and A. Wu, Dalton Trans., 2016, 45, 12653–12660 RSC.
- A. Adenier, M. M. Chehimi, I. Gallardo, J. Pinson and N. Vila, Langmuir, 2004, 20, 8243–8253 CrossRef CAS PubMed.
- J. Marsh, L. Minel, M. G. Barthés-Labrousse and D. Gorse, Appl. Surf. Sci., 1998, 133, 270–286 CrossRef CAS.
- (a) M. Gopiraman, S. G. Babu, Z. Khatri, W. Kai, Y. A. Kim, M. Endo, R. Karvembu and I. S. Kim, Carbon, 2013, 62, 135–148 CrossRef CAS; (b) M. Gopiraman, D. Deng, S. G. Babu, T. Hayashi, R. Karvembu and I. S. Kim, ACS Sustainable Chem. Eng., 2015, 3, 2478–2488 CrossRef CAS.
- (a) C. Bai, Q. Zhao, Y. Li, G. Zhang, F. Zhang and X. Fan, Catal. Lett., 2014, 144, 1617–1623 CrossRef CAS; (b) A. Taher, D.-J. Lee and I.-M. Lee, Synlett, 2016, 27, 2333–2338 CrossRef CAS.
- S. Mannam, S. K. Alamsetti and G. Sekar, Adv. Synth. Catal., 2007, 349, 2253–2258 CrossRef CAS.
- G. Zhang, X. Han, Y. Luan, Y. Wang, X. Wen and C. Ding, Chem. Commun., 2013, 49, 7908–7910 RSC.
- C. Bai, A. Li, X. Yao, H. Liu and Y. Li, Green Chem., 2016, 18, 1061–1069 RSC.
- (a) R. H. Liu, X. M. Liang, C. Y. Dong and X. Q. Hu, J. Am. Chem. Soc., 2004, 126, 4112–4113 CrossRef CAS PubMed; (b) J. L. Long, X. Q. Xie, J. Xu, Q. Gu, L. M. Chen and X. X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- S. Horike, M. Dinca, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854–5855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, digital images, 1H NMR spectra, EDS spectrum, N2 adsorption–desorption isotherms, pore volume vs. diameter curve, XPS spectra, X-ray powder diffraction, effect of catalyst loading and FTIR spectrum. See DOI: 10.1039/c6ra28743c |
| This journal is © The Royal Society of Chemistry 2017 |