
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescence detection of Mn2+, Cr2O72− and nitroexplosives and photocatalytic degradation of methyl violet and rhodamine B based on two stable metal–organic frameworks†
Yu Wu *a,
Jian Wub,
Zhidong Luoc,
Jun Wanga,
Yulong Lia,
Yaoyao Hanc and
Jianqiang Liu*c
*a,
Jian Wub,
Zhidong Luoc,
Jun Wanga,
Yulong Lia,
Yaoyao Hanc and
Jianqiang Liu*c
aInstitute of Functional Materials, Sichuan University of Science & Engineering, Zigong, 643000, PR China. E-mail: wuyuhlj@163.com; Fax: +86-813-5505605; Tel: +86-813-5505605
bGuangxi Key Laboratory of Chemistry and Engineering of Forest Products, Guangxi University for Nationalities, College of Chemistry and Chemical Engineering, Nanning, Guangxi 530006, China
cSchool of Pharmacy, Guangdong Medical University, Dongguan, 523808, China. E-mail: jianqiangliu2010@126.com
First published on 7th February 2017
Abstract
The reaction of Zn(NO3)2·6H2O/Cd(NO3)2·4H2O with a semi-flexible ligand 4′-(4-(3,5-dicarboxylphenoxy)phenyl)-4,2′:6′,4′′-terpyridine (H2dbp) gave two new metal–organic frameworks of [Zn(dbp)]n (1) and [Cd(dbp)(H2O)·2H2O·CH3CN]n (2). Single crystal X-ray diffraction analyses reveal that both 1 and 2 show three-dimensional, two-fold interpenetrating four-connected networks with 44.610.8 topology. The photoluminescence investigation indicates that both 1 and 2 could be prospective candidates for developing luminescence sensors for the sensing of Mn2+, Cr2O72− and nitroaromatic analytes. Furthermore, the photocatalysis properties of 1–2 for the degradation of methyl violet and rhodamine B were examined.
Metal–organic frameworks (MOFs) are a fascinating class of materials that have been extensively studied because of their rich structural chemistry and potential applications in numerous areas, including gas storage and separation, catalysis, and chemical sensing.1–5 The organic ligands containing an aromatic π-conjugated system are subject to excitation, giving rise to photoluminescence (PL) upon irradiation. In addition, the metal components can also contribute to the PL, in which case d10-metal are often involved. Naturally, these luminescent MOFs (LMOFs) are used for the efficient, economic, and portable fluorometers, greatly promoting research and development in this subject area.6–11 Recently, considerable research effort has been focused on the potential applications of MOFs in photocatalysis.12,13 However, the building of MOFs with specific properties still remains a challenge because there are so many factors influencing the resulting polymers.14,15
It has been proposed that aromatic organic ligands play an important role in interacting with the substrate.16,17 On the basis of the previously mentioned points, a pyridine carboxylate ligand of 4′-(4-(3,5-dicarboxylphenoxy)phenyl)-4,2′:6′,4′′-terpyridine (H2dbp), which contains both carboxylate and pyridine donor moieties, was selected, which can help enrich the coordination modes. As far as is known, the MOFs based on H2dbp have been not explored extensively to date.18 Recently, a series of MOFs with a dbp ligand have been designed and synthesized and their degradation of methyl violet (MV) has been examined.18a Continuing the interest in the structures and properties of M-dbp architectures, in this paper, the assembly of H2dbp with zinc/cadmium (Zn2+/Cd2+) centers in two 3D microporous MOFs, [Zn(dbp)]n (1) and [Cd(dbp)(H2O)·2H2O·CH3CN]n (2), are reported, which represent a new category of two-fold interpenetrating four-connected networks in the dbp system. The luminescence sensors and photocatalysis properties for the degradation of MV and rhodamine B (RhB) of 1–2 were evaluated and discussed.
Materials and method
All the other reagents used for the syntheses were commercially available and employed without further purification. Powder X-ray diffraction (PXRD) was performed on a Bruker D8 ADVANCE X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å) at 50 kV, 20 mA with a scanning rate of 6° per min and a step size of 0.02°. Fourier transform infrared (FT-IR) spectra were collected using a potassium bromide pellet were measured using a Nicolet Impact 750 FTIR spectrometer in the range of 400–4000 cm−1. Thermogravimetric analysis (TGA) was performed under a nitrogen (N2) atmosphere from room temperature to 900 °C at a heating rate of 10 °C min−1. The photocatalytic activity studies were carried out using a Shimadzu UV-Vis 2501PC spectrophotometer.The PL sensing was performed as follows: the PL properties of 1–2 were investigated in N,N-dimethylformamide (DMF) emulsions at room temperature using a Shimadzu RF-5301PC spectrofluorophotometer. The 1–2–DMF emulsions were prepared by adding 3 mg of 1–2 powder into 3.00 mL of DMF and then ultrasonically agitating the mixture for 30 min before testing.
The photocatalytic reactions were performed as follows: 50 mg of 1–2 were dispersed in a 50 mL aqueous solution of MV or RhB (10 mg L−1) under stirring in the dark for 30 min to ensure the establishment of an adsorption–desorption equilibrium. Then the mixed solution was exposed to ultraviolet (UV) irradiation from a mercury (Hg) lamp (250 W) and kept under continuous stirring during irradiation for 100 min. Samples (5 mL) of the mixture were removed every 10 min and collected by centrifugation for analysis using UV-Visible (Vis) spectrometer. By contrast, a simple control experiment was also performed under the same conditions without adding any catalysts.
X-ray crystallography
Single crystal X-ray diffraction data were collected on a Bruker SMART APEX X-ray diffractometer that was equipped with graphite monochromated MoKα radiation (λ = 0.71073 Å) by using an ω-scan technique. The intensities were corrected for absorption effects by using the Siemens Area Detector ABSorption correction program (SADABS). The structure was solved using SHELXL-97.19 All the hydrogen atoms were generated geometrically and refined isotropically using a riding model. All non-hydrogen atoms were refined using anisotropic displacement parameters. Crystallographic details and selected bond dimensions for 1–2 are listed in Tables S1 and S2 (ESI†), respectively. CCDC numbers are: 1515961 and 1502038.Syntheses
The two materials were characterized using infrared spectroscopy (IR) and TGA, and the results are shown in Fig. S1 (ESI) and S2 (ESI†). The PXRD patterns indicated that 1–2 remains crystalline upon desolvation (Fig. S3; ESI†). PXRD patterns (Fig. S4a; ESI†) confirmed that the crystallinity of 1 when it was soaked at different pHs of the medium, although it was likely that there were structural and/or symmetry changes, especially at low pHs. However, under different pHs, no major changes to the PXRD patterns of 2 were detected, which confirmed its stability (Fig. S4b; ESI†). To assess the porosity of 1 and 2, the N2 sorption isotherm was performed at 77 K, which displayed a type-I isotherm with the Brunauer–Emmett–Teller surface areas of 201.3 and 10.9 m2 g−1, respectively, and the full details are shown in Fig. S5 (ESI†).
Results and discussion
Crystal structures
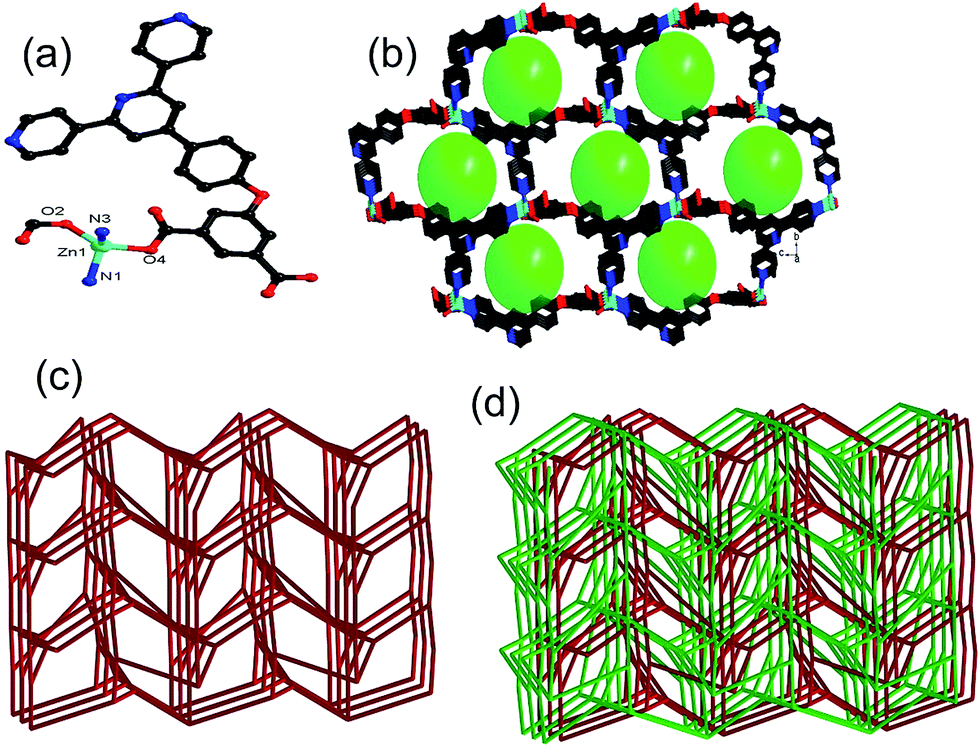 | ||
| Fig. 1 (a) Coordination environments of Zn(II) atoms in 1; (b) ball-and-stick representation of the 3D porous structure in 1; (c) view of 4-connected networks with 44.610.8 topology; (d) view of two-fold interpenetrating net. All the hydrogen atoms are omitted for clarity. | ||
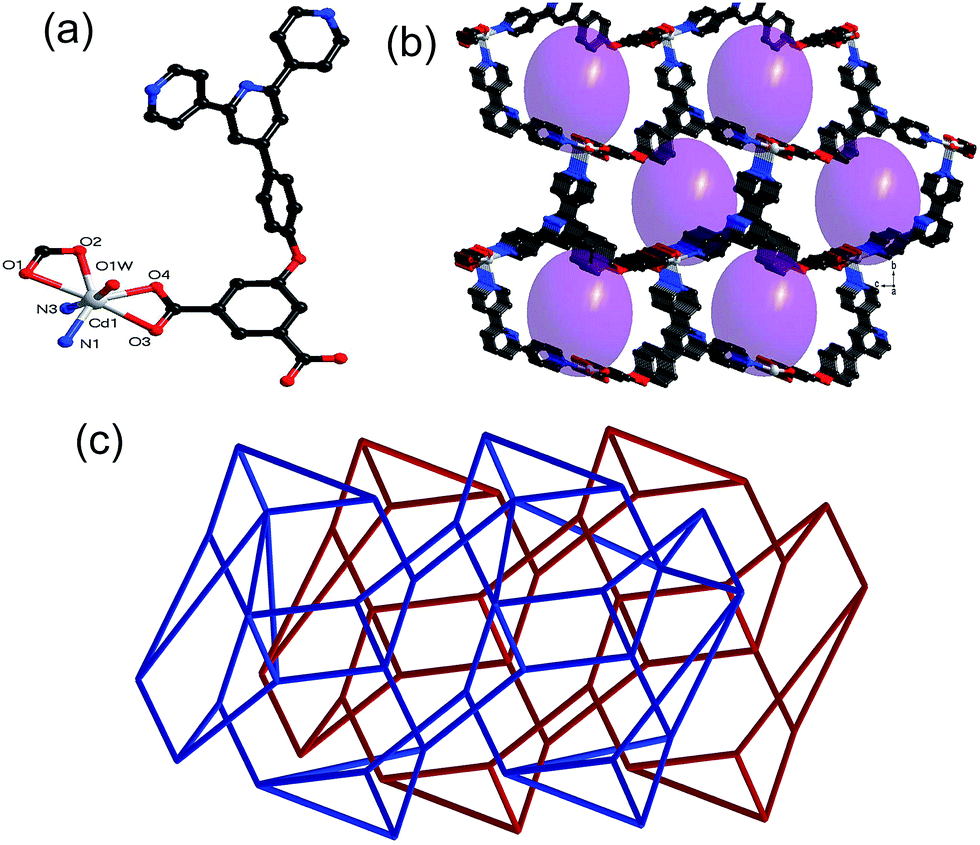 | ||
| Fig. 2 (a) Coordination environments of Cd(II) atoms in 2; (b) ball-and-stick representation of the 3D porous structure in 2; (c) view of two-fold interpenetrating net. All the hydrogen atoms are omitted for clarity. | ||
Luminescence sensing
Because of the existence of available voids in 1–2, their fluorescence responses to small molecules, cations and anions were measured. The PL spectra of 1 and 2 were recorded (Fig. S6; ESI†). It has been reported that the free H2dbp ligand shows emission at 550 nm (λex = 404 nm).18b Compared to the free H2dbp ligand, 1–2 show an emission at 393 and 405 nm (λex = 315 nm), respectively. The features of the luminescent properties of 1–2 are mostly attributed to the emission of the organic linker.22 In order to explore the luminescent responses of 1–2 to metal ions, 1–2 was dispersed in DMF solutions containing different M(NO3)x salts (M = Ag+, Ca2+, Cd2+, Co2+, Hg+, K+, Li+, Mg2+, Mn2+, Na+, Ni2+, Pb2+, Zn2+) as microcrystalline solids for potential luminescent sensing studies. Interestingly, the luminescent intensity of the samples was dependent on the nature of the metal ions. The luminescent properties of Mn+@1/2 are recorded and listed in Fig. 3a and 4a. The intensity of 1–2 is slightly enhanced by the addition of Ca2+, Li+, and Mg2+ (Fig. S7 and S8; ESI†), however, Mn2+ almost quenches the luminescence of 1–2, indicating that there is highly selective sensing of Mn2+ ions by 1–2 through this quenching effect.23 The PXRD patterns of 1–2 indicated their basic frameworks are kept upon dispersion in the metal ion solutions (Fig. S3; ESI†). To quantitatively investigate the response behavior of 1–2 towards Mn2+ ions, the emission spectra of 1–2 in the presence of different concentrations of Mn2+ ions in DMF were measured at room temperature. The luminescent intensity of 1–2 is almost completely quenched at an Mn(NO3)2 concentration of 37.5 × 10−4 and 60 × 10−5 M, respectively. Furthermore, a linear correlation for (I0 − I)/I0 and the concentration of Mn2+ ions was obtained, and following the 3δ/slope, the detection limits of 1.80 × 10−4 M and 5.08 × 10−6 M were calculated for 1 and 2, respectively, (Fig. S8†),18c which is higher relative to reported examples. The luminescent measurements indicate that the different anions have a great influence on the luminescent intensity of 1 and 2. The Cr2O72− ion has the largest quenching effect on the emission of 1–2 (Fig. 3c and 4c). The luminescent intensity of 1–2 decreased to 50% at concentrations of 15 × 10−4 and 10 × 10−5 M (Fig. 3d and 4d), respectively. It has been reported that the Mn2+ and Cr2O72− ions may compete for the absorption of light with the organic molecules and thus reduce the efficiency of energy transfer from ligand to ions.24,25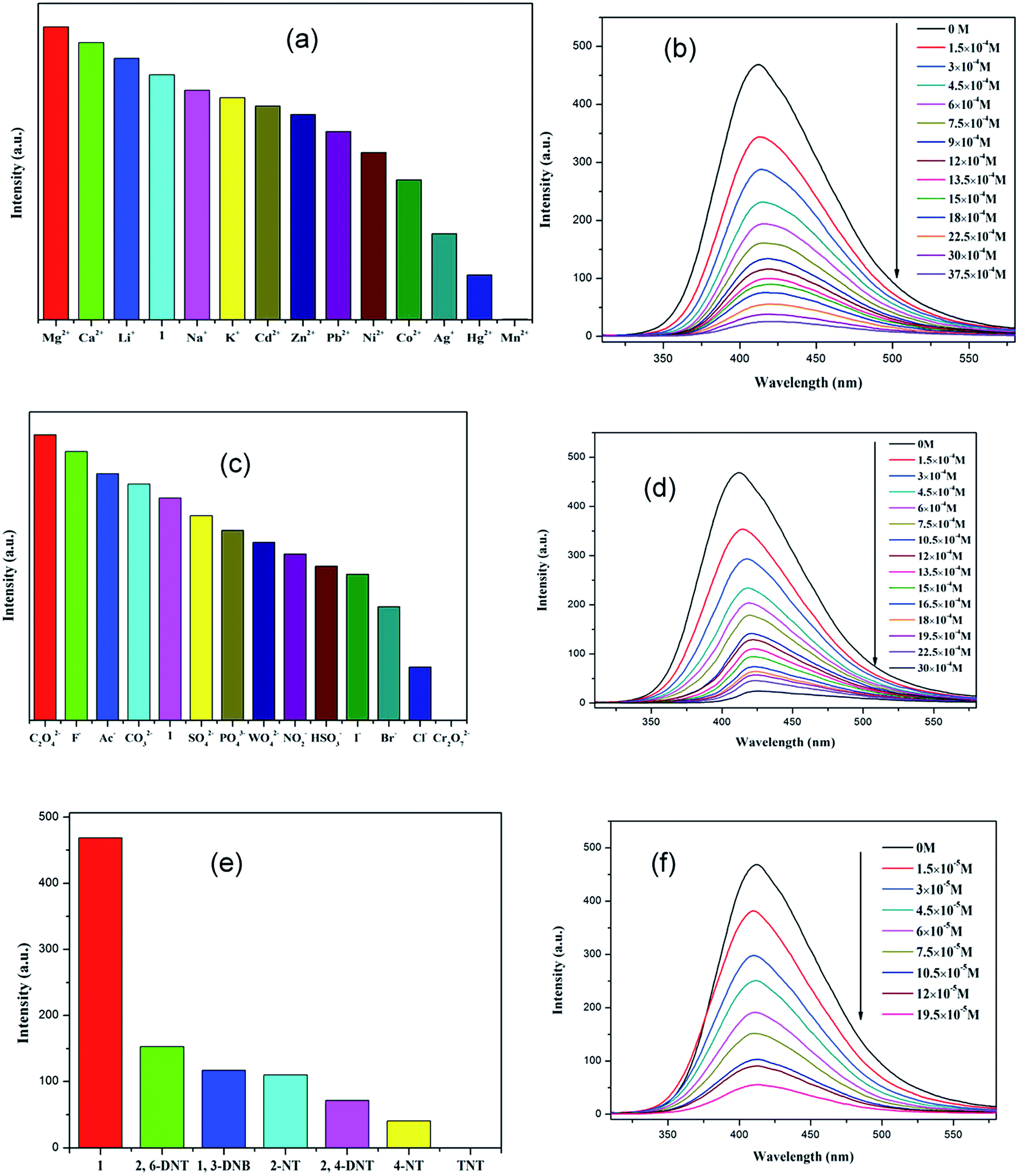 | ||
| Fig. 3 (a), (c) and (e) Emission spectra of 1 in different metal ions, anions and nitro-explosives (excited at 315 nm). (b), (d) and (f) Emission spectra of 1 in different concentrations of metal ions, anions and nitro-explosives in DMF (excited at 315 nm). | ||
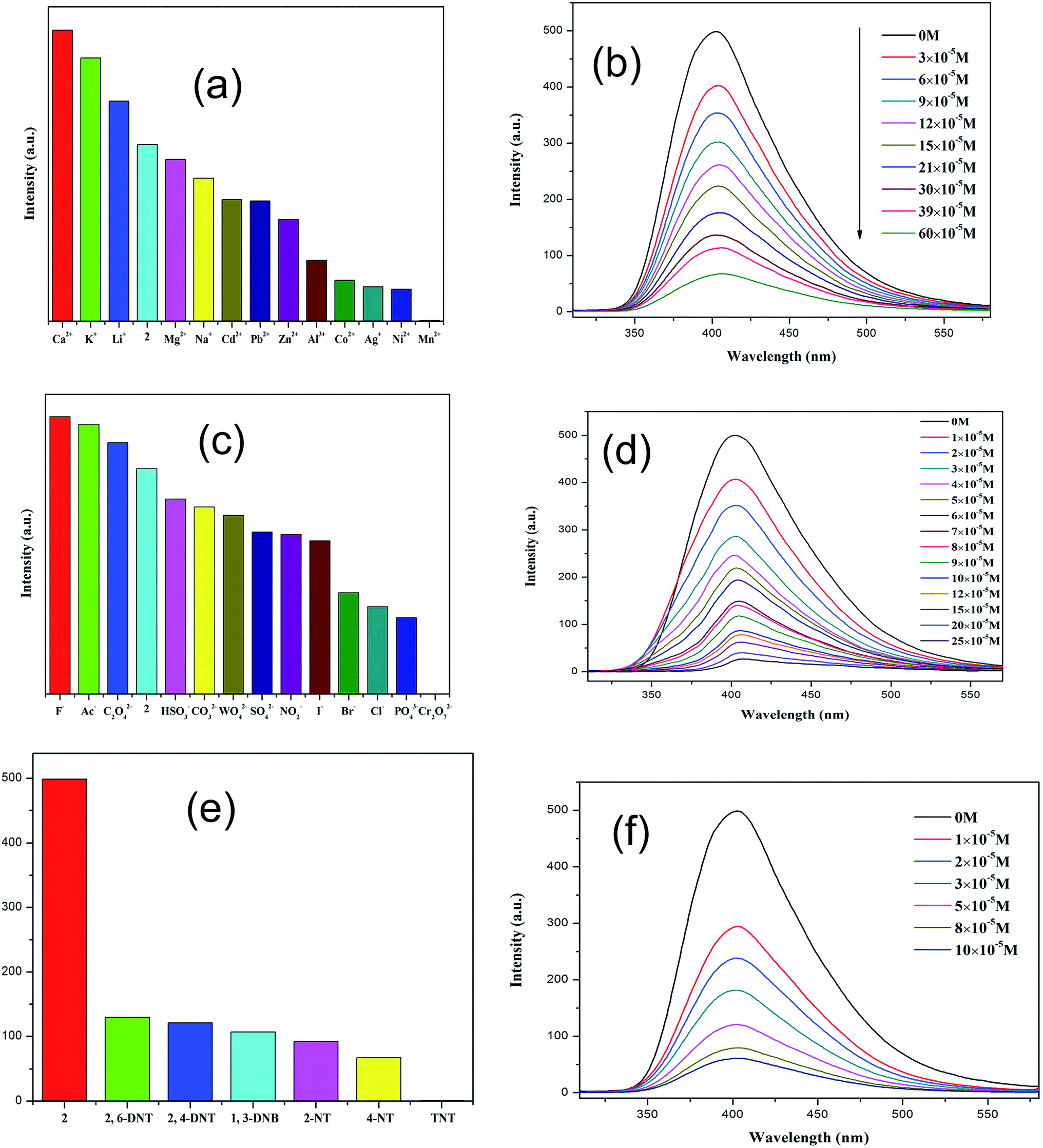 | ||
| Fig. 4 (a), (c) and (e) Emission spectra of 2 in different metal ions, anions and nitro-explosives (excited at 315 nm). (b), (d) and (f) Emission spectra of 2 in different concentrations of metal ions, anions and nitro-explosives in DMF (excited at 315 nm). | ||
The remarkable fluorescence and stability of 1–2 led to the investigation of its potential for fluorescence sensing of nitro-explosives (Fig. 3e and 4e).26–28 Thus, the DMF suspension of 1–2 was chosen for use in the detection of different kinds of nitroexplosives, including 2,4-dinitrotoluene (2,4-DNT), 2,6-dinitrotoluene (2,6-DNT), 2,4,6-trinitrotoluene (TNT), 2-nitrotoluene (2-NT), 4-nitrotoluene (4-NT) and 1,3-dinitrobenzene (1,3-DNB). These six nitro compounds can weaken the PL intensity of 1–2 to different degrees. The order of quenching efficiency is TNT > 4-NT > 2,4-DNT > 2-NT ≈ 1,3-DNB > 2,6-DNT. This efficiency is comparable to that of other MOFs in sensing TNT.9,26 Compared with 2,6-DNT and 1,3-DNB, TNT shows the strongest quenching effect, which may be ascribed to the fact that it has the strongest π-stacking between polymer and analyte. This phenomenon was observed in Zn/Cd–MOFs.26c Therefore, 1 and 2 can be used to distinguish between nitroaromatic molecules with different numbers of –NO2 groups. As shown in Fig. 3f and 4f, with the increase of TNT concentration, the luminescence intensity of 1–2@TNT suspension decreases distinctly. The maximum fluorescent intensity of 1–2 was reduced by 95% upon exposure to 19 × 10−5 and 10 × 10−5 M solutions of TNT. The fluorescence quenching efficiency can be quantitatively explained by the Stern–Volmer (SV) equation: (I0/I) = 1 + Ksv[Q],29,30 where Ksv is the quenching constant (M−1), and [Q] is the molar concentration of the analyte (M). The SV plots for TNT in 1–2 are linear at low concentrations, with a Ksv value of 4.35 × 104 and 1.38 × 105 M−1, respectively, and are among the higher values for MOF-based TNT sensors (Fig. S7 and S8; ESI†).28 The high quenching constant for TNT reveals that 1–2 are excellent sensors for sensitive and selective detection of TNT. As shown in Fig. 3e and 4e, the rates of quenching of the Zn(II) polymer 1 was faster with the analyte than that of Cd(II) polymer 2. Thus, the observed quenching trend is not controlled by a single factor, such as the nature of the analyte molecules, metal centers and relative orbital energies and interactions between coordination polymers and analyte molecules.
In the anion, cation and nitro-explosives used in this research, Mn2+, Cr2O72− and a series of nitro-explosives were all shown to have a strong absorbing range from 250 to 350 nm (Fig. S9; ESI†), whereas other substances detected have no significant absorption band in this range. The strong absorption band of dbp is located at approximately 280 nm, which is largely overlapped by the absorbing band of Mn2+ (Cr2O72− and series of nitro-explosives). Thus, there is competition for the absorption of the light source energy between the analytes and dbp from 250 to 350 nm. Combined with the absorption and luminescent spectra, it was suggested that the energy absorbed by the dbp is transferred to the Mn2+ (Cr2O72− and series of nitro-explosives) analytes, resulting in quenching feature in the luminescence intensity of 1–2. The quenching mechanism obtained here was in agreement with that proposed previously by others.28–31
As is known, fast and easy regeneration methods are two important issues for the recyclable performance. To determine the recyclable performance of 1–2, samples of 1–2 were immersed in an DMF solution of TNT (10−3 M) for 5 minutes to form TNT@1 and TNT@2, and then TNT@1 and TNT@2 were washed several times with DMF. The luminescence intensity of the recycled 1–2 were consistent with that of the original ones, although three runs were performed (Fig. S10; ESI†). The results indicate that 1–2 is still stable and that 1–2 can be recycled by a fast and simple method.
Photocatalytic activities
The dyes, MV and RhB were selected as model organic contaminants for a study of the photocatalytic activities of 1–2. As shown in Fig. 5, the absorption peaks of MV and RhB obviously decrease with increasing of reaction time in the presence of 1–2. The calculated results show that the degradation rate of MV is 72.7% for 1, and 93.0% for 2, whereas for RhB, the degradation rate for 1 and 2 is 81.6% and 88.2%, respectively. In addition, the control experiments on the degradation of organic pollutants were examined using the same reaction conditions but without the catalyst. The degradation rate of MV and RhB was 14.3% and 18.8% within 100 min under UV irradiation without a catalyst. As the previous photocatalytic results show, the photocatalytic performance of 2 is the better one because of its compacted structural features and stability. So, the final structure may influence its photocatalytic activities. In order to evaluate reproducible abilities of the as-synthesized photocatalysts, the repeated photocatalytic degradation with a constant MV/RhB concentration was determined, using complex 2 as an example. The degradation rates of 2 showed no significant reduction when the photocatalysts were used five times in the same procedures (Fig. S11a and b; ESI†), which suggested that the photocatalytic activities have good reproducibility. After photocatalysis, the PXRD patterns of 2 were similar to the simulated one, implying that 2 maintains its structural interlinkage after the photocatalysis reaction (Fig. S3; ESI†). A Cd-based MOF consisting of an amide-inserted flexible multicarboxylate ligand was reported by Wang et al.,32d however, the Cd-based MOF shows a low photocatalytic activity and only 47.9% of the RhB was degraded within 8 h. Wang et al. also explained that its narrow photoresponse region and low quantum yield limit its photocatalytic activity to some extent. An Anderson-type polyoxometalate-based metal–organic complex was hydrothermally synthesized at different pH ranges, and this showed that the removal of RhB dyes was almost negligible, but it showed a good photocatalysis selectivity for MV of 95.3%.32e The UV-Vis-near infrared spectra confirmed that 1–2 can be activated by radiation from the UV and visible light regions (Fig. S11c; ESI†). A possible photocatalytic mechanism for the previously described degradation process was proposed and is given in Scheme S2 (ESI†). When the absorbed energy is equal to or higher than its band gap energy (LUMO and the HOMO) of the materials 1–2, the HOMO seized one electron from one water molecule and then go back to stable state. When the HOMO captured one electron from one water molecule it then goes back to the stable state. The water molecule was oxidized into a ˙OH. Meanwhile, the electron of the LUMO reduced one O2 to one O2− by the combination of electrons (e−), which also changed to ˙OH. Finally, the complete ˙OH radical active works as an oxidizing agent to decompose MV/RhB.32–35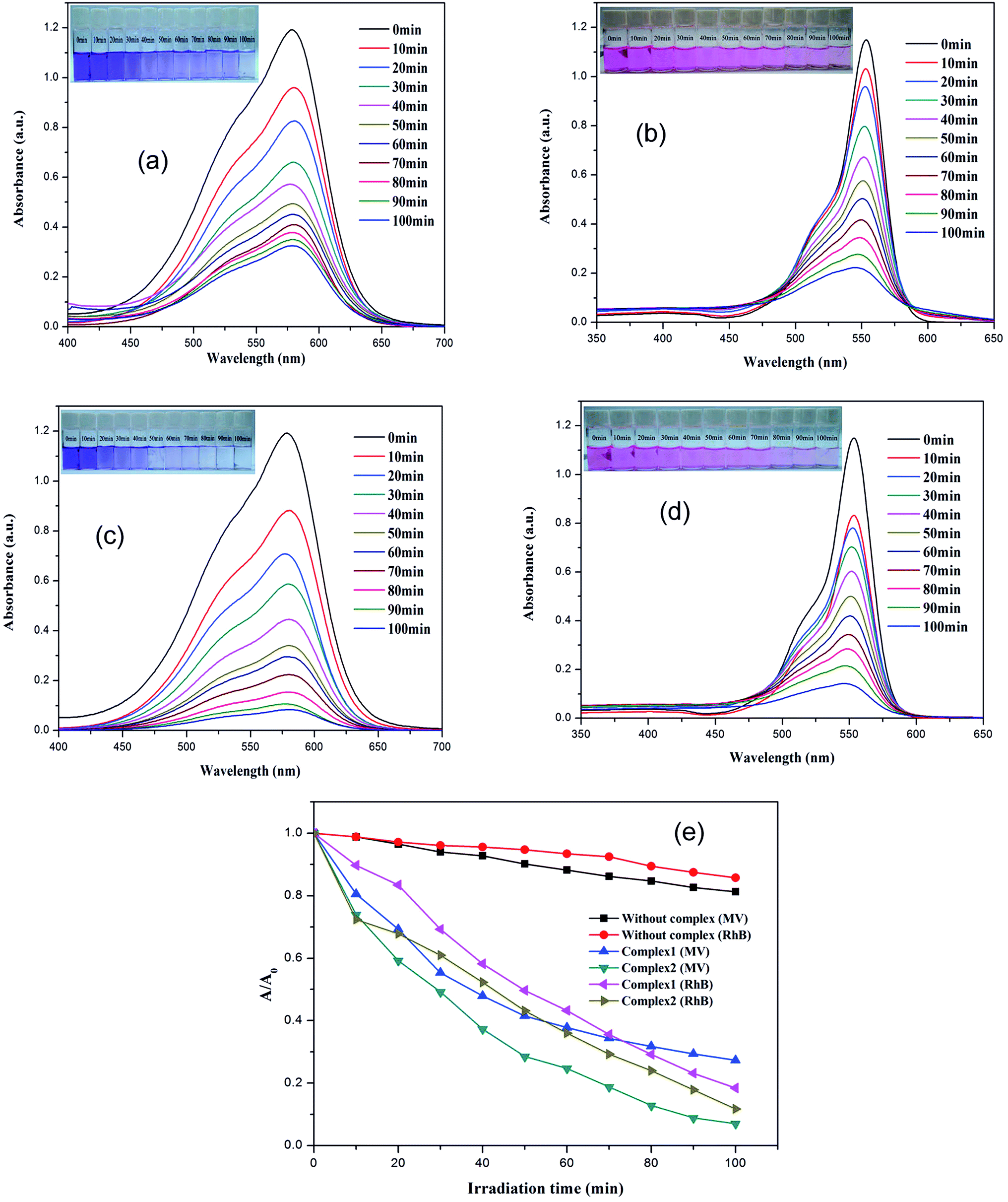 | ||
| Fig. 5 (a–d) UV-Vis absorption spectra of the MV/RhB solution during the decomposition reaction under irradiation with a 500 W Hg lamp, in the presence of 1–2; (e) photodegradation of MV/RhB solution with the change in A/A0 of 1–2, and the control experiments without any catalyst and with metal ions. | ||
In summary, two MOFs of 1–2 were successfully synthesized, and these could be used for the quick and sensitive detection of ions, cations and explosives using a fluorescence quenching technique. High stability and recyclability of 1–2 make them outstanding candidates for photocatalytic reactions. The present work is a promising approach for the design of MOF-based fluorescence sensors and photocatalytic reactions and this will probably be useful for more practical applications in the future.
Acknowledgements
The authors acknowledge financial assistance from National Natural Science Foundation of China (21501124), the Opening Project of Key Laboratory of Green Catalysis of Sichuan Institutes of High Education (No: LYJ1301, LYJ1306), the Education Committee of Sichuan Province (No. 15ZB0222, 15ZB0214), the start-up foundation of Sichuan University of Science & Engineering (No. 2014RC05, 2014RC34, 2015RC26), the Institute of Functionalized Materials (No. 2014PY01, 2015PY03), and the Project of Zigong Science & Technology and Intellectual Property Bureau (No. 2014HX02, 2015HX16), Guangxi Natural Science Foundation (No. 2016GXNSFAA380063), and City Social Science and Technology Development Program of Dongguan (Grant 2016108101005) and Science Foundation funded project of Guangdong Medical University (Z2016001 and M2016023).References
- (a) P. Cui, Y. Ma, H. Li, B. Zhao, J. Li, P. Cheng, P. B. Balbuena and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 18892 CrossRef CAS PubMed; (b) H. Fu, C. Qin, Y. Lu, Z. Zhang, Y. Li, Z. Su, W. Li and E. Wang, Angew. Chem., Int. Ed., 2012, 124, 8109 CrossRef.
- (a) L. Du, Z. Lu, K. Zheng, J. Wang, X. Zheng, Y. Pan, X. You and J. Bai, J. Am. Chem. Soc., 2013, 135, 562 CrossRef CAS PubMed; (b) J. Q. Liu, G. L. Liu, C. Y. Gu, W. C. Liu, J. W. Xu, B. H. Li and W. J. Wang, J. Mater. Chem. A, 2016, 4, 11630 RSC; (c) J. Q. Liu, J. Wu, F. M. Li, W. C. Liu, B. H. Li, J. Wang, Q. L. Li, R. Yadav and A. Kumar, RSC Adv., 2016, 6, 31161 RSC; (d) B. H. Li, J. Wu, J. Q. Liu, C. Y. Gu, J. W. Xu, M. M. Luo, R. Yadav, A. Kumar and S. R. Batten, ChemPlusChem, 2016, 81, 885 CrossRef CAS.
- Y. N. Gong, Y. L. Huang, L. Jiang and T. B. Lu, Inorg. Chem., 2014, 53, 9457 CrossRef CAS PubMed.
- (a) Z. C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC; (b) J. N. Hao and B. Yan, J. Mater. Chem. A, 2015, 3, 4788 RSC; (c) X. Y. Xu, Q. C. Chen, Y. D. Yu and X. C. Huang, Inorg. Chem., 2016, 55, 75 CrossRef CAS PubMed.
- (a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (b) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC; (c) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (d) S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656 RSC.
- (a) C. Y. Sun, X. L. Wang, X. Zhang, C. Qin, P. Li, Z. M. Su, D. X. Zhu, G. G. Shan, K. Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717 Search PubMed; (b) J. Q. Liu, J. Wu, F. M. Li, W. C. Liu, B. H. Li, J. Wang, Q. L. Li, R. Yadav and A. Kumar, RSC Adv., 2016, 6, 31161 RSC.
- (a) J. S. Qin, D. Y. Du, W. L. Li, J. P. Zhang, S. L. Li, Z. M. Su, X. L. Wang, Q. Xu, K. Z. Shao and Y. Q. Lan, Chem. Sci., 2012, 3, 2114 RSC; (b) Y. Q. Lan, S. L. Li, H. L. Jiang and Q. Xu, Chem.–Eur. J., 2012, 18, 8076 CrossRef CAS PubMed; (c) S. L. Li, Y. Q. Lan, H. Sakurai and Q. Xu, Chem.–Eur. J., 2012, 18, 16302 CrossRef CAS PubMed; (d) S. R. Zhang, D. Y. Du, K. Tan, J. S. Qin, H. Q. Dong, S. L. Li, W. W. He, Y. Q. Lan, P. Shen and Z. M. Su, Chem.–Eur. J., 2013, 19, 11279 CrossRef CAS PubMed.
- (a) S. R. Zhang, D. Y. Du, J. S. Qin, S. J. Bao, S. L. Li, W. W. He, Y. Q. Lan, P. Shen and Z. M. Su, Chem.–Eur. J., 2014, 20, 3589 CrossRef CAS PubMed; (b) C. Q. Zhang, L. Sun, Y. Yan, J. Y. Li, X. W. Song, Y. L. Liu and Z. Q. Liang, Dalton Trans., 2015, 44, 230 RSC; (c) C. Q. Zhang, Y. Yan, Q. H. Pan, L. B. Sun, H. M. He, Y. L. Liu, Z. Q. Liang and J. Y. Li, Dalton Trans., 2015, 44, 13340 RSC; (d) L. B. Sun, H. Z. Xing, J. Xu, Z. Q. Liang, J. H. Yu and R. R. Xu, Dalton Trans., 2013, 42, 5508 RSC; (e) C. Q. Zhang, Y. Yan, L. B. Sun, Z. Q. Liang and J. Y. Li, CrystEngComm, 2016, 18, 4102 RSC; (f) A. J. Lan, K. H. Li, H. H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. C. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334 CrossRef CAS PubMed.
- (a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153 CrossRef CAS PubMed; (b) S. Pramanik, Z. Hu, X. Zhang, C. Zheng, S. Kelly and J. Li, Chem.–Eur. J., 2013, 19, 15964 CrossRef CAS PubMed; (c) D. Banerjee, Z. Hu, S. Pramanik, X. Zhang, H. Wang and J. Li, CrystEngComm, 2013, 15, 9745 RSC; (d) B. Gole, A. K. Bar and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 2276 CrossRef CAS PubMed; (e) G. Liu, Y. Qin, L. Jing, G. Wei and H. Li, Chem. Commun., 2013, 49, 1699 RSC; (f) Y. S. Xue, Y. He, L. Zhou, F. J. Chen, Y. Xu, H. B. Du, X. Z. You and B. Chen, J. Mater. Chem., 2013, 1, 4525 RSC; (g) T. K. Kim, J. H. Lee, D. Moon and H. R. Moon, Inorg. Chem., 2013, 52, 589 CrossRef CAS PubMed; (h) C. Zhu, W. Xuan and Y. Cui, Dalton Trans., 2012, 41, 3928 RSC.
- I. H. Park, R. Medishetty, J. Y. Kim, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5591 CrossRef CAS PubMed.
- B. Gole, A. K. Bar and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 13321 CrossRef CAS PubMed.
- (a) X. Li, C. F. Bi, Y. H. Fan, X. Zhang, X. M. Meng and L. S. Cui, Inorg. Chem. Commun., 2014, 50, 35 CrossRef CAS; (b) G. G. Mohamed, M. M. Omar and A. A. Ibrahim, Spectrochim. Acta, Part A, 2010, 75, 678 CrossRef PubMed.
- Z. L. You, Y. Lu, N. Zhang and B. W. Ding, Polyhedron, 2011, 30, 2186 CrossRef CAS.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC.
- (a) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (b) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC; (c) Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- H. Xiang, W. Y. Gao, D. C. Zhong, L. Jiang and T. B. Lu, CrystEngComm, 2011, 13, 5825 RSC.
- (a) D. Li, T. Wu, X. P. Zhou, R. Zhou and X. C. Huang, Angew. Chem., Int. Ed., 2005, 44, 4175 CrossRef CAS PubMed; (b) H. Wu, J. Yang, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2012, 12, 2272 CrossRef CAS; (c) S. C. Chen, R. R. Qin, Z. H. Zhang, J. Qin, H. B. Gao, F. A. Sun, M. Y. He and Q. Chen, Inorg. Chim. Acta, 2012, 390, 61 CrossRef CAS; (d) C. J. Shen, T. L. Sheng, C. B. Tian, Q. L. Zhu and X. T. Wu, Inorg. Chem. Commun., 2012, 17, 142 CrossRef CAS.
- (a) Y. Wu, L. Lu, J. S. Feng, Y. L. Li, Y. C. Sun and A. Q. Ma, J. Solid State Chem., 2017, 245, 213 CrossRef CAS; (b) K. L. Cao, Y. Xia, G. X. Wang and Y. L. Feng, Inorg. Chem. Commun., 2015, 53, 42 CrossRef CAS; (c) H. Xu, H. C. Hu, C. S. Cao and B. Zhao, Inorg. Chem., 2015, 54(10), 4585 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Program for Siemens Area Detector Absorption Corrections, University of Göttingen, Germany, 1997 Search PubMed.
- L. Hou, L. N. Jia, W. J. Shi, Y. Y. Wang, B. Liu and Q. Z. Shi, Dalton Trans., 2013, 42, 3653 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- (a) R. Singh, J. Mrozinski and P. K. Bharadwaj, Cryst. Growth Des., 2014, 14, 3623 CrossRef CAS; (b) T. Wang, C. Zhang, Z. Ju and H. Zheng, Dalton Trans., 2015, 44, 6926 RSC; (c) F. L. Hu, W. Wu, P. Liang, Y. Q. Gu, L. G. Zhu, H. Wei and J. P. Lang, Cryst. Growth Des., 2013, 13, 5050 CrossRef CAS.
- (a) Q. Tang, S. X. Liu, Y. W. Liu, J. Miao, S. J. Li, L. Zhang, Z. Shi and Z. P. Zheng, Inorg. Chem., 2013, 52, 2799 CrossRef CAS PubMed; (b) Y. T. Liang, G. P. Yang, B. Liu, Y. T. Yan, Z. P. Xi and Y. Y. Wang, Dalton Trans., 2015, 44, 13325 RSC.
- X. Y. Dong, R. Wang, J. Z. Wang, S. Q. Zang and T. C. W. Mak, J. Mater. Chem. A, 2015, 3, 641 CAS.
- B. L. Chen, S. C. Xiang and G. D. Qian, Acc. Chem. Res., 2010, 43, 1115 CrossRef CAS PubMed.
- (a) S. R. Zhang, D. Y. Du, J. S. Qin, S. J. Bao, S. L. Li, W. W. He, Y. Q. Lan, P. Shen and Z. M. Su, Chem.–Eur. J., 2014, 20, 3589 CrossRef CAS PubMed; (b) D. Tian, Y. Li, R. Y. Chen, Z. Chang, G. Y. Bu and X. H. Bu, J. Mater. Chem. A, 2014, 2, 1465 RSC; (c) L. Y. Zhang, L. Y. Sun, X. Y. Li, Y. L. Tian and G. Z. Yuan, Dalton Trans., 2015, 44, 401–410 RSC.
- (a) M. E. Germain and M. J. Knapp, J. Am. Chem. Soc., 2008, 130, 5422 CrossRef CAS PubMed; (b) Z. J. Zhang, S. C. Xiang, X. T. Rao, Q. Zheng, F. R. Fronczek, G. D. Qian and B. L. Chen, Chem. Commun., 2010, 46, 7205 RSC; (c) D. X. Ma, B. Y. Li, X. J. Zhou, Q. Zhou, K. Liu, G. Zeng, G. H. Li, Z. Shi and S. H. Feng, Chem. Commun., 2013, 49, 8964 RSC; (d) X. H. Zhou, H. H. Li, H. P. Xiao, L. Li, Q. Zhao, T. Yang, J. L. Zuo and W. Huang, Dalton Trans., 2013, 42, 5718 RSC; (e) G. Y. Wang, L. L. Yang, Y. Li, H. Song, W. J. Ruan, Z. Chang and X. H. Bu, Dalton Trans., 2013, 42, 12865 RSC.
- (a) J. Wang, J. Mei, W. Z. Yuan, P. Lu, A. J. Qin, J. Z. Sun, Y. G. Ma and B. Z. Tang, J. Mater. Chem., 2011, 21, 4056 RSC; (b) W. Wei, X. B. Huang, K. Y. Chen, Y. M. Tao and X. Z. Tang, RSC Adv., 2012, 2, 3765 RSC; (c) S. Ramachandra, Z. D. Popovic, K. S. Schuermann, F. Cucinotta, G. Calzaferri and L. D. Cola, Small, 2011, 7, 1488 CrossRef CAS PubMed.
- (a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed; (b) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- Z. C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- A. K. Chaudhari, S. S. Nagarkar, B. Joarder and S. K. Ghosh, Cryst. Growth Des., 2013, 13, 3716 CAS.
- (a) S. S. Chen, J. Fan, T. Okamura, M. S. Chen, Z. Su, W. Y. Sun and N. Ueyama, Cryst. Growth Des., 2010, 10, 812 CrossRef CAS; (b) H. Y. He, D. Collins, F. G. Dai, X. L. Zhao, G. Q. Zhang, H. Q. Ma and D. F. Sun, Cryst. Growth Des., 2010, 10, 895 CrossRef CAS; (c) G. Li, Z. Lei and Q. M. Wang, J. Am. Chem. Soc., 2010, 132, 17678 CrossRef CAS PubMed; (d) F. Q. Wang, C. F. Dong, Z. C. Wang, Y. R. Cui, C. M. Wang, Y. N. Zhao and G. D. Li, Eur. J. Inorg. Chem., 2014, 6239 CrossRef CAS; (e) X. L. Wang, J. J. Sun, H. Y. Lin, Z. H. Chang, G. C. Liu and X. Wang, RSC Adv., 2016, 6, 110583 RSC.
- (a) H. Yang, X. W. He, F. Wang and J. Zhang, J. Mater. Chem., 2012, 22, 21849 RSC; (b) W. Q. Kan, B. Liu, J. Yang, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2012, 12, 2288 CrossRef CAS.
- (a) J. L. Wang, C. Wang and W. B. Lin, ACS Catal., 2012, 2, 2630 CrossRef CAS; (b) M. C. Das, H. Xu, Z. Y. Wang, G. Srinivas, W. Zhou, Y. F. Ke, V. N. Nesterov, G. D. Qian and B. L. Chen, Chem. Commun., 2011, 47, 11715 RSC.
- (a) D. X. Li, C. Y. Ni, M. M. Chen, M. Dai, W. H. Zhang, W. Y. Yan, H. X. Qi, Z. G. Ren and J. P. Lang, CrystEngComm, 2014, 16, 2158 RSC; (b) K. X. Wang and J. S. Chen, Acc. Chem. Res., 2011, 44, 531 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1502038 and 1515961. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28728j |
| This journal is © The Royal Society of Chemistry 2017 |