DOI:
10.1039/C6RA26983D
(Paper)
RSC Adv., 2017,
7, 10407-10414
Reductive removal of gaseous nitrous oxide by activated carbon with metal oxide catalysts
Received
18th November 2016
, Accepted 2nd February 2017
First published on 7th February 2017
Abstract
The efficient reductive decomposition of gaseous nitrous oxide (N2O) from industrial effluents is of practical importance in abating greenhouse gas emissions. In the present study, active carbon (AC) has been chosen as both a reductive agent and a catalyst support. Dozens of AC-based catalysts with different kinds and amounts of metal oxide have been prepared under various conditions and characterized. The performances of Cu-containing AC have been studied at varying gas flow rates, Cu contents, and calcination temperatures. N2O in a gas mixture (42% N2O, 58% N2) was found to be completely removed by Cu-loaded AC (9 wt% Cu, calcined at 400 °C) at 325 °C with a GHSV of 2293 h−1. This process is viable by virtue of the low cost of AC and easier manipulation and process control in comparison with alternative methods employing reductive gases such as hydrogen and ammonia.
1. Introduction
Nitrous oxide (N2O) is a greenhouse gas with a global warming potential value of about 310. As a by-product of the manufacture of several chemical products, such as adipic acid and nitric acid, its atmospheric concentration has increased significantly over the last decades, and continues to increase by 0.2–0.3% per year.1 This inexorable trend has attracted a great deal of attention for the development of an efficient method for the removal of N2O from industrial exhausts, especially for the tail gas of the adipic acid production process using HNO3 as oxidizing agent, where the N2O content is as high as 38% along with N2 48.1%, O2 4.4%, CO2 9.5%, and NO 0.03% (in wt%).2
A direct decomposition process is generally used for gas effluents with low N2O concentrations,3 which operates at high temperatures (>500 °C) with various catalysts, e.g., hydrotalcite-like materials,4 metal-containing zeolites,5–7 or supported metal oxides,8–11 e.g. CaO–Co3O4,12 and NiLaOx.13 The decomposition of N2O is composed of three steps, viz., adsorption, cleavage of the N–O bond, and desorption of O2, of which the desorption of O2 is considered as the rate-limiting step. To promote O2 desorption, some reductive substances, e.g., CH4, NH3,14 H2,15 and CO,16 have been used to instantly consume the in situ generated O2. However, these reductive gases are expensive and/or explosive, and their flow rate needs dynamic control according to the composition of the gas stream, which complicates the process. Activated carbon (AC), as an inexpensive and active solid reducing agent, can overcome these drawbacks, and is thus worthy of study for the reductive removal of N2O. In fact, coal char is effective in reducing N2O and NO to N2 at combustion temperature,17 and AC loaded with alkali metal oxides shows better performance. Both Na and K can catalyze the reaction between carbon and N2O, and K is better than Na.18,19 The reaction between N2O and C may also be catalyzed by Ni and Pt, and further promoted by K, as reported by Gonçalves et al.20,21
To date, little research has been conducted to compare the performances of different metal oxides supported on AC for the removal of N2O under otherwise identical conditions. In this paper we are focused on the treatment of the tail gas of adipic acid production process through catalytic reduction of N2O using AC as a solid reducing agent. For this purpose, we made a comparative study on the catalytic performances of different metal oxides for the reduction of N2O on AC, and CuO is found to be the most efficient one. Based on catalyst characterization, a structure–activity relationship has been elucidated, and a reaction mechanism is proposed.
2. Experimental
2.1 Chemical materials
The AC used was a commercial coconut-shell-derived carbon (Tangshan Hua Neng Technology Carbon Co., Ltd., Tangshan, China). All metal nitrates, i.e. Cu(NO3)2·3H2O, Ce(NO3)3·6H2O, Ni(NO3)2·6H2O, Zn(NO3)2·6H2O, Co(NO3)2·6H2O, Fe(NO3)3·9H2O, and Mn(NO3)2, were of analytical reagent grade (Beijing Chem. Co., Ltd., Beijing, China, purity ≥ 99.0%) and were used as received. All aqueous solutions were prepared with deionized water.
2.2 Pre-treatment of AC
The AC was firstly ground into particles of 10–20 mesh, boiled with deionized water for 15 min, and then washed five times with deionized water. The resultant particles were placed in an oven and dried at 110 °C for 24 h.
2.3 Preparation of AC-supported metal oxide catalysts
Certain amounts (0.1–11 wt%, mass fraction of metal atom in AC) of metal nitrate were weighed and dissolved in water (300 mL), AC (30 g) was added to each metal nitrate solution, and the mixtures were stirred magnetically for 24 h at room temperature. They were then placed in an oven and dried at 90 °C for 72 h until the water had completely evaporated. The resultant AC was transferred to a muffle furnace and calcined at 200, 300, or 400 °C for 2 h. Hereinafter, the obtained catalysts are designated as xM-AC-T, where M represents the supported metal, x is the mass fraction of metal atoms in the AC, and T is the calcination temperature.
2.4 Catalyst characterization
Fourier-transform infrared (FTIR) spectra in the range ν = 4000–400 cm−1 were recorded at room temperature on a Nicolet 6700 spectrometer. Spectra were collected after 32 scans at 4 cm−1 resolution. The spectra were acquired from KBr pellets composed of 1 mg of the samples and 100 mg of KBr dried at 200 °C for 24 h. X-ray diffraction (XRD) patterns of the calcined samples were recorded at room temperature on a D/Max 2500 (VB2+ PC) diffractometer using Cu-Kα radiation, operated at 40 kV and 200 mA. The patterns were recorded over a 2θ range of 10–90° with a 0.02° step size. Simultaneous thermogravimetric analysis (TGA) and derivative thermogravimetric analysis curves were recorded on a TGA/DSC1/1100 SF apparatus at a heating rate of 10 °C min−1 over the range 25–700 °C in air. X-ray photoelectron spectroscopy (XPS) spectra were recorded on an ESCALAB 250 apparatus with a scanning electron spectroscopy for chemical analysis (ESCA) microscope equipped with an Al monochromatic X-ray source (300 W), employing a beam diameter of 2 mm in CAE analyzer mode. All binding energies were referenced to the C 1s peak at 285 eV. The individual components were obtained by curve fitting.
2.5 N2O removal experiments
A stainless steel tubular reactor of Φ5 (i.d.) × 650 mm (effective length 200 mm) was used to investigate the activity of metal oxides supported on AC for N2O removal at atmospheric pressure. Catalyst (2.0 g) was loaded into the reactor each time, and the gas mixture (42% N2O, 58% N2) was fed at rates varying from 50 to 150 mL min−1 by means of a V10FA mass flow controller. The reaction temperature was controlled through a proportion integral derivative (PID)-regulated oven. To compare the catalytic performances of different transition metal oxides, the reaction temperature was varied from 300 to 550 °C in steps of 50 °C. To study the effect of the Cu-loading on the performance of the AC-supported Cu catalysts, the reaction temperature was varied from 300 to 550 °C in steps of 25 °C. Furthermore, the reaction temperature was varied from 150 to 325 °C in steps of 25 °C to evaluate the activities of the catalysts calcined at different temperatures. In all of the experiments, temperature was increased at a rate of 5 °C min−1. At any given temperature, the reaction duration was 1 h. The concentration of N2O in the outlet gas was monitored online by means of a TM GC7700 gas chromatograph equipped with a TCD and a Porapak Q (6 m × 3 mm) column.
3. Results and discussion
3.1 Characterization of the catalysts
Structural characteristics of AC. The AC support used here is a coconut-shell-derived carbon material, which is commercially available. Its structure has been characterized by many researchers22–24 using N2 adsorption–desorption method, and the structural parameters were determined as follows, i.e. BET specific area 797 ± 45 m2 g−1, pore size 2.03 ± 0.07 nm, and total pore volume 0.402 ± 0.02 m3 g−1. Obviously, the AC has a larger pore volume and specific area with rich porosity and micropores, which is much higher than that of coal chars and comparable with many commercial AC sorbents.25–27 The pore size and surface area is known to be instrumental for the mass transfer and reactivity for a heterogeneous reaction, and a higher surface area and porosity is helpful for enhancing the dispersion of the catalysts and accessibility of the AC and thus the reduction rate of N2O. In short, higher surface area favors the catalytic reactivity of N2O reduction on AC.
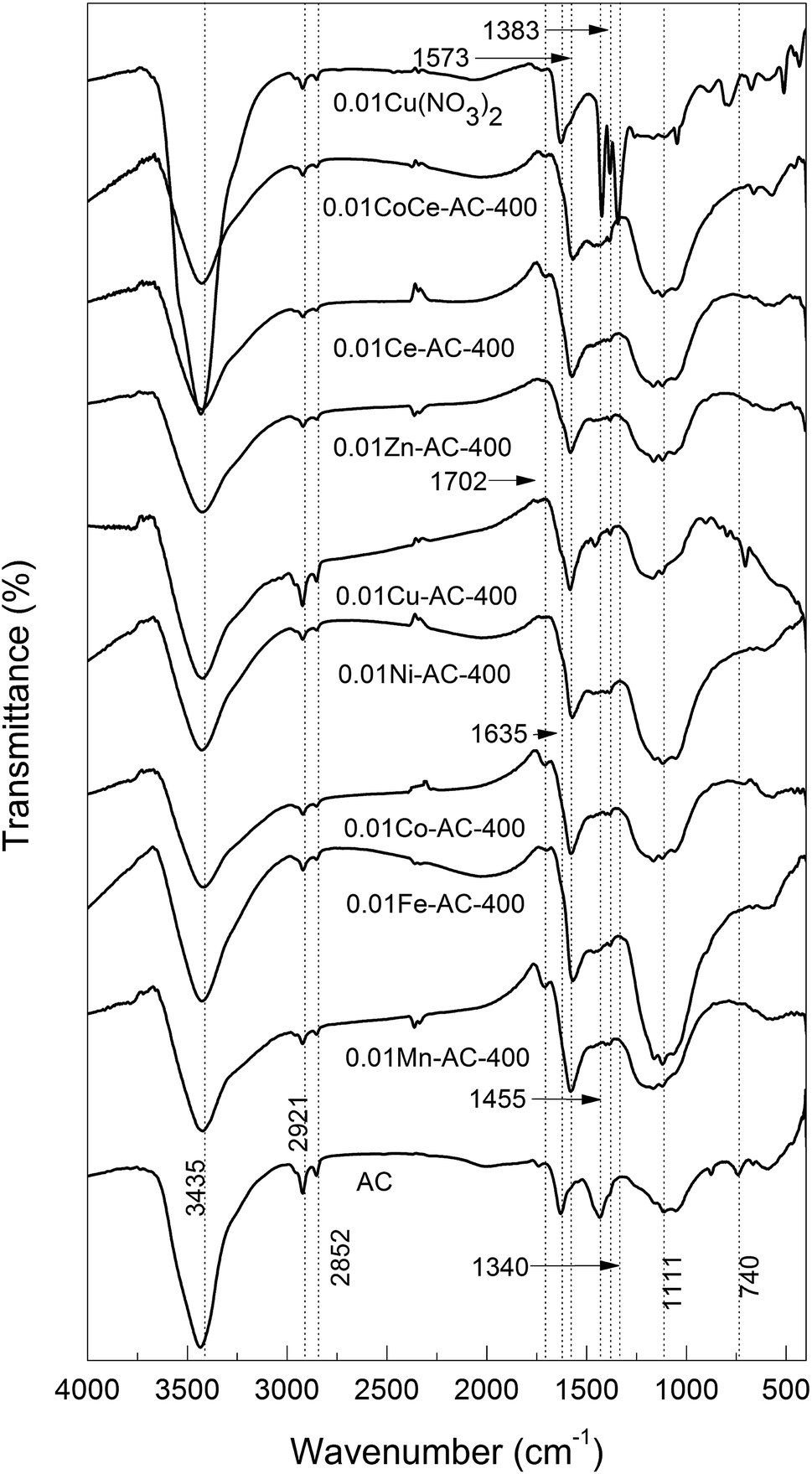
FTIR characterization. FTIR spectra of the pristine AC and metal-oxide-loaded AC samples are shown in Fig. 1. All samples showed an absorption at ν ≈ 3435 cm−1, which could be ascribed to the stretching vibration of O–H28,29 due to the presence of water. The water contents of all samples were similar, except for AC-supported Cu(NO3)2, as evidenced by the similar intensities of their O–H peaks. Furthermore, all of the samples showed two bands centered at ν = 2921 and 2852 cm−1 due to asymmetric and symmetric C–H stretching vibrations in aliphatic CH, CH2, and CH3 groups.29,30 A scissor vibration of a CH2 group is responsible for an absorption peak at ν = 1455 cm−1, which was no longer seen for the metal-oxide-loaded AC samples. For AC-supported Cu(NO3)2, two bands were seen at ν = 1383 and 1340 cm−1 attributable to nitrate (ν = 1410–1340 cm−1),30 which disappeared in the calcined ACs. From the above results, we can deduce that: (1) all nitrates were decomposed to the corresponding metal oxides upon calcination at 400 °C; (2) the structural change of AC is virtually irrelevant to the metal species.
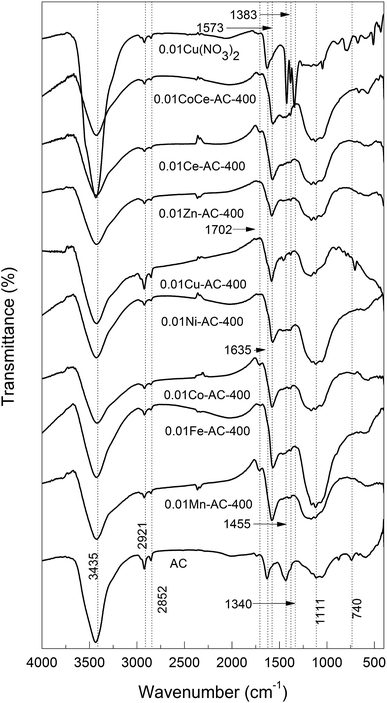 |
| | Fig. 1 FTIR spectra of pristine AC and metal-oxide-loaded samples. | |
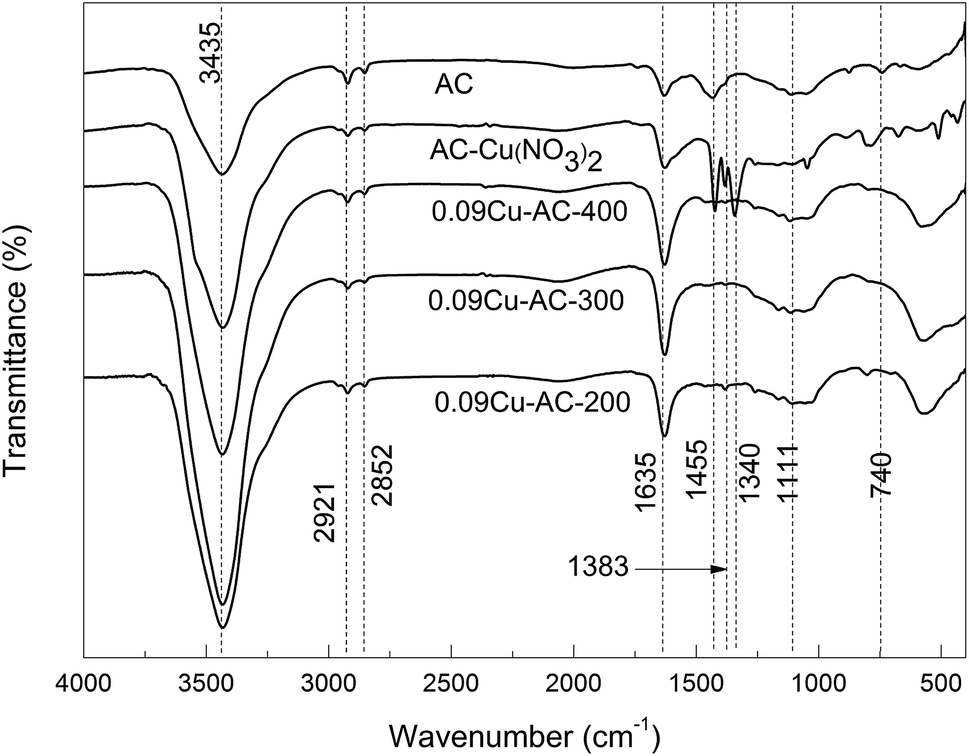
Fig. 2 shows the FTIR spectra of Cu-loaded AC samples calcined at 200, 300, and 400 °C, respectively. It is evident that the loaded Cu(NO3)2 was completely converted to CuO in all of the calcined samples.
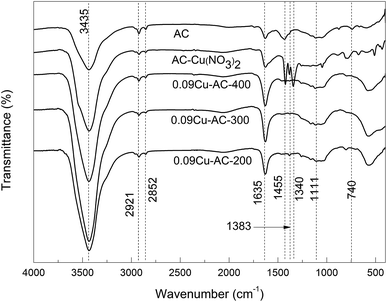 |
| | Fig. 2 FTIR spectra of AC with CuO calcined at different temperatures. | |
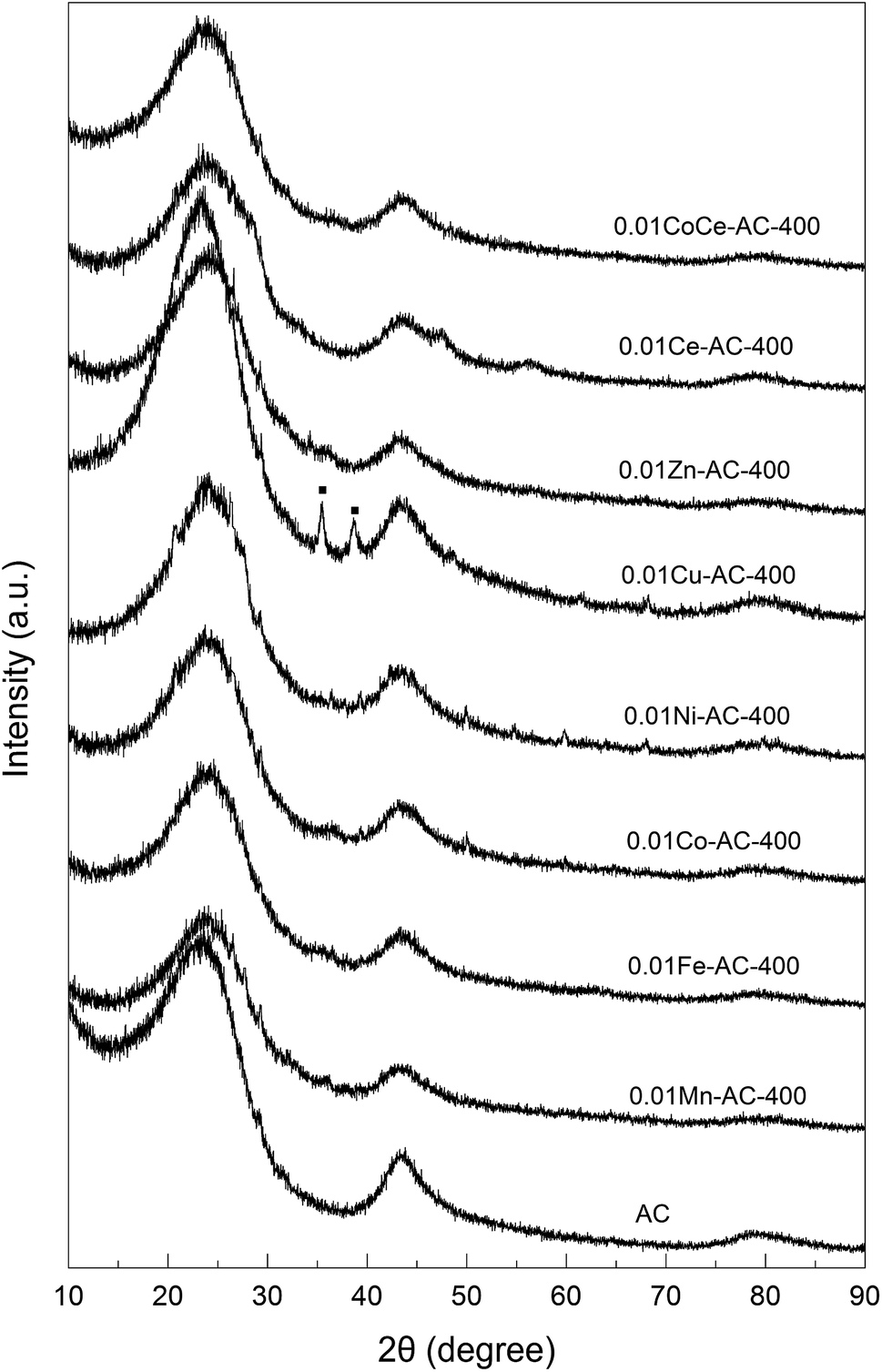
XRD characterization. The XRD patterns of the pristine and metal-oxide-loaded AC samples are shown in Fig. 3. The pristine AC showed two broad peaks, viz. at 2θ ≈ 23° and 43°, respectively, which are characteristic of amorphous carbon.31 These two peaks were also present in the patterns of all of the metal-oxide-loaded AC samples, indicating retention of the amorphous structure of AC in the modified AC catalysts. The pattern for 0.01Cu-AC-400 features two distinct peaks at 35° and 38°, which can be ascribed to CuO (JCPDS).32 Except for 0.01Cu-AC-400, the diffraction peaks of other metal oxides on the AC samples were virtually unobservable. This may indicate amorphous structures of these metal oxides due to their poor crystallinity at a calcination temperature of 400 °C. Chmielarz et al. studied cobalt-containing hydrotalcite-like materials, and found that an amorphous structure of the metal oxide was formed when the calcination temperature was 600 °C, but that the crystallinity increased significantly with temperature.33 Therefore, the poor crystallinity of some of the present metal oxides on AC may have resulted from the lower calcination temperature.
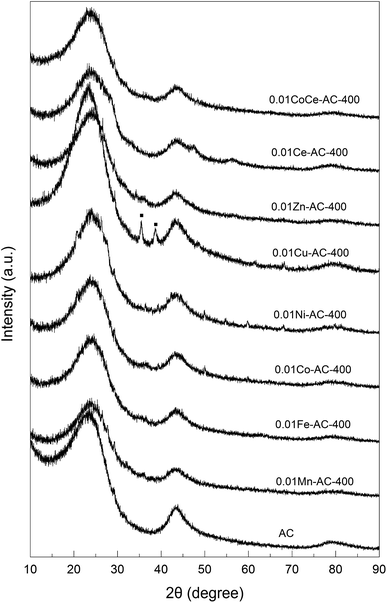 |
| | Fig. 3 XRD patterns of pristine AC and metal-oxide-loaded samples. | |
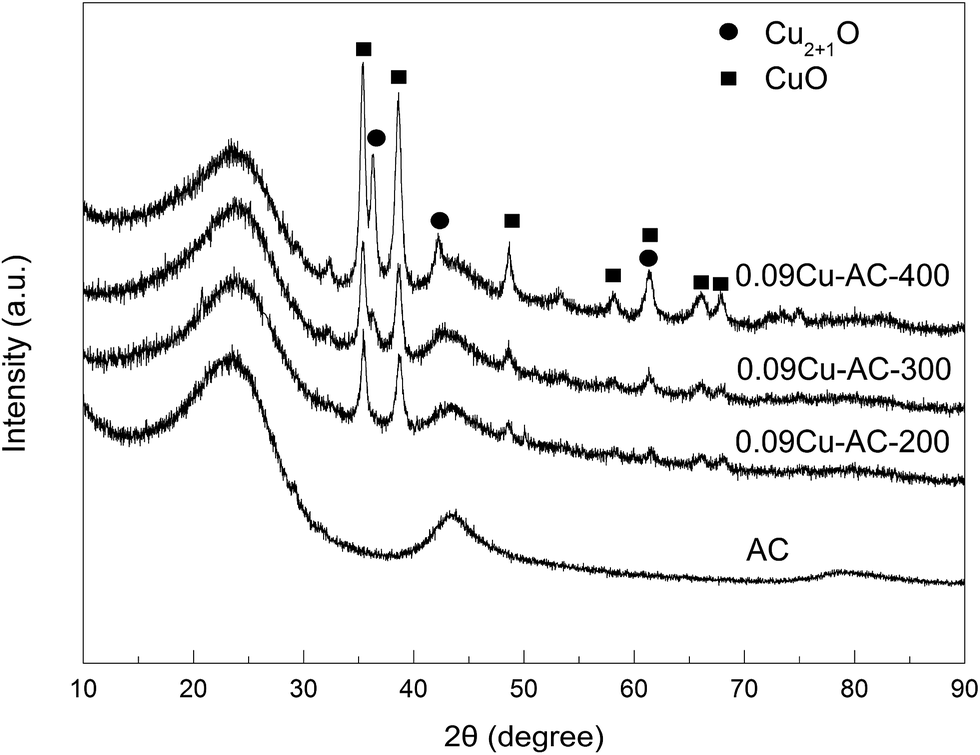
XRD patterns of the Cu-loaded AC samples calcined at 200, 300, and 400 °C are presented in Fig. 4. Several peaks attributable to CuO and Cu2O were observed,32,34 and the intensities of these peaks increased steadily with increasing calcination temperature, implying increasing crystallinity of the copper oxides. Additionally, the content of Cu2O increased with increasing calcination temperature, suggesting that a certain amount of CuO was reduced by AC at 400 °C.
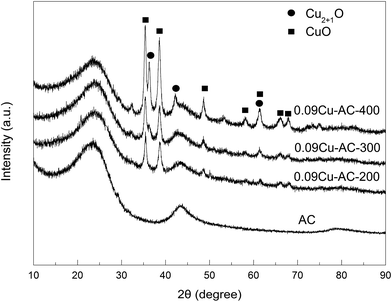 |
| | Fig. 4 XRD patterns of AC-supported CuO calcined at different temperatures (Cu2+1O is a crystal mixture of CuO and Cu2O). | |
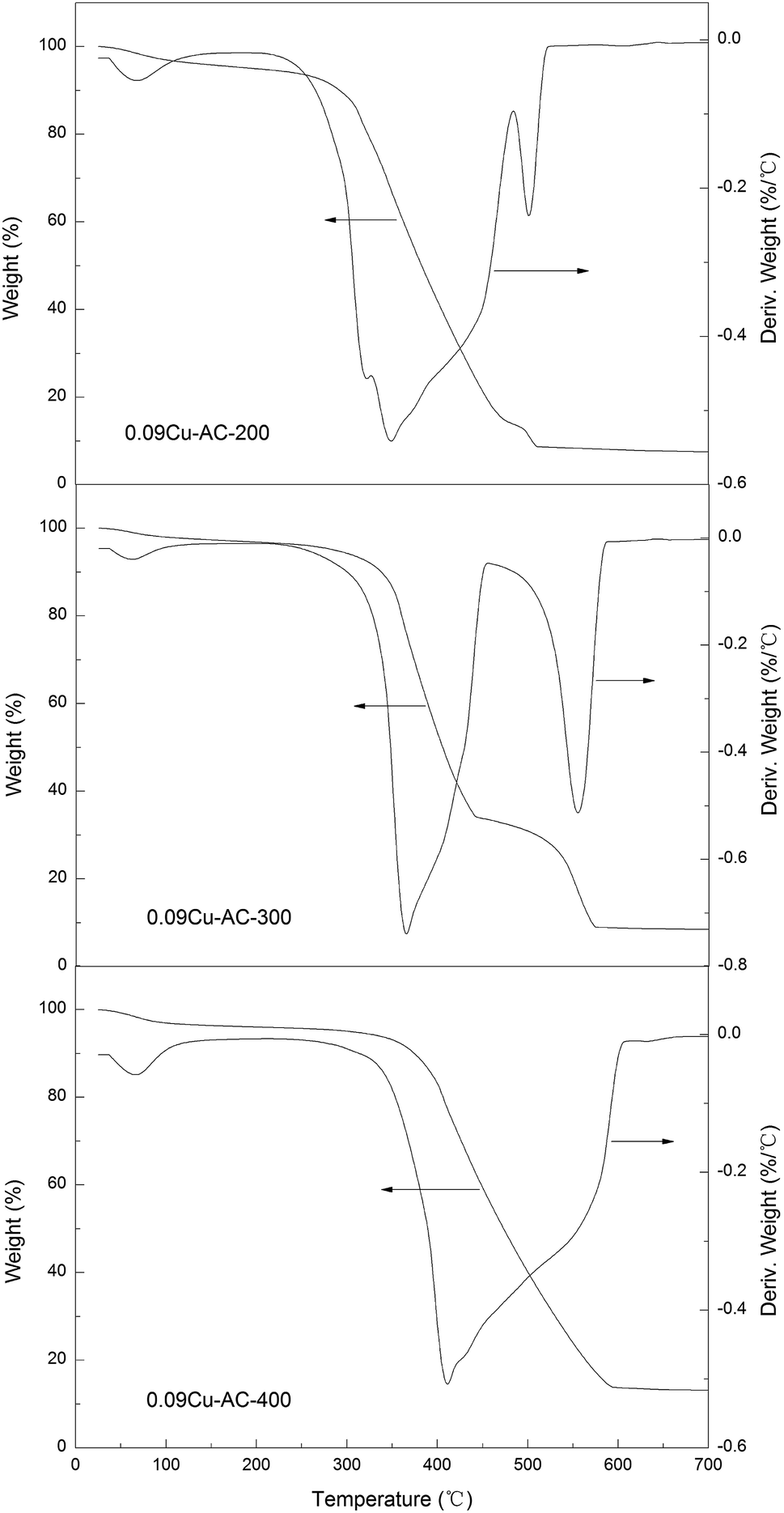
Thermal analysis. TGA results pertaining to three 0.09Cu-AC samples calcined at 200, 300, and 400 °C are presented in Fig. 5. It can be seen that each of the samples showed a small peak before 100 °C due to the evaporation of water. Based on the derivative weight curves, the initiating temperatures of carbon oxidation by air are about 280, 335, and 375 °C for 0.09Cu-AC-200, 0.09Cu-AC-300, and 0.09Cu-AC-400, respectively, and the corresponding temperatures for complete carbon oxidation are about 522, 587, and 607 °C. The reactivity of the metal-oxide-loaded carbon samples decreases with increasing calcination temperature for the Cu-loaded AC catalysts, which may be attributed to the decreasing accessibility of carbon to oxygen due to the blanketing effect of the well-developed metal oxide crystalline layer on the surface of the AC. The results also indicate that AC can also be oxidized by the coexistent O2 in the gas stream at about 375 °C, indicating that the coexistent O2 may impose a negative influence on the reduction of N2O, as discussed latter.
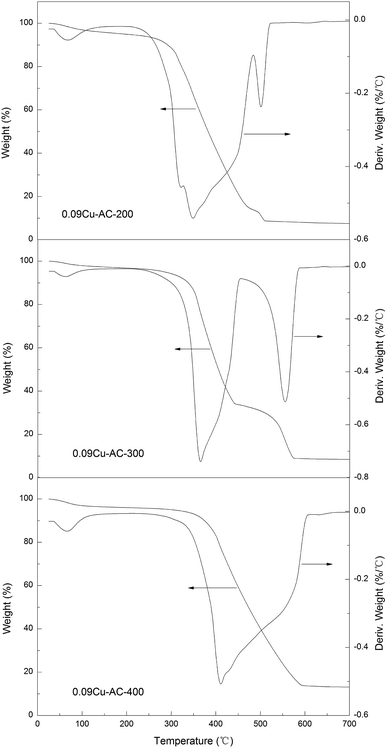 |
| | Fig. 5 TGA curves of AC-supported CuO calcined at 200 °C, 300 °C, and 400 °C. | |
3.2 Comparison of catalytic activities of different metal oxides
The catalytic activities of different metal oxides were evaluated in the tubular reactor using 2 g of each catalyst at a gas flow rate of 100 mL min−1, and the results are shown in Table 1. It can be seen that N2O was not decomposed over the pristine AC below 350 °C, but its decomposition increased significantly with increasing temperature. For example, the conversion of N2O increased drastically from 6% at 400 °C to 100% at 550 °C. Moreover, different metal oxides exhibited different activities for the conversion of N2O. Specifically, 0.01Cu-AC-400 and 0.01Ni-AC-400 completely removed N2O at 400 °C and 450 °C, respectively. The products were exclusively N2 and CO2, as befits the occurrence of the redox reaction between AC and N2O according to eqn (1):
Table 1 Dependence of conversion of N2O on temperature
| Pristine/metal-loaded ACs |
N2O conversion (%) |
| 300 °C |
350 °C |
400 °C |
450 °C |
500 °C |
550 °C |
| AC |
0 |
0 |
6 |
16 |
39 |
100 |
| 0.01Mn-AC-400 |
2 |
3 |
13 |
49 |
100 |
— |
| 0.01Fe-AC-400 |
1 |
2 |
17 |
81 |
94 |
100 |
| 0.01Co-AC-400 |
3 |
13 |
35 |
66 |
100 |
— |
| 0.01Ni-AC-400 |
3 |
5 |
29 |
100 |
— |
— |
| 0.01Cu-AC-400 |
4 |
18 |
100 |
— |
— |
— |
| 0.01Zn-AC-400 |
0 |
1 |
5 |
28 |
100 |
— |
| 0.01Ce-AC-400 |
3 |
4 |
8 |
30 |
100 |
— |
| 0.01CoCe-AC-400 |
4 |
17 |
35 |
61 |
100 |
— |
As evidenced by the FTIR and XRD results, the major difference between the catalysts lay in the loaded metal species, while the structure of the AC remained essentially the same. Therefore, the reactivity differences of the catalysts can be mainly attributed to the different activities of the supported metal oxides for the redox reaction of AC and N2O. The catalytic activities decreased in the order Cu > Co > Ni > Fe > Mn > Ce > Zn. This order conforms to that reported by Campa et al. for the reductive removal of N2O with CH4, where the order was Cu > Co > Mn.35 Therefore, 0.01Cu-AC-400 proved to be the most active catalyst for N2O removal among all those studied, and was deemed worthy of further study.
3.3 Reactivity of Cu-based ACs under varying conditions
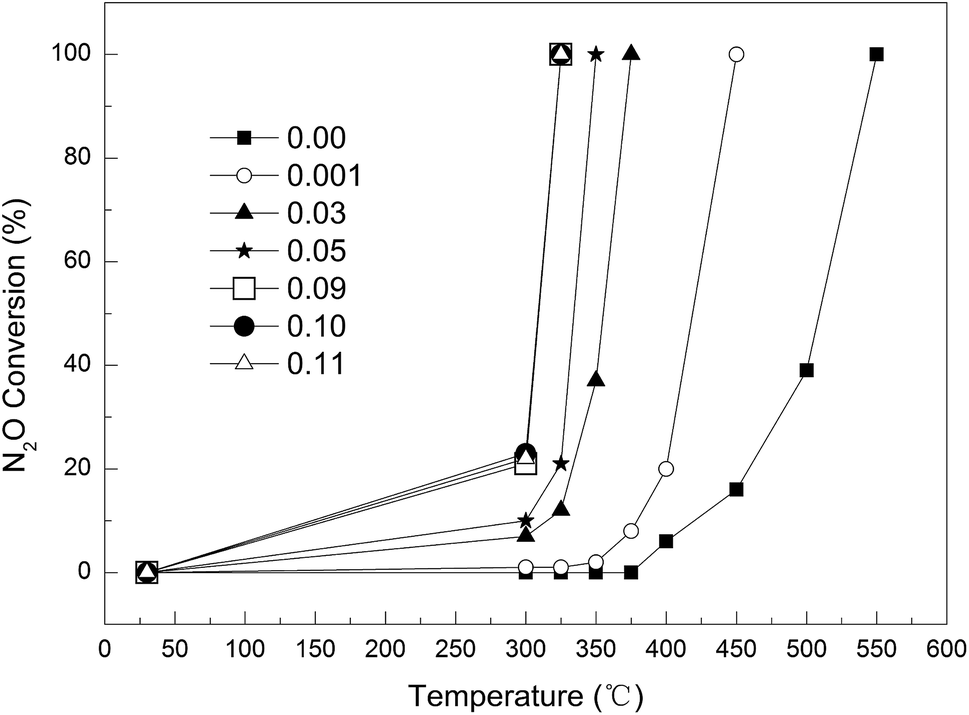
Effect of Cu loading on the catalytic activity. As shown in Fig. 6, the Cu loading had a significant impact on the removal of N2O. The complete decomposition temperature of N2O decreased greatly with increasing Cu loading, e.g., from 450 °C to 325 °C as the Cu loading was increased from 0.1 wt% to 9 wt%. At any given temperature, e.g. 300 °C, the conversion increased with increasing Cu loading up to 9 wt%, beyond which the Cu loading had little further influence on the activity. Therefore, the optimal Cu loading was determined as 9 wt%.
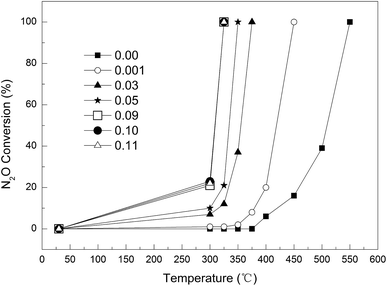 |
| | Fig. 6 Effect of Cu loading on N2O conversion. | |
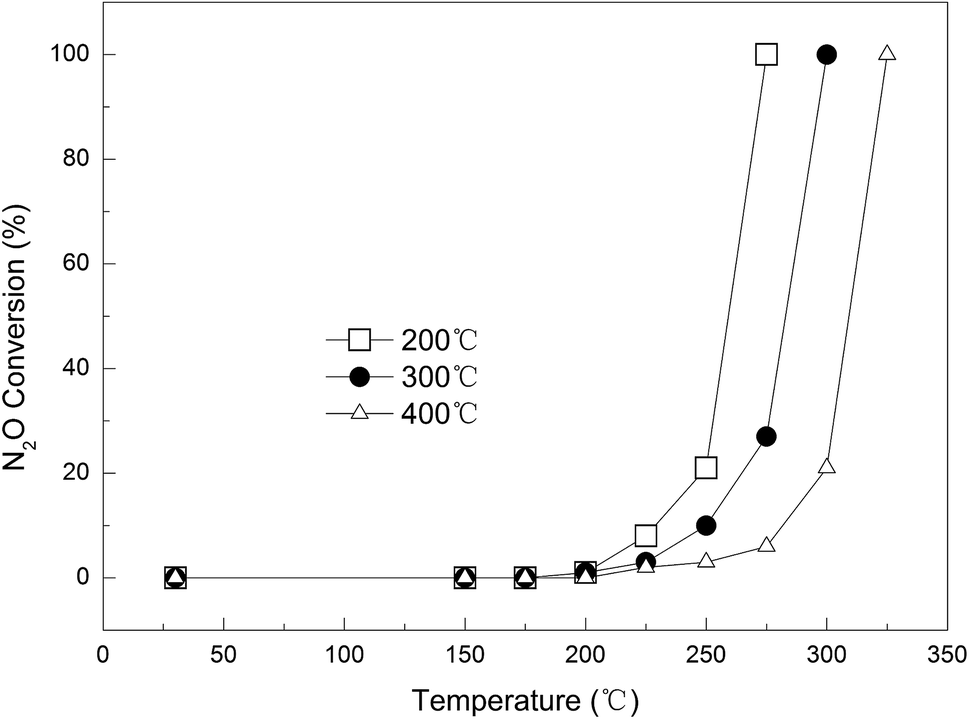
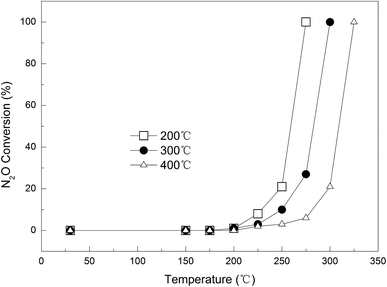
Effect of calcination temperature. Three Cu-loaded AC samples with different calcination temperatures (200, 300, and 400 °C) were prepared, namely 0.09Cu-AC-200, 0.09Cu-AC-300, and 0.09Cu-AC-400, and their catalytic performances are compared in Fig. 7. Evidently, the calcination temperature had a significant effect on the catalytic reduction of N2O, and a higher calcination temperature was unfavorable for the catalytic reaction of AC and N2O. Specifically, the complete removal temperature of N2O increased steadily from 275 °C for 0.09Cu-AC-200, to 300 °C for 0.09Cu-AC-300, and to 325 °C for 0.09Cu-AC-400. This phenomenon has also been observed by Chmielarz et al.,33 who ascribed it to the changing crystal form and the decreasing surface area with increasing calcination temperature. This explanation is consistent with the increasing crystallinity of copper oxides for 0.09Cu-AC-200, 0.09Cu-AC-300, and 0.09Cu-AC-400, as evidenced by the XRD patterns in Fig. 4.
 |
| | Fig. 7 Dependence of N2O conversion on calcination temperature. | |
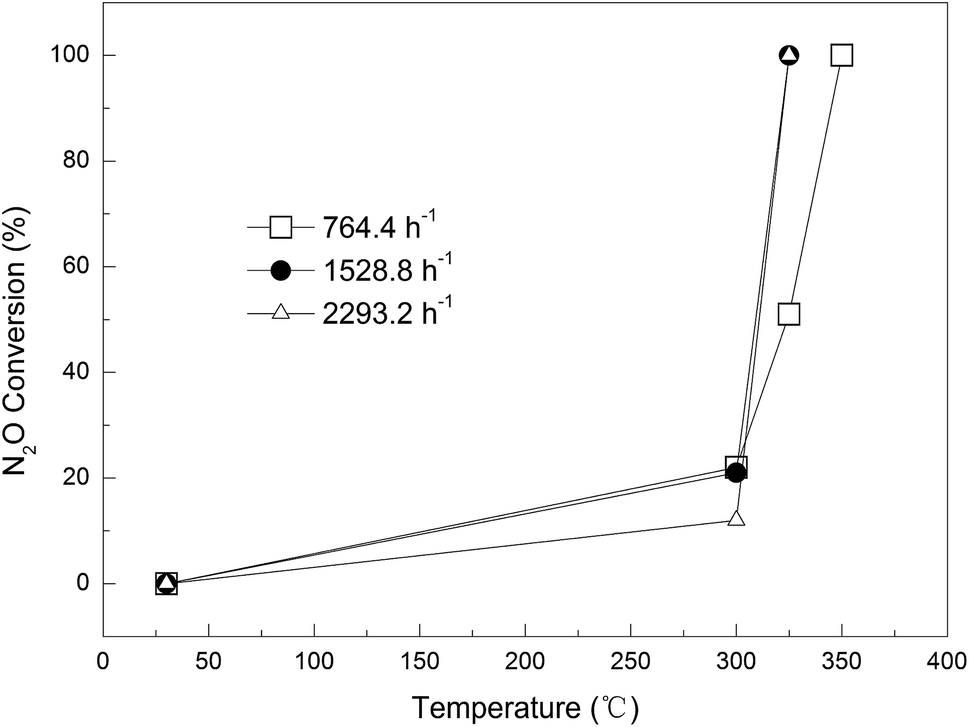
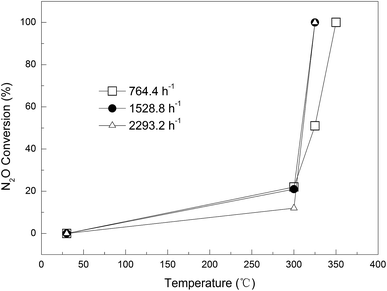
Effect of gas flow rate. The influence of gas flow on the N2O removal efficiency was studied for 0.09Cu-AC-400 at three different flow rates, namely 50, 100, and 150 mL min−1, corresponding to GHSV (gas hourly space velocity; ratio of flow rate of N2O and effective volume of reactor) values of 764.4, 1528.8, and 2293.2 h−1. The results are plotted in Fig. 8. At a lower reaction temperature of 300 °C, the conversion rate of N2O decreased with increasing gas flow rate, being restricted by the reaction kinetics. The conversion rates of N2O at the above flow rates were 22%, 21%, and 12%, respectively. However, when the temperature was increased to 325 °C, the conversion rate of N2O surged to 100% at gas flow rates of 100 and 150 mL min−1, but the N2O conversion was only 51% at a gas flow rate of 50 mL min−1. This phenomenon could be attributed to the exothermic nature of the reaction and the fast reaction kinetics at a higher temperature. As the flow rate of N2O is increased, more reaction heat is released due to the efficient oxidation of carbon by N2O, which raises the temperature of the AC, and thereby accelerates the reaction dramatically. On the other hand, increasing gas flow rate can shorten the retention time, and thus result in incomplete reduction of N2O. Therefore, the reaction temperature and retention time synergistically determine the effect of flow rate on N2O conversion. The appropriate GHSV was identified as 2293.2 h−1.
 |
| | Fig. 8 Effect of gas flow rate on N2O conversion. | |
3.4 Catalytic mechanism
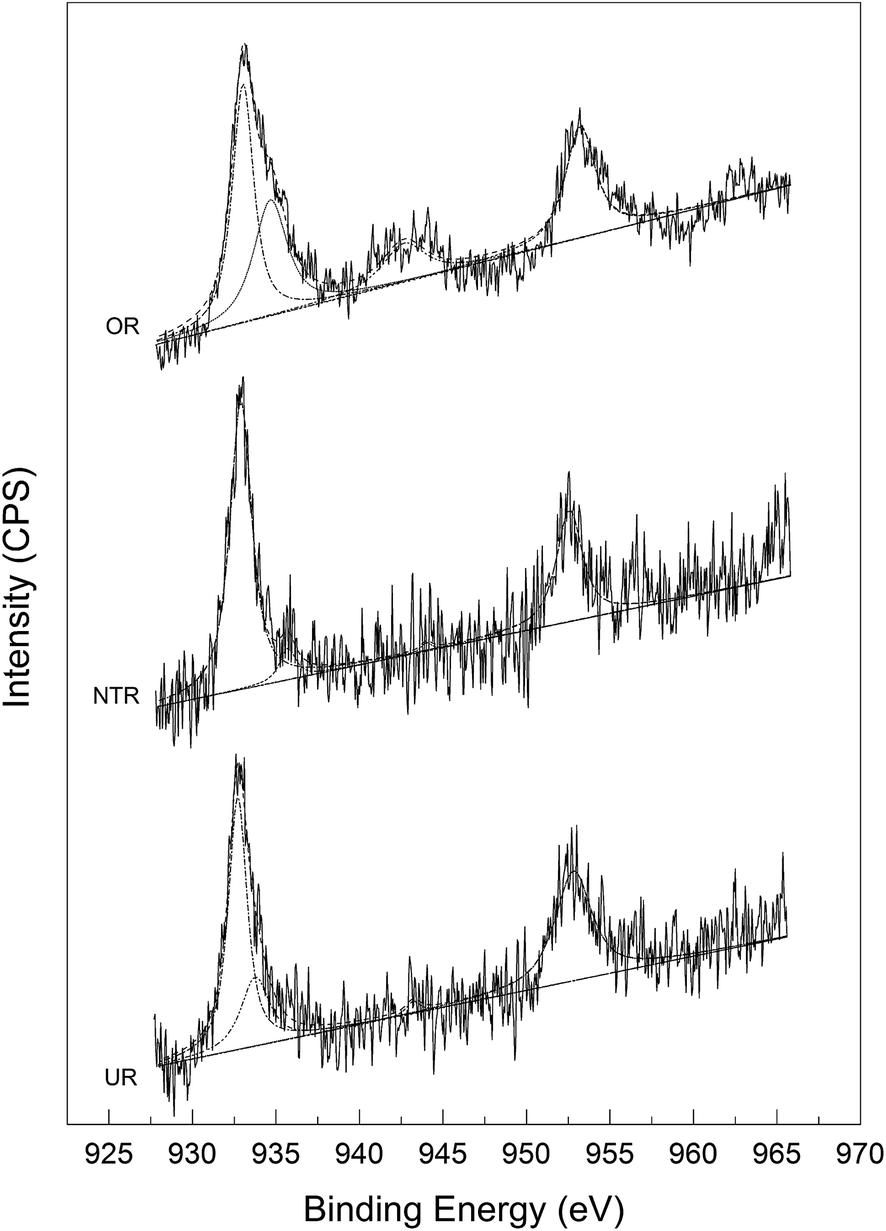
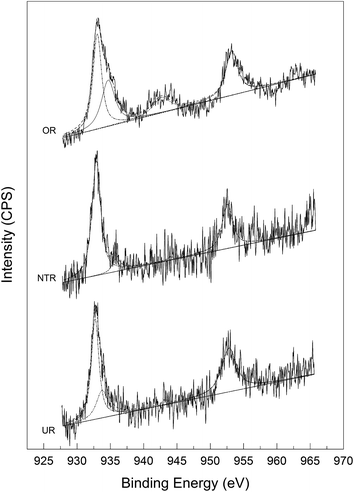
The decomposition process of N2O is believed to involve its redox reaction with carbon under catalysis by the loaded metal oxide, as is manifested by the consumption of AC and the presence of CO2 in the gas effluent. However, this process may be achieved in two different ways, i.e., catalytic decomposition of N2O, forming O2 and in situ oxidation therewith of the carbon support, or reduction of copper oxides by carbon to Cu metal, followed by the in situ oxidation of Cu by N2O. To identify the reaction mechanism, we studied the XPS patterns of three different reagents, namely, the original reactant (OR, 0.03Cu-AC-200), the nitrogen-treated reactant (NTR, obtained by heating OR at 400 °C in N2 for 5 h), and the used reactant (UR, obtained by heating the NTR at 400 °C in N2O for 2 h). Fig. 9 shows the Cu 2p XPS spectra.
 |
| | Fig. 9 Cu 2p XPS patterns of OR, NTR, and UR. | |
As can be seen from the XPS spectra, the primary and shoulder peaks appear at binding energies (B.E.) of around 933 and 943 eV, respectively, for Cu 2p3/2. As shown in Table 2, the B.E. values of the primary peaks for NTR and UR were slightly lower than that for OR, implying a lower valence of Cu in NTR and UR. This might be attributed to the reduction of CuO by carbon at a high temperature of 400 °C. All of the samples showed a shake-up satellite peak at around 943 eV, which is characteristic of Cu2+. Meanwhile, the weakened satellite peaks for NTR and UR imply decreased amounts of Cu2+ and increased amounts of Cu+ or Cu0 therein. The intensity ratios of the satellite and primary peaks for Cu 2p3/2 are shown in Table 2. This ratio is about 0.55 for standard CuO and 0 for a Cu0 phase.36 As can be seen in Table 2, NTR showed the lowest valence of Cu, UR showed a slightly higher valence, and OR showed the highest valence.
Table 2 Cu 2p high-resolution XPS spectra
| Sample |
Cu 2p3/2 mpa B.E. |
Cu 2p3/2 spb B.E. |
Splitting energyc (eV) |
Isp/Impd |
| “mp” denotes the main peak. “sp” denotes the shake-up satellite peak. “splitting energy” is the energy difference between mp and sp. “Isp/Imp” is the intensity ratio of sp and mp. |
| OR |
933.1 |
942.7 |
9.6 |
0.21 |
| NTR |
932.9 |
944.0 |
11.1 |
0.03 |
| UR |
932.7 |
943.3 |
10.6 |
0.04 |
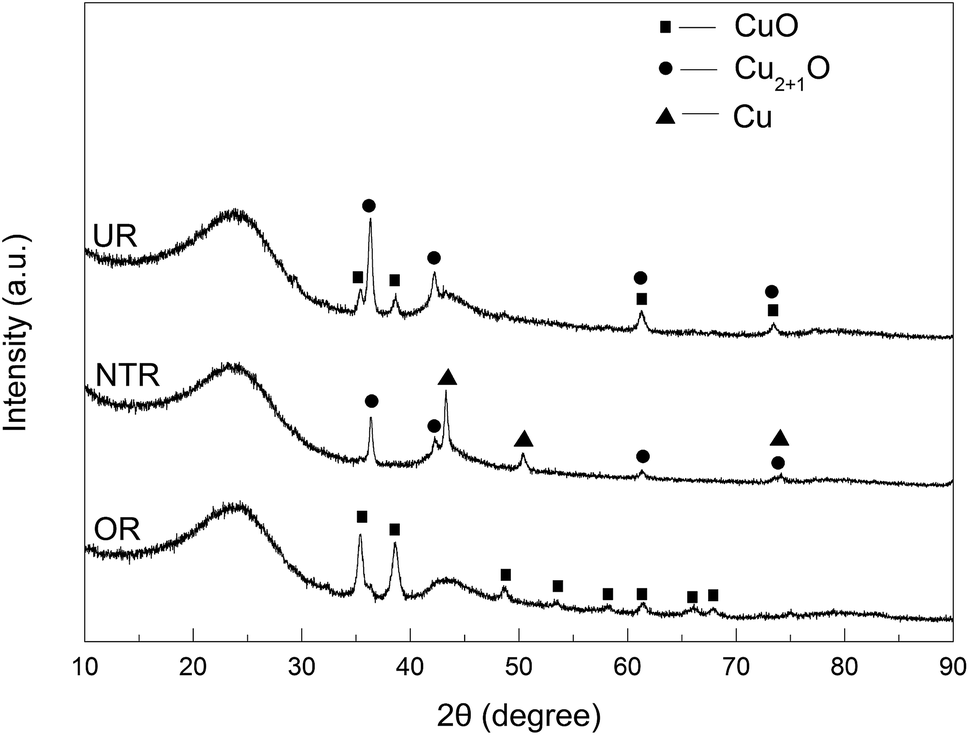
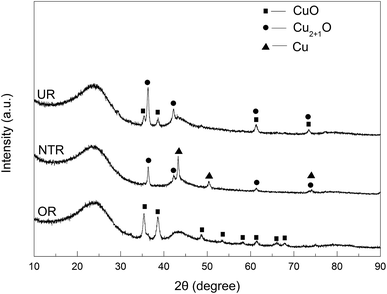
In addition to the XPS results, the above samples were also analyzed by XRD. As is evident from Fig. 10, CuO peaks were only seen for OR; they were no longer seen for NTR due to complete conversion to Cu2O and Cu. Supposedly, at a high temperature of 400 °C under nitrogen, the CuO component in the OR sample was successively reduced by AC to Cu2O and Cu, such that the amount of Cu became dominant. In contrast, only CuO and Cu2O were found in UR, and no elemental Cu could be detected. This may be the net result of the oxidation of Cu by N2O and the reduction of CuO by carbon when NTR was heated to 400 °C in N2O atmosphere. This phenomenon is consistent with earlier investigations by Carabineiro et al.,37,38 and Zhu et al.19
 |
| | Fig. 10 XRD patterns of loaded ACs in different statuses. | |
Based on our experimental evidence and above analysis, the catalytic mechanism of CuO for the reductive removal of N2O by AC is assumed to proceed as follows:
| | |
4CuO + C → 2Cu2O + CO2
| (2) |
| | |
2Cu2O + C → 4Cu + CO2
| (3) |
| | |
2Cu + N2O → Cu2O + N2
| (4) |
| | |
Cu2O + N2O → 2CuO + N2
| (5) |
In this process, CuO is involved in transfer of the O atom from N2O to carbon, accelerating the overall redox reaction between N2O and AC. In short, AC in the present process serves as a consumptive solid reductant for reducing N2O to nitrogen, while CuO functions as a catalyst for the reaction between N2O and carbon.
3.5 Applicability and challenge of the present process
Catalytic reduction of N2O may be achieved at mild conditions by using AC as solid reducing agent and CuO as catalyst. This process is especially suitable for the treatment of tail gas of the adipic acid production process with high concentration of N2O, and is advantageous due to its safety, ease of control, low operating temperature (300–400 °C), and lower cost of AC. And even the industrial waste AC may be also applicable, which further reduces the raw material cost and makes a resource use of another industrial waste. However, some challenges should be considered and tackled properly, e.g. competitive oxidation of O2 with N2O and emission of toxic metal oxide nanoparticles from the exhaust gas. In fact, such competitive oxidation is inevitable in all reductive removal processes of N2O with varying reducing agents, such as H2, CO, and CH4. However, the reactivity of N2O with AC is higher than that of O2, as manifested by its lower reaction temperature on 0.09Cu-AC-400, i.e. 300 °C for N2O (Fig. 6) and about 375 °C for air (O2) (Fig. 5). Therefore, the inhibition effect of O2 for the reduction of N2O may be quite low at appropriate conditions, for example, the presence of O2 was found to have no influence on the N2O-char reactivity at higher temperature.39,40 In contrast, at lower temperature of 320 °C and lower N2O content (0.3% N2O in helium gas), the coexistent 1.5% O2 may lead to a 20% lowering of reactivity of N2O on Cu2O-AC catalyst25 due to the blocking effect of O2 for the active sites of AC.26 As a reducing agent, AC will be oxidized and consumed gradually, and the residue toxic metal oxides may be emitted from the exhaust gas to the atmosphere. However, this problem may be solved through conventional technical approaches. For example, by using a moving-bed reactor, most of the ultrafine metal oxide particles will be retained in the reactor due to the filtering effect of the granular AC catalyst, some of the particles can be collected as sediment in the baffled heat exchanger for cooling the high temperature exhaust gas to ambient temperature, and finally, the ultrafine ashes can be further removed to reach the environmental standard by wet or electrostatic dust collector.
4. Conclusions
N2O can be completely decomposed by AC at 550 °C through a redox reaction between AC as reductant and N2O as oxidant. The decomposition reaction is greatly enhanced by some transition metal oxide catalysts, among which CuO has proved to be the best one. Complete removal of N2O was achieved by 0.09Cu-AC-200 at a much lower temperature of 275 °C. The catalytic mechanism of CuO has been studied by XPS and XRD, which would seem to involve the reduction of CuO to Cu2O and Cu by AC, and oxidation of Cu to Cu2O and CuO by N2O. In this process, CuO is involved in transfer of the O atom from N2O to carbon, accelerating the overall redox reaction between N2O and AC.
References
- M. A. Zamudio, S. Bensaid, D. Fino and N. Russo, Ind. Eng. Chem. Res., 2011, 50, 2622–2627 CrossRef CAS.
- N. Li, Chem. Ind. Eng. Prog., 2007, 26, 1659–1661 CAS.
- Y. Shen, C. Li, Y. Tang and S. Zhu, RSC Adv., 2015, 5, 13212–13219 RSC.
- S. Kannan, Appl. Clay Sci., 1998, 13, 347–362 CrossRef CAS.
- J. K. Lee, Y. J. Kim, H. J. Lee, S. H. Kim, S. J. Cho, I. S. Nam and S. B. Hong, J. Catal., 2011, 284, 23–33 CrossRef CAS.
- B. M. Abu-Zied, Microporous Mesoporous Mater., 2011, 139, 59–66 CrossRef CAS.
- B. H. Chen, N. Liu, X. Y. Liu, R. D. Zhang, Y. P. Li, Y. X. Li and X. L. Sun, Catal. Today, 2011, 175, 245–255 CrossRef CAS.
- T. P. Gaidey, Russ. J. Appl. Chem., 2009, 82, 1689–1705 CrossRef.
- V. G. Komvokis, M. Marti, A. Delimitis, I. A. Vasalos and K. S. Triantafyllidis, Appl. Catal., B, 2011, 103, 62–71 CrossRef CAS.
- S. S. Kim, S. J. Lee and S. C. Hong, Chem. Eng. J., 2011, 169, 173–179 CrossRef CAS.
- M. Hussain, D. Fino and N. Russo, J. Hazard. Mater., 2012, 211, 255–265 CrossRef PubMed.
- Q. Zhang, X. Tang, P. Ning, Y. Duan, Z. Song and Y. Shi, RSC Adv., 2015, 5, 51263–51270 RSC.
- C. Li, Y. Shen, S. Zhu and S. Shen, RSC Adv., 2014, 4, 29107–29119 RSC.
- T. Xu, C. Wang, X. Wu, B. Zhao, Z. Chen and D. Weng, RSC Adv., 2016, 6, 97004–97011 RSC.
- M. Mihet, V.-M. Cristea, P.-S. Agachi, A.-M. Cormos and M. D. Lazar, RSC Adv., 2016, 6, 89259–89273 RSC.
- P. Nematollahia and M. D. Esrafili, RSC Adv., 2016, 6, 59091–59099 RSC.
- J. Rodriguez-Mirasol, A. C. Ooms, J. R. Pels, F. Kapteijn and J. A. Moulijn, Combust. Flame, 1994, 99, 499–507 CrossRef CAS.
- Z. H. Zhu and G. Q. Lu, J. Catal., 1999, 187, 262–274 CrossRef CAS.
- Z. H. Zhu, G. Q. Lu and R. T. Yang, J. Catal., 2000, 192, 77–87 CrossRef CAS.
- F. Gonçalves and J. L. Figueiredo, Appl. Catal., B, 2004, 50, 271–278 CrossRef.
- F. Gonçalves and J. L. Figueiredo, Appl. Catal., B, 2006, 62, 181–192 CrossRef.
- Y. Chen, Z. Hu, X. Wang, G. Zhao, Y. Liu and W. Liu, Acta Phys.-Chim. Sin., 2008, 249, 1589–1596 Search PubMed.
- J. Sun, H. Wang and B. Sun, Chin. J. Environ. Eng., 2011, 5, 261–266 CAS.
- C. Wang, Y. Liu, X. Zheng, Z. Wen, A. Liu, X. Xu and Z. Hu, Gongneng Cailiao, 2010, 9, 1520–1528 Search PubMed.
- Z. H. Zhu, L. R. Radovic and G. Q. Lu, Carbon, 2000, 38, 451–464 CrossRef CAS.
- Y. H. Li, G. Q. Lu and V. Rudolph, Chem. Eng. Sci., 1998, 53, 1–26 CrossRef CAS.
- S. A. Carabineiro, F. B. Fernandes, A. M. Ramos, J. Vital and I. F. Silva, Catal. Today, 2000, 57, 305–312 CrossRef CAS.
- A. N. Zhou, X. L. Ma and C. S. Song, Appl. Catal., B, 2009, 87, 190–199 CrossRef CAS.
- Y. F. Jia and K. M. Thomas, Langmuir, 2000, 16, 1114–1122 CrossRef CAS.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, Wiley, 2004 Search PubMed.
- W. N. R. W. Isahak, M. W. M. Hisham and M. A. Yarmo, J. Chem., 2013, 2013, 1–6 CrossRef.
- J. Co, P. D. Standards, Powder Diffraction File, ASTM, Newtown Square, PA, USA, 2004, pp. 48–1548 Search PubMed.
- L. Chmielarz, M. Rutkowska, P. Kuśtrowski, M. Drozdek, Z. Piwowarska, B. Dudek, R. Dziembaj and M. Michalik, J. Therm. Anal. Calorim., 2011, 105, 161–170 CrossRef CAS.
- J. Co, P. D. Standards, Powder Diffraction File, ASTM, Newtown Square, PA, USA, 2004, pp. 05–0667 Search PubMed.
- M. C. Campa, V. Indovina and D. Pietrogiacomi, Appl. Catal., B, 2012, 111, 90–95 CrossRef.
- A. Patel, P. Shukla, T. Rufford, S. Wang, J. Chen, V. Rudolph and Z. H. Zhu, Appl. Catal., A, 2011, 409, 55–65 CrossRef.
- S. A. Carabineiro, F. B. Fernandes, R. J. C. Silva, J. S. Vital, A. M. Ramos and I. M. Fonseca, Catal. Today, 2008, 133, 441–447 CrossRef.
- S. A. Carabineiro, F. B. Fernandes, J. S. Vital, A. M. Ramos and I. M. Fonseca, Appl. Catal., B, 2005, 59, 181–186 CrossRef CAS.
- I. Gulyurtlu, H. Esparteiro and I. Cabrita, Fuel, 1994, 73, 1098–1102 CrossRef CAS.
- J. Rodriguez-Mirasol, A. C. Ooms, J. R. Pels, F. Kapteijn and J. A. Moulijn, Combust. Flame, 1994, 99, 499–507 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab