
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An amphiphilic non-viral gene vector prepared by a combination of enzymatic atom transfer radical polymerization and enzymatic ring-opening polymerization†
Xinghuo Wanga,
Wenjing Yunb,
Wei Jianga,
Ding Wangb,
Ling Zhang*b and
Jun Tang *a
*a
aDepartment of Polymer Science, Chemistry College, Jilin University, Changchun 130012, People's Republic of China. E-mail: chemjtang@jlu.edu.cn; Fax: +86-431-88498179; Fax: +86-431-85632348
bDepartment of Pathophysiology, Basic Medical College, Jilin University, Changchun 130021, People's Republic of China. E-mail: zhangling3@jlu.edu.cn
First published on 3rd February 2017
Abstract
Biocatalysts, such as enzymes, create a smart platform for polymerization and materials used in biomedical science. Deuterohemin-b-Ala-His-Thr-Val-Glu-Lys (DhHP-6), a peroxidase mimic, acts as a bioinspired catalyst in an ATRP process under moderate temperature for the polymerization of the oil soluble monomer, glycidyl methacrylate (GMA), which has potential to be a successful non-viral gene vector. A successful gene vector should combine high transfection efficiency with low toxicity. Herein, we report a ‘green method’ used to obtain a series of block polymer (PCL-b-PGMA), synthesized using ε-caprolactone (CL) and glycidyl methacrylate (GMA) from a double head initiator HEBiB, which was catalyzed by two enzymes over two steps in order to minimize the residual metal catalyst present in the final products obtained from the ATRP process. The hydrophilic amine moiety (ethanolamine, EA) was employed to decorate the pendant epoxide groups of PCL-b-PGMA to obtain a series of promising gene vectors with different molecular weights.
1. Introduction
Reversible deactivation radical polymerization (RDRP) is a robust polymerization method that has been developed over the last few decades, which has gained significant popularity.1–3 Atom transfer radical polymerization (ATRP), as one of the RDRP methods, is a versatile tool used to synthesize various types of functional polymer materials and displays numerous advantages, including precise control over the molecular weight, specific structure and narrow PDI.4,5 However, to achieve an equilibrium between the active and dormant species, the inevitable utilization of transition metal species (Mtn) in the ATRP process caused the presence of residual metal catalysts in the final products, which restricts their use in biomedical and electronic applications.6 Accordingly, reducing the amount of catalyst is attractive in the ATRP process.7,8 Several versatile methods have been successfully employed to overcome this paramount challenge, such as simultaneous reverse and normally initiated (SR&NI) ATRP,9 initiators for continuous activator regeneration (ICAR) ATRP10,11 and activators regenerated by electron transfer (ARGET) ATRP,12–14 likewise, catalysis using an enzyme has also attracted attention.15–18 Enzymes are selective and efficient catalysts that are harvested from living organisms found in nature and have been discovered to be efficient catalysts for several different types of polymerizations in vitro.19 Thus, it is very appealing to construct biomedical materials using an ATRP process catalyzed by an enzyme.18 Moreover, we envisage that non-viral gene vectors may be prepared via an enzymatic ATRP process and used to explore their biomedical applications.Polymer gene vectors have emerged as more promising gene delivery candidates and have displayed numerous amazing advantages over the other types of gene vectors including low cytotoxicity, safety profile, and easy-tailored structure, which have been applied in gene therapy.20–23 Poly(glycidyl methacrylate) (PGMA), an easily modified polymer, gives access to functional groups, such as amine moieties, through reaction with the pendant epoxy group.24–26 Xu's group has synthesized a series of polycations based on PGMA decorated with ethanol amine (EA),27–31 which can also be used to further link to the PCL and PEGMA segments to obtain a better transfection efficiency for enhancing cellular uptake and biocompatibility.26 Employing a hydroxyl coating on the polycations to induce serum absorption is a creative approach when compared to PEGylation.32 Thus, we believe PCL-b-PGEA will show excellent properties when used as gene vectors. However, polycations based on PGMA are generally synthesized via Cu-mediated processes and it is necessary and urgent to combine enzymatic ROP and enzymatic ATRP to make an alternative way to synthesize polycations.
To avoid using transition metal catalysts in polymer gene vectors, our group has synthesized a series of water soluble polymers via enzymatic ATRP, as previously reported.33,34 Herein, we report a series of amphiphilic block polymers with different molecular weight synthesized from a double head initiator HEBIB26 utilizing mimic enzyme catalysts. Finally, the pendant epoxy groups were decorated with amine moieties to obtain non-viral polycations that were evaluated for gene delivery. The synthesized vector has the ability to bind to pDNA at N/P = 0.75 with particle sizes from 200 nm to 300 nm at the optical transfection efficiency. The amphiphilic block polymer (PCL-b-PGEA) is comparable to PEI 25 kDa in a transfection efficiency assay using in serum-conditioned 293T cells and possessed a relatively lower cytotoxicity when compared to PEI 25 kDa, which may due to the combination of enzyme catalysis and the good biocompatibility of the polymer gene vectors. More importantly, enzyme or mimic enzyme catalysis can basically reduce the use of transition metal catalysis in ATRP and provide a promising platform for synthesizing materials.
2. Experimental section
2.1 Materials
Most of the reagents were of analytical grade and used without further purification unless otherwise noted. Glycidyl methacrylate (GMA) was purchased from Sigma-Aldrich and passed through a column of basic alumina to remove inhibitors before use. ε-Caprolactone (ε-CL) was purchased from Sigma-Aldrich, stored over P2O5 in a desiccator and used without further purification. Novozym 435 (Candida antarctica lipase B immobilized on acrylic resin, CALB, Novozymes) was dried under a vacuum before use. Ethanol amine (EA), potassium bromide (KBr) and ascorbic acid (Asc) were purchased from Tianjin Guangfu Fine Chemical Research Institute and used as received. 2-Hydroxyethyl 2-bromoisobutyrate (HEBiB) was synthesized according to a literature procedure.35 Toluene was dried by refluxing with Na/benzophenone ketyl for 24 h. DhHP-6 was obtained as a gift sample from the College of Life Science, Jilin University (Changchun, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich. Dulbecco's modified eagle's medium (DMEM) was purchased from Hyclone (USA). Fetal bovine serum (FBS) was purchased from biological industries (Israel). 293T cells and GFP plasmid were kindly provided and preserved by the Basic Medical College, Jilin University (Changchun, China).2.2 Polymer characterization
Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker Avance III NMR spectrometer (400 MHz) using CDCl3 and d6-DMSO as solvents. Chemical shifts (in ppm) were reported downfield using tetramethylsilane as an internal standard. Gel permeation chromatography (GPC) was performed on a Malvern instrument (Viscotek T5000, Viscotek T3000 and Viscotek T1000 org GPC/SEC column thermostated to 35 °C and calibrated by linear polystyrene standards). Tetrahydrofuran (THF) was used as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. The detection was calibrated with polystyrene standards with narrow molecular weight distribution. Fourier transform infrared spectra (FT-IR) were recorded on a Shimadzu FTIR 8400S spectrometer. The complexes' diameters and surface zeta potential were measured using a Zeta sizer 3000 (Malvern, UK).2.3 Novozym 435 catalyzed polymerization of ε-caprolactone using a bifunctional ATRP initiator
Novozym 435 (150 mg), HEBiB (300 mg) and anhydrous toluene were placed into a 50 mL round-bottom flask, and ε-caprolactone (3 mL) was added into the flask, followed by immersion in an oil bath and maintained at 80 °C under magnetic stirring for 48 h. Finally, the reactions were terminated by adding an excess of dichloromethane and removing the enzymes by filtration. The solvent was removed by rotary evaporation to obtain a concentrated polymer solution, followed by precipitation in cold methanol 3 times. The cold methanol should be stored at −30 °C for several hours in order to precipitate all of the polymer. The polymer was dried under vacuum overnight to give a white solid powder. The structure was determined by 1H NMR spectroscopy, and the molecular weight was analyzed via GPC using THF as the mobile phase.2.4 DhHP-6 catalyzed ATRP of GMA using the macroinitiator PCL-Br
PCL-b-PGMA was prepared via atom transfer radical polymerization (ATRP) of GMA in DMF/H2O using the macroinitiator PCL-Br. In a typical polymerization, GMA (0.6169 g), DhHP-6 (1.8 mg), PCL-Br (Mn = 2375) and DMF were introduced to a branch-necked flask, followed by purging with nitrogen for at least 30 min. Finally, 0.1 mL of ascorbic acid stock solution (containing 6.16 mg ascorbic acid) was added into the reaction mixture and the branch-necked flask was immersed in an oil bath at 30 °C. After the desired reaction time, the reaction mixture was diluted with dichloromethane and passed through neutral alumina to remove the catalyst as much as possible and then rotary evaporated to eliminate the eluent. The solution was poured into a large excess of cold ether. Finally, the polymers were dried under a vacuum for 24 h. For the kinetics study, samples were taken from the flask at timed intervals for 1H NMR and GPC analysis.2.5 Synthesis of PCL-b-PGEA under catalyst free conditions
The procedures followed by Xu's etc. previous work.24 Briefly, PCL-b-PGMA (50 mg) was dissolved in 5 mL of DMF, and then the polymer solution was added dropwise into the EA solution containing 2 mL of DMF. After reacting for 7 h in a 60 °C oil bath, the reaction mixture was poured into a large excess of ether to precipitate the product. The product was centrifuged to separate it from ether, followed by dialysis (molecular weight cut-off: 3500 g mol−1) against 5 L of distilled water for 5 days prior to lyophilization.2.6 Preparation of the polyplexes
Different molecular weight PCL-b-PGEA polymer solutions (in 150 mM PBS) were mixed with pDNA at different w/w ratios (weight ratios of polymer relative to pDNA), as previously reported.32 Then, the solutions were diluted to obtain solution of the complexes at a certain volume (30 μL) using PBS, followed by vortexing for 30 s and incubation for 20 min at 25 °C.2.7 Agarose gel electrophoresis assay
The ability of the polymer binding pDNA was evaluated using a gel mobility shift assay. The GFP plasmids were diluted to 1 μg μL−1 using doubly distilled water and PCL-b-PGEA with different molecular weights were formulated to 1 μg μL−1 in PBS. Then, the 1 μg plasmid samples were placed into the different vectors mixed in a total system of 10 μL and incubated for 20 min at room temperature. The electrophoretic experiments were carried out at various N/P ratios and the binding rate was we observed using 1% agarose gel electrophoresis with TAE buffer for 20 min at 120 V.2.8 Particle size and surface zeta potential measurements under serum-free conditions
The particle size and surface zeta potential were measured using a Zetasizer Nano ZS90 instrument at 25 °C. The polyplexes were incubated for 20 min by adding 1 μg of plasmid to 30 μL of PBS (150 mM) at different w/w ratios, followed by the addition of doubly distilled H2O to a certain volume (1.2 mL) for size and surface zeta potential measurements. Data are shown as the mean ± standard deviation (SD) based on three independent experiments.2.9 Cell culture
293T cells were cultured with Dulbecco's modified eagle medium (DMEM) containing 10% fetal bovine serum under the conditions of 37 °C and 5% CO2.2.10 Cytotoxicity assay
To detect the cytotoxic effects of PCL-b-PGEA on 293T cells, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay was performed using a microplate reader at 490 nm with PEI 25 kDa as the positive control. 293T cells were cultured in 96-well plates at a density of 1 × 104 for 24 hours in 100 μL of DMEM containing 10% FBS. Cells were grown in different concentrations of gradient PCL-b-PGEA (final w/w ratio of 5, 10, 15, 20, 25) and MTT was added into each well 20 μL solution (5 mg mL−1, i.e. 0.5% MTT) 24 h later. MTT was incubated for 4 h and 150 μL of DMSO was added to each well. Finally, the absorbance of each well in the enzyme immunoassay analyzer was measured at an OD of 490 nm. Data are shown as the mean ± standard deviation (SD) based on three independent experiments.2.11 In vitro serum-conditioned gene transfection assay
The transfection of GFP mediated by PCL-b-PGEA60 and PCL-b-PGEA142, respectively was evaluated in 293T cells and PEI 25 kDa was used as a positive control. Cells were seeded into 96-well plates at a density of 1 × 104, cultured in DMEM and 10% fetal bovine serum and incubated in 5% CO2 in 37 °C for 24 h. Cells were washed twice with PBS before transfection. GFP plasmids were transfected at 0.3 μg per well. The transfection efficiency solutions of PCL-b-PGEA60 and PCL-b-PGEA142 were added to the cell wells, followed by incubation for 48 h and the fluorescence was observed with an inverted fluorescence microscope (Yokogawa, Japan). The percentage of the GFP positive cells was determined using flow cytometry (BD FACSCalibur). Data are shown as the mean ± standard deviation (SD) based on three independent experiments.3. Results and discussion
Deuterohemin-b-Ala-His-Thr-Val-Glu-Lys (DhHP-6) is composed of an iron porphyrin center and six amino acid residues, which can act as an ATRP catalyst33 and the oligopeptide gives an approach to synthesize hybrid catalysts and increase the solubility. The enzyme exhibits low cytotoxicity when compared to that of transition metal catalysts, and therefore, developing an ATRP catalyzed by an enzyme or enzyme mimic provides a promising platform for applications of polymer materials in the biomedical field. As presented in Scheme 1, PCL-b-PGMA was synthesized over two steps from the double head initiator HEBiB via a dual enzyme mediated process. The macroinitiator (PCL-Br) was prepared by polymerizing ε-caprolactone initiated from HEBiB in toluene, followed by purification, and the block polymer was synthesized in DMF using PCL-Br and GMA catalyzed by DhHP-6 at a mild temperature. Finally, the epoxy groups on the block polymer were decorated with EA under catalyst-free conditions.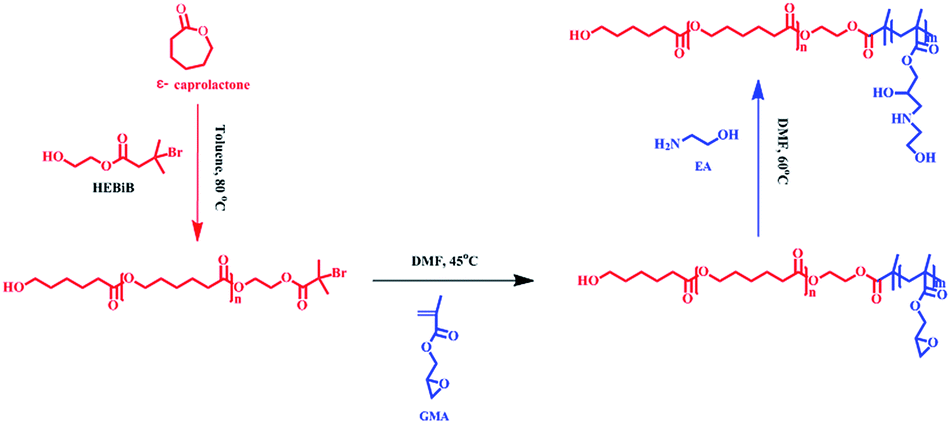 | ||
| Scheme 1 The preparation process for PCL-b-PGEA from the double head initiator HEBiB using a combination of a ring-opening reaction and ATRP mediated by a dual enzyme. | ||
3.1 Characterization of the polymer structure
The representative structures of PCL-Br and PCL-b-PGMA were first characterized by 1H NMR spectroscopy, as shown in Fig. 1. For PCL-Br (Mn = 2400, PDI = 1.22, Table 1), the signals at δ = 3.64 (E) and 1.93 (G) ppm correspond to the methylene adjacent to the hydroxy group and methyl adjacent to Br, respectively. Moreover, the ratio of the two peaks integrals was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.66, which indicates that the end functionality was 73%. As shown in Fig. 2b, PCL-b-PGMA was prepared from the macroinitiator PCL-Br via an enzymatic ATRP process. The new peaks at δ = 3.26 (d) ppm and δ = 2.86 and 2.66 (e) ppm were mainly attributed to the epoxy groups of the PGMA segment. Moreover, the signals at δ = 4.34 and 3.84 (c) ppm were assigned to the methylene group adjacent to the epoxy group. The ratio of the peak integrals of c, d, and e is about 2:1:2, which demonstrates that the epoxy groups were retained during the AGET ATRP process.36 These peaks clearly indicate that a well-defined structure of PCL-b-PGMA was synthesized via the dual enzyme mediated process. The GPC data for PCL-Br, PCL-b-PGMA60 and PCL-b-PGMA142 are also shown in Fig. S1–S3† and Table 1, respectively. After being decorated with ethanol amine (EA) via the ring-opening reaction under catalyst free conditions, the 1H NMR spectrum of PCL-b-PGEA was characterized in d6-DMSO. The peaks at δ = 4.34 and 3.84 ppm (c in Fig. 1b) corresponding to the methylene link to the ester group and epoxy groups were shifted to one peak δ = 3.83 ppm (c in Fig. 1c). The new peak at δ = 3.52 ppm (d) was ascribed to the proton of the methane link to the hydroxyl group.24 The GPC and 1H NMR data confirmed that the macroinitiator and block polymer were successfully synthesized.
:2.66, which indicates that the end functionality was 73%. As shown in Fig. 2b, PCL-b-PGMA was prepared from the macroinitiator PCL-Br via an enzymatic ATRP process. The new peaks at δ = 3.26 (d) ppm and δ = 2.86 and 2.66 (e) ppm were mainly attributed to the epoxy groups of the PGMA segment. Moreover, the signals at δ = 4.34 and 3.84 (c) ppm were assigned to the methylene group adjacent to the epoxy group. The ratio of the peak integrals of c, d, and e is about 2:1:2, which demonstrates that the epoxy groups were retained during the AGET ATRP process.36 These peaks clearly indicate that a well-defined structure of PCL-b-PGMA was synthesized via the dual enzyme mediated process. The GPC data for PCL-Br, PCL-b-PGMA60 and PCL-b-PGMA142 are also shown in Fig. S1–S3† and Table 1, respectively. After being decorated with ethanol amine (EA) via the ring-opening reaction under catalyst free conditions, the 1H NMR spectrum of PCL-b-PGEA was characterized in d6-DMSO. The peaks at δ = 4.34 and 3.84 ppm (c in Fig. 1b) corresponding to the methylene link to the ester group and epoxy groups were shifted to one peak δ = 3.83 ppm (c in Fig. 1c). The new peak at δ = 3.52 ppm (d) was ascribed to the proton of the methane link to the hydroxyl group.24 The GPC and 1H NMR data confirmed that the macroinitiator and block polymer were successfully synthesized.
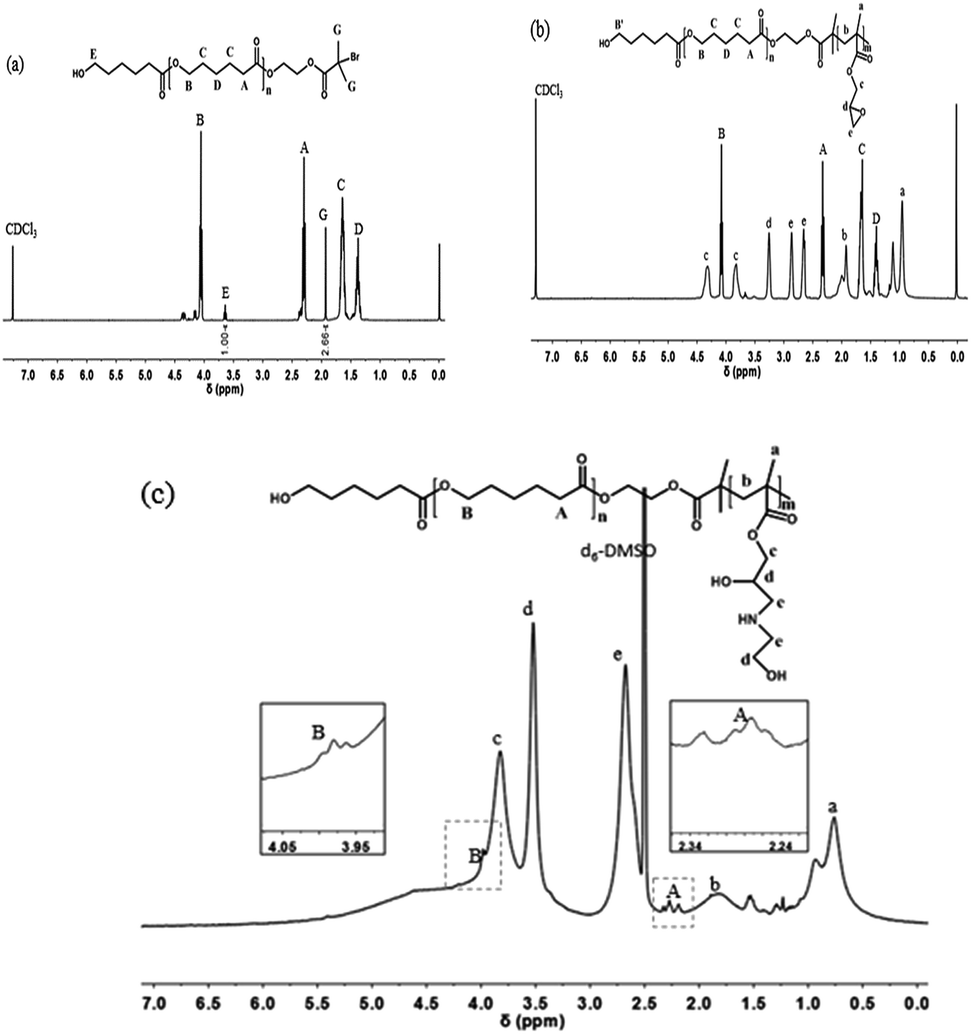 | ||
| Fig. 1 The 1H NMR (400 MHz) spectra of (a) PCL-Br (b) PCL-b-PGMA60 and (c) PCL-PGEA142. | ||
| Sample | Mna (kg mol−1) | PDIa | CL unitsa | GMA unitsa |
|---|---|---|---|---|
| a Determined from the GPC results.b Synthesized by N435 catalyzed using a molar feed ratio [ε-caprolactone]:[HEBiB] = 19:1 at 80 °C in 15 mL of ε-caprolactone/toluene (1/3, v/v).c Synthesized by DhHP-6 catalyzed using a molar feed ratio [GMA]:[PCL-Br]:[DhHP-6]:[Asc] = 124:1:1:0.5 at 45 °C in 2 mL of DMF. |
||||
| PCL-Brb | 2.4 | 1.22 | 21 | |
| PCL-b-PGMA60c | 10.8 | 1.35 | 21 | 60 |
| PCL-b-PGMA142c | 22.5 | 1.41 | 21 | 142 |
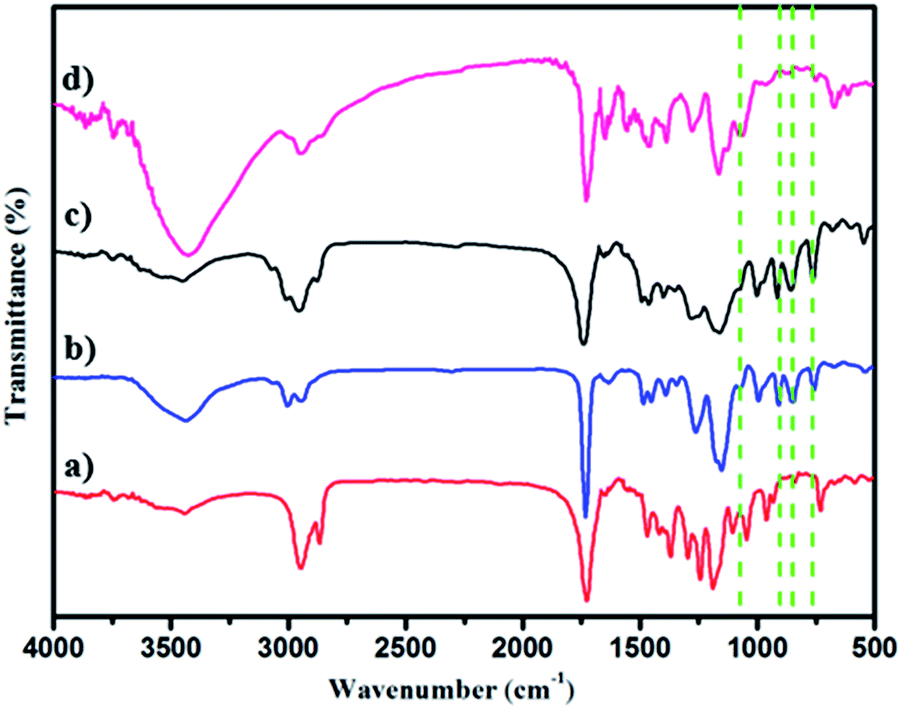 | ||
| Fig. 2 The FT-IR spectra of (a) PCL-Br (b) PGMA (c) PCL-b-PGMA and (d) PCL-b-PGEA. | ||
The polymer structures were also ascertained by FT-IR spectroscopy (Fig. 2a–d). The peaks (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1730 cm−1 and 1188 cm−1 in Fig. 2a indicating PCL-Br was well prepared using the enzyme catalyst. PGMA (Mn = 15000, PDI = 1.69) was also synthesized and characterized as the control as shown in Fig. 2b and showed peaks at 1729 cm−1, 1253 cm−1 and 1147 cm−1 when compared with PCL-Br (Fig. 2a). Moreover, PCL-b-PGMA with different molecular weights was synthesized and the FT-IR spectrum of the block polymer PCL-b-PGMA (Fig. 2c) was also shown to further confirm the structure. The appearance of a peak for vOH at 1120 cm−1 (Fig. 2d) and the disappearance of the peaks of the epoxy groups, at 900 cm−1 and 845 cm−1 showed that the epoxy groups were almost completely opened by nucleophilic reagent EA.
O) at 1730 cm−1 and 1188 cm−1 in Fig. 2a indicating PCL-Br was well prepared using the enzyme catalyst. PGMA (Mn = 15000, PDI = 1.69) was also synthesized and characterized as the control as shown in Fig. 2b and showed peaks at 1729 cm−1, 1253 cm−1 and 1147 cm−1 when compared with PCL-Br (Fig. 2a). Moreover, PCL-b-PGMA with different molecular weights was synthesized and the FT-IR spectrum of the block polymer PCL-b-PGMA (Fig. 2c) was also shown to further confirm the structure. The appearance of a peak for vOH at 1120 cm−1 (Fig. 2d) and the disappearance of the peaks of the epoxy groups, at 900 cm−1 and 845 cm−1 showed that the epoxy groups were almost completely opened by nucleophilic reagent EA.
3.2 Kinetic study of the polymerization of GMA using the macroinitiator under DhHP-6 catalysis
To gain further insight into the mechanism of the polymerization, the kinetics of the polymerization under DhHP-6 catalysis were investigated. Due to the ATRP being a controlled/“living” radical polymerization (CRP) technique, we studied the relationships between the reaction time and ln([M]0/[M]) and the monomer conversion with the number of average molecular weight (Mn) and polydispersity (PDI). From Fig. 3a, the resulting semilogarithmic kinetic plot of ln([M]0/[M]) vs. reaction time changed linearly during the reaction, which was in accordance with the ATRP features and the conversion ranged from 27.75% to 63.69%. The first time point of number average molecular weight of the block polymer was 7.7 kg mol−1 with a low PDI of 1.18 (Fig. 3b) and the molecular weight increased linearly with the monomer conversion during the entire polymerization reaction, which reached 24.5 kg mol−1 in 2 h; selected GPC traces are shown in Fig. S4.†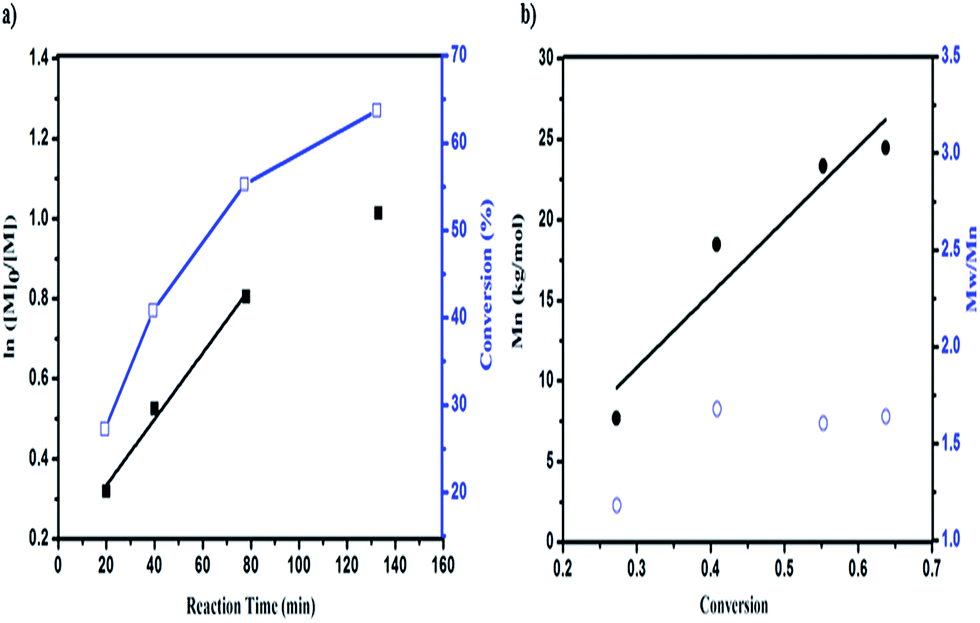 | ||
| Fig. 3 The DhHP-6 catalyzed AGET ATRP of GMA using the macroinitiator PCL-Br. (a) First-order kinetic plots for semilogarithmic kinetic plot of ln([M]0/[M]) (■) and conversion (□) as a function of reaction time. (b) Mn (●) and Mw/Mn (○) as a function of the monomer conversion. Note: [M]:[I]:[C]:[Asc] = 124:1:1:0.5. | ||
However, due to the poor solubility and low deactivation rate of hemin in organic solvents, only poor control could be achieved.38 Accordingly, we constructed a block polymer (PCL-b-PGMA) via an activator generated by electron transfer (AGET) ATRP process mediated by DhHP-6. In addition, a small amount of reducing agent, such as ascorbic acid (Asc), created the possibility of an ATRP process catalyzed by Fe(III), which could start the polymerization reaction and allow it to tolerate a limited amount of air in the system.37
Although the PDI was in the range from 1.18 to 1.64, this ATRP process could be controlled through enzyme self-deactivation by the approach of adding an enzyme mimic catalyst in an equal molar ratio to the initiator, which removed the need to add KBr into the reaction system in order to enhance the control in the aqueous system.18 Therefore, GMA can be polymerized by PCL-Br in a relatively well-controlled fashion mediated by an enzyme mimic (DhHP-6). Significantly, employing a low toxic catalyst, which expands the material's use in biomedical applications such as in gene delivery, may show better properties.
3.3 The block polymer employed as a polymer gene vector
Condensing pDNA into small polyplexes using a cationic polymer is a prerequisite for delivering genes to target cells. Polymer/DNA complexes are formed by electrostatic interactions and hydrophobic interactions, which were characterized through gel retardation (Fig. 4). Both PCL-b-PGEA60 and PCL-b-PGEA142 exhibited a high ability to package pDNA with complete condensation at N/P = 0.75, suggesting a good binding ability.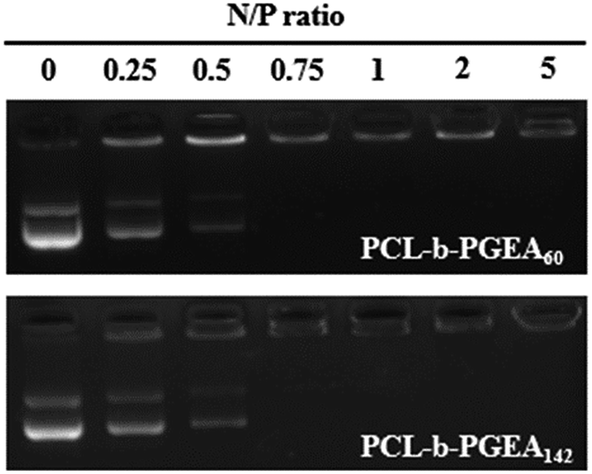 | ||
| Fig. 4 Agarose gel electrophoreses assay of PCL-b-PGEA60 and PCL-b-PGEA142. | ||
The particle sizes and surface zeta potential after packing pDNA were also evaluated by dynamic light scattering (DLS) analysis under serum free conditions (Fig. 5). It was reported that, due to enhanced permeability and retention effect (EPR),38 an appropriate particle size with a mild positive potential was beneficial for efficient transfection. The sizes of the polymer/pDNA complexes decreased upon increasing the w/w ratios, indicating that a larger amount of polycations can compact pDNA into tighter polyplexes with a diameter range from 200 nm to 300 nm, a suitable hydrodynamic size for endocytosis (Fig. 5a). The transmission electron microscopy (TEM) image of the polyplexes of PCL-b-PGEA142 at w/w = 10 is shown in Fig. S5.†
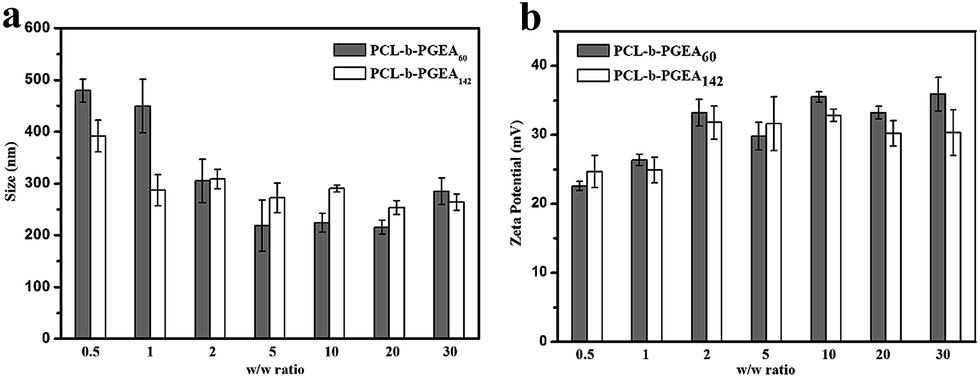 | ||
| Fig. 5 The particle size and surface zeta potential of PCL-b-PGEA60 and PCL-b-PGEA142 in serum free condition at various w/w ratios. Data are shown as the mean ± SD (n = 3). | ||
Moreover, a positive surface zeta potential will allow the vectors to bind to electronegative pDNA and attach to the cell membrane easily via electrostatic interactions. As presented in Fig. 5b, the polymer complexes at all the w/w ratios have a positive potential, which is greater than 20 mV and stabilized at 35 mV, and may exhibit good cellular uptake due to the negative charge of the cell membrane. In addition, the hydroxyl groups outside the complexes may construct a loose structure with a limited affinity to pDNA through electrostatic interactions and may release pDNA in a more elegant way when compared to strong binding affinity polycations, such as PEI.32 The hydroxyl barrier may also coat on the surface of complexes and play a vital role to dilute the charges in order to reduce serum adsorption.
The cell viability as a function of various w/w ratios is an important factor to evaluate a gene vector. As presented in Fig. 6, the MTT assay illustrated the cytotoxicity increased upon increasing the w/w ratio and accordingly, the cytotoxicity was dose-dependent. As reported, the small molecular weight vector (PCL-b-PGEA60) exhibits a relatively lower cytotoxicity than the larger one (PCL-b-PGEA142). PEI 25 kDa was also evaluated at w/w = 3, and the cytotoxicity was found to be larger than that of PCL-b-PGEA at almost all the w/w ratios studied, demonstrating that this type of vector synthesized by enzyme catalyzed shows low cytotoxicity.
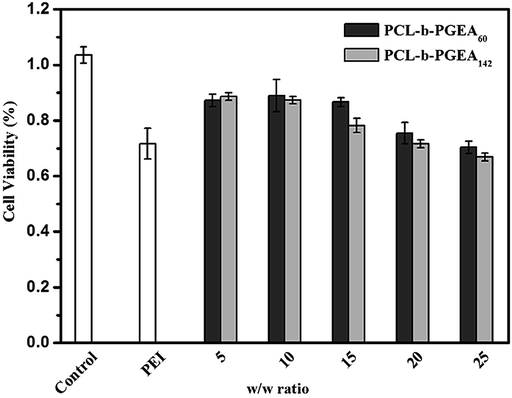 | ||
| Fig. 6 The cell viability of the polymer/pDNA complexes at different w/w ratios for PCL-b-PGEA60 and PCL-b-PGEA142, respectively PEI 25 kDa was used as the control at w/w = 3. | ||
An in vitro gene transfection assay was performed in serum-conditioned 293T cells using green fluorescent protein (GFP) as the gene reporter to visualize the gene expression under fluorescence microscopy. Fig. 7 shows the gene transfection efficiency of PCL-b-PGEA60 and PCL-b-PGEA142 at different w/w ratios, which were compared with PEI 25 kDa at an optical w/w ratio of 3. A strongest fluorescence signal was observed for PCL-b-PGEA60 at a w/w ratio of 25 and for PCL-b-PGEA142 at a w/w ratio of 10; however, PCL-b-PGEA60 only exhibited a very low gene transfection efficiency at w/w = 10. The results reflect that the high molecular weight polycation vector has a stronger ability to condense pDNA into a small complex, which exhibits a higher gene efficiency at the same N/P ratio. The gene transfection efficiency decreased as the w/w ratio was increased due to the higher cytotoxicity at high w/w ratios. As a result, a successful gene vector must exhibit a high gene transfection efficiency with minimum cytotoxicity. Moreover, flow cytometry analysis quantified the serum-conditioned gene transfection efficiency in 293T cells at optical w/w ratios of the two gene vectors in comparison with that of PEI 25 kDa (Fig. S4†).
 | ||
| Fig. 7 GFP expression under fluorescence microscopy mediated by PCL-b-PGEA60 and PCL-b-PGEA142 at various w/w ratios in 293T cells, PEI 25 kDa was used as control at w/w = 3. | ||
4. Conclusion
In conclusion, by combining enzymatic ROP with enzymatic ATRP processes, amphiphilic block polymers were successfully synthesized using a macroinitiator (PCL-Br). Kinetic studies showed that comparative control can be observed during the enzymatic ATRP process. To the best of our knowledge, it is also the first time that materials synthesized using ATRPase have been used in the biomedical field, which can act as an efficient non-viral gene vector for gene delivery in serum-conditioned 293T cells. When compared with PEI, the higher gene transfection efficiency under fluorescence microscopy and lower cytotoxicity proved that this ‘green method’ for polymer construction is promising and attractive. We believe that this method will make a good basis for synthesizing biomaterials mediated by enzymes or enzyme mimics.Acknowledgements
This research was supported by the National Science Foundation of China (No. 21074042, 50773028).Notes and references
- K. Matyjaszewsk and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T.-K. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- G. Barre, D. Taton, D. Lastecoueres and J.-M. Vincent, J. Am. Chem. Soc., 2004, 126, 7764–7765 CrossRef CAS PubMed.
- K. Matyjaszewski, T. Pintauer and S. Gaynor, Macromolecules, 2000, 33, 1476–1478 CrossRef CAS.
- J. Gromada and K. Matyjaszewski, Macromolecules, 2001, 34, 7664–7671 CrossRef CAS.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Y. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- L. Mueller, W. Jakubowski, W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 6464–6472 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS PubMed.
- H. C. Dong and K. Matyjaszewski, Macromolecules, 2008, 41, 6868–6870 CrossRef CAS.
- K. Min, H. F. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- Y.-H. Ng, F. D. Lena and C. L. L. Chai, Polym. Chem., 2011, 2, 589–594 RSC.
- Y.-H. Ng, F. D. Lena and C. L. L. Chai, Chem. Commun., 2011, 47, 6464–6466 RSC.
- B. Kalra and R. A. Gross, Biomacromolecules, 2000, 1, 501–505 CrossRef CAS PubMed.
- A. Simakova, M. Mackenzie, S. E. Averick, S. Park and K. Matyjaszewski, Angew. Chem., Int. Ed., 2013, 52, 12148–12151 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 581–593 CrossRef CAS PubMed.
- A. Aied, U. Greiser, A. Pandit and W. X. Wang, Drug Discovery Today, 2013, 18, 1090–1098 CrossRef CAS PubMed.
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671–681 CrossRef CAS PubMed.
- S. K. Samal, M. Dash, S. V. Vlierberghe, D. L. Kaplan, E. Chiellini, C. V. Blitterswijk, L. Moronid and P. Dubruel, Chem. Soc. Rev., 2012, 41, 7147–7194 RSC.
- F. J. Xu, M. Y. Chai, W. B. Li, Y. Ping, G. P. Tang, W. T. Yang, J. Ma and F. S. Liu, Biomacromolecules, 2010, 11, 1437–1442 CrossRef CAS PubMed.
- M. Benaglia, A. Alberti, L. Giorgini, F. Magnonia and S. Tozzia, Polym. Chem., 2013, 4, 124 RSC.
- H. Wei, L. R. Volpatti, D. L. Sellers, D. O. Maris, I. W. Andrews, A. S. Hemphill, L. W. Chan, D. S. H. Chu, P. J. Horner and S. H. Pun, Angew. Chem., Int. Ed., 2013, 52, 5377–5381 CrossRef CAS PubMed.
- R.-Q. Li, Y. N. Wu, Y. Zhi, X. C. Yang, Y. L. Li, J. Du and F.-J. Xu, Adv. Mater., 2016, 28, 7204–7212 CrossRef CAS PubMed.
- X.-C. Yang, Y.-L. Niu, N.-N. Zhao, C. Mao and F.-J. Xu, Biomaterials, 2014, 3873–3884 CrossRef CAS PubMed.
- R.-Q. Li, H.-Q. Song and F.-J. Xu, Polym. Chem., 2015, 6, 6208–6218 RSC.
- H. Hu, H.-Q. Song, B.-R. Yu, Q. Cai, Y. Zhu and F.-J. Xu, Polym. Chem., 2015, 6, 2466–2477 RSC.
- Y. J. Yang, Y. Qi, M. Zhu, N. N. Zhao and F. J. Xu, Nano Res., 2016, 9, 2531–2543 CrossRef CAS.
- X.-H. Luo, F.-W. Huang, S.-Y. Qin, H.-F. Wang, J. Feng, X.-Z. Zhang and R.-X. Zhuo, Biomaterials, 2011, 32, 9925–9939 CrossRef CAS PubMed.
- H. Zhou, W. Jiang, N. An, Q. P. Zhang, S. D. Xiang, L. P. Wang and J. Tang, RSC Adv., 2015, 5, 42728–42735 RSC.
- H. Zhou, X. Wang, J. Tang and Y.-W. Yang, Polymers, 2016, 8, 277–287 CrossRef.
- W. Jakubowski, J.-F. Lutz, S. Slomkowski and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1498–1510 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139–4146 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Macromol. Rapid Commun., 2011, 32, 1710–1715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28650j |
| This journal is © The Royal Society of Chemistry 2017 |